

Abstract
Purpose
Cholangiocarcinoma is a fatal tumor with limited therapeutic options. We have reported that calmodulin antagonists tamoxifen and trifluoperazine induced apoptosis in cholangiocarcinoma cells. Here, we determined the effects of tamoxifen on tumorigenesis and the molecular mechanisms of tamoxifen-induced apoptosis.
Experimental Design
Nude mice xenograft model of cholangiocarcinoma was used and tamoxifen was given i.p. and intratumorally. Cholangiocarcinoma cells were used to characterize molecular mechanisms of tamoxifen-induced apoptosis in vitro.
Results
I.p. or intratumoral injection of tamoxifen decreased cholangiocarcinoma tumorigenesis by 40% to 80% in nude mice. In cells isolated from tumor xenografts, tamoxifen inhibited phosphorylation of AKT (pAKT) and cellular FLICE like inhibitory protein (c-FLIP). Immunohistochemical analysis further showed that pAKT was identified in all nontreated tumors but was absent in tamoxifen-treated tumors. In vitro, tamoxifen activated caspase-8 and caspase-10, and their respective inhibitors partially blocked tamoxifen-induced apoptosis. Overexpression of c-FLIP inhibited tamoxifen-induced apoptosis and enhanced tumorigenesis of cholangiocarcinoma cells in nude mice, whereas deletion of the calmodulin-binding domain on c-FLIP restored the sensitivity to tamoxifen and inhibited tumorigenesis. With two additional cholangiocarcinoma cell lines, we confirmed that the expression of FLIP is an important factor in mediating spontaneous and tamoxifen-induced apoptosis.
Conclusions
Thus, tamoxifen inhibits cholangiocarcinoma tumorigenesis in nude mice. Tamoxifen-induced apoptosis is partially dependent on caspases, inhibition of pAKT, and FLIP expression. Further, calmodulin-FLIP binding seems to be important in FLIP-mediated resistance to tamoxifen. Therefore, the present studies support the concept that tamoxifen may be used as a therapy for cholangiocarcinoma and possibly other malignancies in which the calmodulin targets AKT and c-FLIP play important roles in the tumor pathogenesis.
Cholangiocarcinoma is a highly malignant neoplasm originating from cholangiocytes of the intrahepatic and/or extrahepatic biliary system. Extensive surgical resection is the only effective therapy available for long-term survival as conventional chemotherapy and radiotherapy are generally ineffective. The overall survival following diagnosis of unresectable cholangiocarcinoma is <12 months (1) and the 5-year survival for the patients undergoing surgical resection ranges from 0% to 40% (2). Over the last few years, there has been a marked increase in the incidence and mortality from cholangiocarcinoma, warranting an increasing need for effective strategies to prevent and treat this lethal tumor (3). In search of an efficient chemotherapy for cholangiocarcinoma, we identified that tamoxifen, a potent calmodulin antagonist (4, 5), induces apoptosis in cholangiocarcinoma cells in vitro and inhibits tumorigenesis in vivo in nude mice xenografts (6). We showed that the apoptosis-inducing abilities of tamoxifen in cholangiocarcinoma cells seem to be associated with its role as a calmodulin antagonist (7). In the present studies, we further determined the molecular mechanisms responsible for tamoxifen-induced apoptosis in cholangiocarcinoma and its therapeutic potential in nude mice xenografts.
Tamoxifen was first identified as an antifertility drug in the early 1960s. It was approved in the United Kingdom, for the first time, as an anticancer agent (8). Today, it is one of the most widely used anticancer drugs worldwide, is relatively cheap, and is reported to have few side effects (9, 10). Over the last two decades, substantial effort has been focused on characterizing the molecular mechanisms behind the effects of tamoxifen, including tamoxifen-induced apoptosis. Various pathways such as protein kinase C, TGF-β, AKT, MAPK, c-myc, and calmodulin have been implicated in tamoxifen-induced effects on human cancer cell lines (11). Further, it is now clear that tamoxifen can activate genomic (estrogen receptor–mediated) as well as nongenomic (estrogen receptor–independent) pathways in dose-dependent and cell type–specific manners (4, 11, 12). Our laboratory has been focusing on the calmodulin-dependent aspect of tamoxifen biology in a cholangiocarcinoma tumor model using a human cholangiocarcinoma cell line that lacks estrogen receptors (7). We have reported previously that calmodulin antagonists, tamoxifen and trifluoperazine, induced apoptosis in cholangiocarcinoma cells in a Fas-related manner (7, 13). In this report, we further characterized the modulation of calmodulin signaling by tamoxifen in both cholangiocarcinoma cells in vitro and a mouse xenograft model in vivo.
Calmodulin is an important Ca2+-binding protein that has been well conserved through evolution and mediates various signaling pathways induced by Ca2+ (14, 15). Calmodulin interacts with a variety of proteins in Ca2+-dependent and Ca2+-independent manners (16). With increases in local Ca2+ concentration, it undergoes a conformational change and interacts with its target proteins such as calmodulin-dependent kinase II, calcineurin, and nitric oxide synthase to mediate various downstream Ca2+ signaling events (17, 18). An increase in Ca2+ is shown to be important in cellular proliferation as well as apoptosis, and calmodulin is believed to be important in mediating these signaling events (14, 19). Among the wide variety of proteins interacting with calmodulin, we identified that two prosurvival molecules, AKT and cellular FLICE like inhibitory protein (c-FLIP), are regulated by tamoxifen in cholangiocarcinoma culture and tumor xenografts.
c-FLIP is largely described as an antiapoptotic protein that interferes with activation of initiator caspases, thus inhibiting the downstream apoptotic signaling (20, 21). It is overexpressed in various cancer cell lines and is associated with resistance to chemotherapeutic agents (22–24). Recently, we identified a direct Ca2+-dependent interaction between calmodulin and FLIP that is regulated by changes in intracellular Ca2+ levels (25). AKT, also called protein kinase B, is another important prosurvival molecule that has been shown to interact with calmodulin, possibly facilitating its translocation to the plasma membrane (26). Classically, AKT signaling is initiated by binding of a growth factor to a receptor tyrosine kinase, leading to activation of phosphatidylinositol 3-kinase (27–29). Activated phosphatidylinositol 3-kinase leads to the generation of phosphatidylinositol 3,4,5-triphosphate (PIP3), which then interacts with AKT and anchors the AKT kinase to the plasma membrane where it undergoes phosphorylation and activation by various kinases (30, 31). Thus, translocation of AKT to the plasma membrane is an important step in its activation. Calmodulin was shown to compete with PIP3 for binding with AKT in a Ca2+-dependent manner such that elevations in Ca2+ concentrations disrupted the interaction between AKT and phosphoinositides (26).
In the investigations presented here using cholangiocarcinoma as a model, we showed that tamoxifen, as a potent calmodulin antagonist, inhibits phosphorylation of AKT and the expression of c-FLIP. Furthermore, tamoxifen is effective in decreasing tumor size in a mouse xenograft model probably by modulating calmodulin/FLIP signaling pathways.
Materials and Methods
Cell culture, antibodies, and reagents
The cholangiocarcinoma cell line Sk-ChA-1 was kindly provided by Dr. A. Knuth (Ludwig Institute for Cancer Research, London, United Kingdom). Stable cholangiocarcinoma clones overexpressing wild-type and mutant FLIP with deletion of the calmodulin-binding domain (ΔFLIP) were generated using lentiviral FLIPL expression vector as described previously (25). ΔFLIP protein was generated by deleting the calmodulin-binding region (amino acids 197–213) from FLIPL as described previously (25). Cells were grown in RPMI 1640 (Invitrogen) supplemented with penicillin (5 units/mL), streptomycin (5 μg/mL), and 10% heat-inactivated fetal bovine serum. Cholangiocarcinoma cell lines MZ-ChA-1 and HuCCT-1 were kindly provided by Dr. Gregory J. Gores (Mayo Clinic, Rochester, MN) and maintained as previously described (32, 33).
Antibodies include anti-FLIP, NF6 (Alexis Corp.), anti–caspase-8, anti–caspase-10, anti–caspase-3, anti–caspase-9, anti-AKT, anti–phospho-AKT (Cell Signaling), and anti–glyceraldehyde-3-phosphate dehydrogenase (Research Diag., Inc.). The monoclonal antibody to calmodulin was developed as described previously (34). Anti–vascular endothelial growth factor (VEGF)-A and VEGF-C antibodies, as well as horseradish peroxidase–conjugated goat anti-mouse and bovine anti-rabbit IgG antibodies were purchased from Santa Cruz Biotechnology, Inc.
Mouse xenograft model
The animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham, Birmingham, AL. Eight-week-old athymic (nu/nu) BALB/c mice (both male and female, Charles River Laboratories) were used for tumor inoculation. Briefly, cholangiocarcinoma cells (5 × 106 in 200 μL PBS per site) were inoculated s.c. into the flanks of mice. Tamoxifen was purchased from Calbiochem. After 1 wk, mice were randomly assigned into two groups. Tumors in one group were treated with tamoxifen (15 μmol/L in 100 μL volume per tumor site) injections intratumorally and tumors in the other group were injected with PBS as control. For i.p. injections, 0.1 mg tamoxifen in 0.1 mL oil was given i.p. for 3 consecutive days followed by 1 d of rest for 2 wk. Tumors were measured every 3 to 4 d and volumes were determined using the formula volume = length × width2/2.
Culture of xenograft tumor-derived cells
Tumors were removed from mice aseptically, minced into pieces, and placed in growth medium containing collagenase I for 30 min at 37°C followed by mashing through a cell strainer (100 μmol/L, BD Pharmingen). The cell suspension was washed twice with PBS and cultured in RPMI 1640.
Immunohistochemical analysis
Tumors were placed in buffered formalin and embedded in paraffin. Slides were deparaffinized, rehydrated, and heated in 10 μmol/L citrate buffer (pH 6.0) for 40 min using a steamer. The slides were blocked with 10% normal rabbit serum for 30 min, washed with PBS, and incubated with anti–phosporylated-AKT-1 antibody (Santa Cruz Biotechnology) for 1 h at room temperature. Biotinylated anti-rabbit IgG was applied for 30 min followed by 30 min of incubation with a polymer (Envision plus, DAKO). Slides used as negative controls did not receive the incubation with primary antibodies. After rinsing, slides were treated with diaminobenzidine chromogen solution and counterstained with routine H&E. Brown staining in >10% of the cells was considered a positive stain.
Assessment of apoptosis
Annexin V–FITC and propidium iodide staining was done using an apoptosis detection kit (BD Biosciences) and analyzed by flow cytometry. For studies involving caspase inhibitors, cells were pretreated with caspase inhibitor for 2 h before addition of tamoxifen. The percentage of cells that were Annexin V positive and propidium iodide negative were considered to be apoptotic cells.
Western blot analysis
Protein extracts were isolated from cells using lysis buffer containing 100 mmol/L Tris-HCl (pH 8.0), 150 mmol/L NaCl, 1% SDS, 10% glycerol, 1% Triton X, 5 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 10 mmol/L sodium fluoride, 1 mmol/L sodium orthovanadate, 1 mmol/L β-glycerophosphate, and protease inhibitor cocktail tablets (Roche). Concentrations of protein were determined with the bicinchoninic acid kit (Sigma). Proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane as described previously (35). For calmodulin, the membranes were fixed in 0.2% glutaraldehyde in TBS for 30 min before the blocking step. Membranes were incubated with primary antibodies overnight at 4°C. Horseradish peroxidase–conjugated secondary antibodies in the blocking buffer were incubated for 2 h at room temperature. Signals were detected using Immobilon Western chemiluminescent horseradish peroxidase substrate (Millipore) detection kit.
Thiazolyl blue tetrazolium bromide assay
Proliferation of cholangiocarcinoma cells in culture was analyzed using thiazolyl blue tetrazolium bromide reagent (Sigma-Aldrich) for a 4-d period. Briefly, 10,000 cells were seeded in 200 μL of complete RPMI 1640 per well in a 96-well plate. Thiazolyl blue tetrazolium bromide was added on days 0 (after allowing the attachment of cells overnight), 2, and 4 to the final concentration of 0.5 mg/mL and incubated at 37°C for 2 h. The medium was removed and the crystals were dissolved in DMSO followed by measuring the absorbance at 570 nm.
Statistical analysis
Results are expressed as means ± SE. Differences between two groups were identified with Student’s t tests. Significance was defined as P < 0.05.
Results
Tamoxifen inhibited tumorigenesis of cholangiocarcinoma cells and phosphorylation of AKT in nude mice xenografts
We have reported that calmodulin antagonists tamoxifen and trifluoperazine induce apoptosis in human cholangiocarcinoma cells in vitro in a Fas-related manner and probably through calmodulin-dependent pathways (7). To test its efficacy in vivo, we injected tamoxifen i.p. (0.1 mg/mouse) in nude mice bearing cholangiocarcinoma xenografts and found that it reduced tumor size by 82% after 2 weeks as shown in Fig. 1A [tumor volume (mm3), control = 208 ± 27, tamoxifen = 37 ± 5, n = 6, P < 0.0005]. Similar studies with i.p. tamoxifen injections over 6 weeks consistently showed that the cholangiocarcinoma xenografts were significantly reduced in size with tamoxifen treatment (6). In addition, the effects of tamoxifen (daily intratumoral injection, 100 μL of 15 μmol/L stock) on the tumor progression were monitored for 27 days. The tumor volumes were measured every 3 to 4 days and mice were sacrificed after 4 weeks of treatment. As shown in Fig. 1B, tamoxifen inhibited tumor growth, resulting in a significant decrease in tumor size compared with control group injected with PBS (at the end point, control = 523 ± 58 mm3 and tamoxifen = 312 ± 22 mm3, n = 10, P = 0.009). The inset in Fig. 1B shows a representative photograph of the cholangiocarcinoma xenograft growth with PBS (control) and tamoxifen treatment after 4 weeks. Thus, both i.p. as well as intratumoral injections of tamoxifen were found to be effective in reducing cholangiocarcinoma xenografts growth in nude mice model.
Fig. 1.

Tamoxifen inhibits cholangiocarcinoma tumorigenesis and phosphorylation of AKT in nude mice. Cholangiocarcinoma cells (SK-ChA-1) were inoculated s.c. into the flanks of 8-week-old athymic (nu/nu) BALB/c mice. After 6 d, mice were randomly assigned into two groups. A, 0.1mL oil as a control or 0.1mg tamoxifen (TMX) in 0.1mL oil was given i.p. in each mouse daily as described in Materials and Methods and tumors were measured after 1wk of treatment. Columns, mean (n = 6, P < 0.05); bars, SE. B, tumors in one group were injected with tamoxifen (100 μL tamoxifen stock of 15 μmol/L) and tumors in the other group were injected with PBS as control. Points, mean tumor volume (n = 10, *P < 0.05); bars, SE. Inset, picture of tumor-bearing mice in the control (left) and tamoxifen-treated (right) groups; arrows, tumors. C, H&E staining of a section from a PBS-treated (a) and a tamoxifen-treated tumor (b); immunohistochemical staining of pAKTon a PBS-treated (c) and a tamoxifen-treated tumor section (d; magnification, ×400).
We have previously showed that AKT signaling plays an important role in cholangiocarcinoma pathogenesis (35). Therefore, we characterized the effects of tamoxifen on phosphorylation of AKT in xenografts that were treated with tamoxifen or PBS. As shown in Fig. 1C, a and b, H&E stain for control and tamoxifen-treated tumors show the tumor cells arranged in a glandular pattern. The cells in the control section are dysplastic, showing enlarged nuclei with altered nucleocytoplasmic ratio and showed prominent nucleoli. The tamoxifen-treated cells are less dysplastic. Previously, we reported that phosphorylation of AKT was increased in human cholangiocarcinoma tumors but not in adjacent non-neoplastic bile duct epithelium (35). Consistently, immunohistochemical analysis of mice xenografts showed that pAKT, seen predominantly in the cytoplasm as brown stain, was detected in PBS-treated tumors (Fig. 1C, c) but not in tamoxifen-treated tumors (Fig. 1C, d). Further, 27 ± 11% cells were found to stain positive for pAKT in PBS-treated group whereas tamoxifen-treated tumors did not show any staining for pAKT (n = 7). Thus, tamoxifen inhibited phosphorylation of AKT in cholangiocarcinoma xenografts in nude mice.
Tamoxifen inhibited phosphorylation of AKT and FLIP expression in tumor cells from mouse cholangiocarcinoma xenografts
To further characterize the effects of tamoxifen on cholangiocarcinoma, cells were isolated from cholangiocarcinoma xenografts. As expected, tamoxifen induced apoptosis in tumor cells from mouse xenografts (Fig. 2A). Western blot analysis of lysates from these cells further confirmed that tamoxifen inhibited phosphorylation of AKT and expression of FLIP in these cells but the expression of calmodulin and Fas was unaffected by tamoxifen as shown in Fig. 2B. Thus, inhibition of pAKT was observed in vitro as well as in vivo in cholangiocarcinoma xenografts in nude mice.
Fig. 2.

Apoptosis in response to tamoxifen in tumor cells isolated from cholangiocarcinoma xenografts from nude mice. Cells from cholangiocarcinoma xenografts were treated with or without tamoxifen (10 μmol/L) for 24 h. A, apoptosis was determined by AnnexinV/propidium iodide staining. Columns, mean of five independent experiments done in duplicate; bars, SE. B, Western blot analysis for calmodulin, Fas, FLIP, AKT, and phospho-AKT. Tamoxifen treatment decreased phospho-AKT and FLIP expression but did not affect CaM and Fas levels. Representative blot from three identical experiments.
Tamoxifen-mediated apoptosis involves activation of caspase-10, caspase-8, caspase-9, and caspase-3 and inhibition of FLIP expression in cholangiocarcinoma cells
We determined the involvement of caspases, including caspase-10, caspase-8, caspase-3, and caspase-9, in tamoxifen-induced apoptosis. As seen in Fig. 3A, tamoxifen induced a 3.9-fold increase in apoptosis in vitro in cholangiocarcinoma cells compared with control. It activated caspase-10 and caspase-8, both of which are initiator caspases as seen in Fig. 3B. Further, consistent with our previous reports, tamoxifen also activated caspase-9 and caspase-3 (13). By contrast, an antiapoptotic protein, c-FLIP, was down-regulated by tamoxifen. We also characterized the effect of tamoxifen in two additional cholangiocarcinoma cell lines, MZ-ChA-1 and HuCCT-1 (32, 33). We found that >24% of HuCCT-1 underwent spontaneous apoptosis, compared with 7.7% of MZ-ChA-1, after 24 hours of incubation. However, tamoxifen induced the apoptosis of the MZ-ChA-1 cell by 3-fold, but did not affect the apoptosis of HuCCT-1 cells (Fig. 3C). We further determined the expression and activation of FLIP, AKT, and caspases in MZ-ChA-1 and HuCCT-1 cells (Fig. 3D). The expression of pAKT is higher in the MZ-ChA-1 cells compared with that in HuCCT-1 cells under basal conditions (Fig. 3D). Inhibition of pAKT by tamoxifen was shown in the MZ-ChA-1 cells, but not HuCCT-1 cells. Tamoxifen inhibited FLIP expression in MZ-ChA-1 cells. The expression of FLIP was not detectable in the HuCCT-1 cells (Fig. 3D). Activation of caspase-8, caspase-9, and caspase-10 was induced by tamoxifen in MZ-ChA-1 cells, but not HuCCT-1 cells. In HuCCT-1 cells, partial activation of the caspases was shown in the control cells, which is consistent with the high levels of basal apoptosis in these cells. Taken together, our observations showed that the expression of pAKT and FLIP regulates cholangiocarcinoma cell survival and tamoxifen executes its apoptotic effect on cholangiocarcinoma cells through the calmodulin/FLIP axis and activation of caspases.
Fig. 3.

Analysis of proapoptotic and antiapoptotic markers in response to tamoxifen treatment of cholangiocarcinoma cells. Cholangiocarcinoma cells (SK-ChA-1) were treated with 20 μmol/L tamoxifen for 24 h. A, apoptosis was analyzed by AnnexinV/propidium iodide staining and expressed as fold difference compared with control. Columns, mean (n = 6, P < 0.05); bars, SE. B, Western blot analysis: activation of caspase-8, caspase-10 (seen as a decrease in precursor form), caspase-3, and caspase-9; inhibition of phospho-AKT; and c-FLIP down-regulation. Cholangiocarcinoma cells MZ-ChA-1and HuCCT-1were treated with 20 μmol/L tamoxifen for 24 h. C, apoptosis was analyzed by Annexin V/propidium iodide staining and expressed as percentage of Annexin V – positive and propidium iodide – negative staining cells. Columns, mean (n = 3, P < 0.05); bars, SD. D, Western blot analysis of AKT, phospho-AKT, and c-FLIP as well as caspase-8, caspase-9, and caspase-10.
Caspase inhibitors partially blocked tamoxifen-induced apoptosis
To determine the requirement of caspase-8 and caspase-10 in tamoxifen-induced apoptosis, cholangiocarcinoma cells were pretreated with the respective caspase inhibitors for 2 hours followed by tamoxifen treatment for 24 hours. As shown in Fig. 4A and C, tamoxifen-induced apoptosis was partially blocked by both caspase-8 (43 ± 2%) and caspase-10 (36 ± 5%) inhibitors. Further, caspase-3 activation was blocked by both of these inhibitors, suggesting that both caspase-8 as well as caspase-10 are required for tamoxifen-induced caspase-3 activation in cholangiocarcinoma cells. Western blot analysis, shown in the right of Fig. 4B and D, shows that caspase-8 and caspase-10 inhibitors effectively blocked tamoxifen-induced activation of the respective caspases. Further, we confirmed that the pan-caspase inhibitor, zVAD, also caused partial inhibition of tamoxifen-induced apoptosis as shown previously (13), suggesting that tamoxifen-induced apoptosis is only partially caspase dependent. However, the caspase inhibitors may not be entirely specific and the specificity can be diminished with increased concentrations and prolonged incubation times (36, 37). Nevertheless, it can be concluded from these studies using caspase inhibitors that tamoxifen induces apoptosis in caspase-dependent and caspase-independent manners.
Fig. 4.
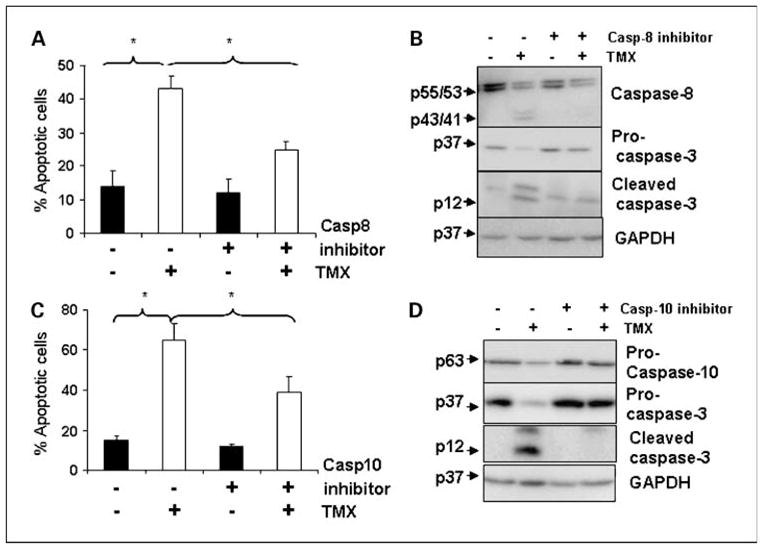
Effect of caspase-8 and caspase-10 inhibitors on tamoxifen-induced apoptosis in cholangiocarcinoma cells. Cholangiocarcinoma cells were pretreated with or without (A) caspase-8 (50 μmol/L) and (C) caspase-10 inhibitors (100 μmol/L) for 2 h before addition of tamoxifen (20 μmol/L) for 24 h and apoptosis determined. B and D, the whole-cell lysates from these cells were analyzed by Western blot for caspase-8, caspase-10, caspase-3, c-FLIP, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Columns, mean (n = 3, *P < 0.05); bars, SE.
Role of FLIP and calmodulin/FLIP binding in cholangiocarcinoma tumorigenesis and tamoxifen-induced apoptosis
Increased expression of FLIP has been identified in numerous cancer cell lines and associated with resistant phenotypes of various cancers, including cholangiocarcinoma (38–41). We found that tamoxifen inhibited activation of AKT as well as FLIP expression (Figs. 2 and 3). AKT is a calmodulin-binding protein (25). Recently, we reported that calmodulin interacts directly in a Ca2+-dependent manner with c-FLIPL (25, 26). Thus, we asked two questions: (a) Does FLIP expression modulate the apoptotic response to tamoxifen? and (b) If so, is it dependent on FLIP binding with calmodulin? Cholangiocarcinoma cells stably overexpressing wild-type (WT) FLIPL, ΔFLIPL (mutant with deletion of calmodulin-binding domain) or LacZ (control) proteins were generated as described previously (25) and the effects of tamoxifen on apoptosis of these cells were determined by Annexin/propidium iodide staining. As shown in Fig. 5A, FLIP over-expression decreased the sensitivity of cholangiocarcinoma cells to tamoxifen, which was restored by deletion of calmodulin-binding domain from overexpressed FLIPL. The effects of WT and ΔFLIP overexpression on tamoxifen-induced apoptosis were confirmed by using another known calmodulin antagonist, W-7. Thus, WT FLIP overexpression decreased W-7–induced apoptosis, which was restored after deletion of the calmodulin-binding region from FLIPL. These results confirm that tamoxifen-induced apoptosis involves modulation of calmodulin signaling in cholangiocarcinoma (Fig. 5B). Further, as shown in Fig. 5C, disruption of calmodulin-FLIP binding also resulted in decreased cell proliferation compared with WT FLIP-expressing cholangiocarcinoma cells in which calmodulin-FLIP binding is intact. Thus, overexpression of FLIPL seems to be an important regulator of tamoxifen-induced apoptosis and calmodulin binding to FLIPL is critical for mediating the antiapoptotic/prosurvival effects of FLIP.
Fig. 5.

Role of FLIP and CaM/FLIP binding in cholangiocarcinoma tumorigenesis and tamoxifen-induced apoptosis. Wild-type FLIPL, ΔFLIPL, and LacZ proteins were overexpressed in cholangiocarcinoma cells. A, the cells were treated with 20 μmol/L tamoxifen for 24 h followed by staining with Annexin V/propidium iodide to assess apoptotic cell death. Columns, mean (n = 3); bars, SE. B, the cells were treated with the calmodulin antagonistW-7 (50 μmol/L) for 24 h followed by assessment of apoptotic cell death. Columns, mean (n = 4); bars, SE. C, the cells were seeded in a 96-well plate and analyzed for cell accumulation using thiazolyl blue tetrazolium bromide reagent, added on days 0, 2, and 4. Results are from three independent experiments done in quadruplet. D, 2 × 106 LacZ, WT FLIP, and ΔFLIP cells were injected in the flanks of 6- to 7-week-old nude mice and the tumor growth was followed over 18 d. The tumor volumes were calculated as described in Materials and Methods. Columns, mean (n = 6, *P < 0.05 compared with control in each group); bars, SE.
Tumorigenesis of WT- and ΔFLIP-overexpressing cholangiocarcinoma cells was characterized in a nude mice xenograft model. Cholangiocarcinoma cells expressing LacZ, WT FLIP, and ΔFLIP were injected in the flanks of 6- to 7-week-old athymic nude mice. The tumor volumes were measured every 3 to 4 days for 18 days. As shown in Fig. 5D, at day 7, no significant differences were observed in tumorigenesis of LacZ, WT, and ΔFLIP cells. WT FLIP cells formed significantly bigger tumors than control LacZ and ΔFLIP cells, whereas ΔFLIP cells formed the smallest tumors at day 18 (tumor volumes in mm3, control LacZ = 124 ± 12, WT FLIP = 170 ± 25, ΔFLIP = 82 ± 15). At day 14, ΔFLIP cells grew significantly smaller tumors than both LacZ and WT FLIP cells (LacZ = 99 ± 9, WT FLIP = 101 ± 16, ΔFLIP = 62 ± 11), supporting the concept that the interaction between calmodulin and FLIP is important in cholangiocarcinoma pathogenesis.
Discussion
Tamoxifen is an extensively tested and widely used drug worldwide. It is inexpensive and has a low-side-effect profile, thus providing a very attractive therapeutic option (9, 10). Cholangiocarcinoma, a highly lethal tumor arising from biliary epithelium, has a poor 5-year survival rate and no effective therapy (1, 2). With increasing incidence of this disease, it is imperative that novel strategies are identified to prevent and treat this deadly disease. Our group previously showed that tamoxifen induces apoptosis in cholangiocarcinoma cells, both in vitro and in vivo in a mouse xenograft model (6, 7, 13). Further, we showed that other calmodulin antagonists, W-7 and trifluoperazine, also induced apoptosis in human cholangiocarcinoma cells (7). In addition, the apoptosis-inducing abilities of calmodulin antagonists were found to correlate with the sensitivity of cholangiocarcinoma cells to Fas-mediated apoptosis. Cross-talk between Fas and calmodulin pathways was further substantiated by our identification of a direct binding between calmodulin and Fas receptor (42).
The apoptosis-inducing ability of tamoxifen in human cholangiocarcinoma cell lines seem to be estrogen receptor independent, as these cells lack estrogen receptors (7). In addition, we found that tamoxifen did not affect the expression of VEGF-A or VEGF-C in all three cholangiocarcinoma cell lines tested (data not shown), suggesting that tamoxifen may affect cholangiocarcinoma cell growth independent of modulating the angiogenic factors VEGF-A and VEGF-C, which have been well shown by Mancino et al. to regulate cholangiocarcinoma tumorigenesis in response to estrogen stimulation (43). We proposed that tamoxifen induces apoptosis in cholangiocarcinoma cells by modulating calmodulin signaling. Calmodulin is an important Ca2+-binding protein that binds with a variety of proteins in response to increases in intracellular Ca2+ and mediates a variety of signaling pathways ranging from proliferation, differentiation, and apoptosis (17, 18). In neocortical cell cultures, Ca2+-calmodulin was shown to be important in mediating cell survival signals of the brain-derived neurotrophic factor by affecting the phosphatidylinositol 3-kinase–AKT pathway (44). Mutants of calmodulin with disabled Ca2+-binding domains and calmodulin antagonists were shown to inhibit activation of AKT and significantly decreased long-term cell survival, suggesting a pivotal role for calmodulin in AKT-mediated cell survival. Furthermore, calmodulin has recently been shown to interact with AKT and the interaction is believed to modulate the membrane translocation of this kinase by interfering with the AKT-phosphoinositide binding that is important for membrane translocation and subsequent phosphorylation of AKT (26). Thus, calmodulin seems to modulate the AKT signaling pathway via its direct interaction with AKT. Consistently, in our studies, we have shown that the calmodulin antagonist tamoxifen inhibited the phosphorylation of AKT and resulted in increased apoptosis of cholangiocarcinoma cells. We also found that tamoxifen down-regulated the expression of an antiapoptotic molecule, c-FLIP, which we have recently shown to be a calmodulin-binding protein that regulates Fas-mediated apoptosis (25). Inhibition of pAKT has also been associated with decreased expression of c-FLIP in various tumors but the mechanism is not clearly understood (35, 45). It not known whether tamoxifen-mediated decreased expression of c-FLIP observed concurrently with inhibition of pAKT is regulated at the transcriptional level or at the protein level through proteosomal degradation. AKT and FLIP are both calmodulin-binding proteins; therefore, the apoptosis-inducing ability of tamoxifen is likely to be related to the modulation of calmodulin signaling pathways.
AKT is a prosurvival protein and it is up-regulated in several tumors (46, 47). We recently showed that AKT is activated in cholangiocarcinoma cells that are resistant to Fas-induced apoptosis and inhibition of AKT using dominant-negative AKT enhances the sensitivity of these cells to Fas-mediated apoptosis (35). Inhibition of pAKT has been linked to FLIP down-regulation in several cancer cell lines (35, 45). In this report, we found that the expression of pAKT and FLIP is critical for the spontaneous apoptosis of cholangiocarcinoma cells under basal condition (Fig. 3D). We also showed that FLIP expression is an important determinant of the response to tamoxifen, as FLIP-undetectable HuCCT-1 cells were not responsive to tamoxifen (Fig. 3C) and FLIP overexpression reduced the sensitivity of cholangiocarcinoma cells to tamoxifen (Fig. 5A). Thus, for the first time, our studies provide evidence that FLIP expression could be an important determinant of the response to chemotherapy in cholangiocarcinoma. Because tamoxifen-mediated inhibition of AKT and FLIP is related to the calmodulin signaling, the next logical step would be separately modulating AKT, calmodulin, or FLIP and assessing responses to tamoxifen. Because AKT and calmodulin are involved in the regulation of a variety of signaling pathways and cell processes, we decided to target FLIP, specifically the calmodulin-FLIP binding, to assess its effect on tamoxifen-mediated apoptosis. The expression of WT FLIP in cholangiocarcinoma cells decreased their sensitivity to tamoxifen-induced apoptosis, but when calmodulin-FLIP binding was compromised by overexpressing a FLIP mutant that lacked calmodulin-binding region, the sensitivity to tamoxifen was restored (Fig. 5A). These results were confirmed with another calmodulin antagonist, W-7 (Fig. 5B), further supporting the involvement of calmodulin in FLIP-mediated resistance to calmodulin antagonist–induced apoptosis. Further, disrupted calmodulin-FLIP binding decreased cell survival and decreased tumorigenicity in nude mice compared with WT FLIP–overexpressing cells with intact calmodulin-FLIP binding (Fig. 5C and D). Therefore, calmodulin-FLIP binding seems to be important in mediating prosurvival effects of c-FLIP and exerting resistance to tamoxifen in FLIP-overexpressing cholangiocarcinoma cells. Our data support the concept that tamoxifen alone, or coupled with a drug specifically targeting calmodulin-FLIP binding, could provide an effective combination therapy for lethal tumors in which FLIP expression is known to impair response to therapy.
With a variety of signaling pathways modulated by calmodulin and its recently described cross-talk with the Fas pathway, it seems to provide a potential therapeutic target for cholangiocarcinoma. Interestingly, calmodulin binding with both Fas and FLIP is Ca2+ dependent but their calmodulin binding sites are structurally different. Although calmodulin binding with Fas is mediated via a classic 1–5–10 calmodulin-binding motif (42), its binding with FLIP is mediated by a nonclassic motif (25). Thus, there may be differences in affinity between calmodulin-Fas and calmodulin-FLIP binding in basal and activated states. Calmodulin binding with these proteins may confer structural stability and/or conformational changes required for downstream signaling events. It is likely that targeting each of these specific interactions could provide efficient ways to modulate apoptotic sensitivity of cancer cells and novel targets in therapy.
We propose a model for tamoxifen-induced apoptosis in cholangiocarcinoma cells operating through calmodulin. As shown in Fig. 6, tamoxifen induces activation of caspase-8 and caspase-10, leading to activation of caspase-3 and at the same time it also inhibits prosurvival signals by inhibiting phosphorylation of AKT and down-regulating FLIP expression. Importantly, calmodulin has been shown to interact directly with Fas and thus the Fas-calmodulin interaction may compete with Fas-FADD interaction to interfere with the death-inducing signaling complex assembly. Further, calmodulin-FLIP binding adds another arm to this inhibitory effect of calmodulin on Fas-induced apoptosis by allowing increased FLIP recruitment into the death-inducing signaling complex that further interferes with caspase activation. Thus, tamoxifen, by virtue of its anticalmodulin properties, may be able to diminish the inhibitory effects of calmodulin on Fas-induced apoptosis and regulate the calmodulin signaling pathway via modulation of c-FLIP and AKT signaling to induce apoptosis and inhibit tumorigenesis in cholangiocarcinoma.
Fig. 6.

Proposed model of tamoxifen-induced apoptosis. A model depicting the regulation of calmodulin (CaM) and Fas signaling by tamoxifen. Calmodulin interacts with Fas, c-FLIP, and AKT. Tamoxifen interferes with these interactions (arrows) to promote apoptosis and inhibits prosurvival signals mediated by AKT and c-FLIP.⇢, activation; ⊣, inhibition.
Translational Relevance
The work presented here explores the potential use of tamoxifen as a therapy for cholangiocarcinoma, a fatal disease with very limited therapeutic options. Using a cholangiocarcinoma xenograft model in nude mice, we show that i.p. as well as intratumoralinjections of tamoxifen significantly decrease cholangiocarcinoma tumorigenesis in a nude mice model. Tamoxifen is a widely used, well-tolerated, and low-cost drug. The studies presented here highlight the molecular mechanisms involved in tamoxifen-induced apoptosis in vitro and confirm the therapeutic effect of tamoxifen in a xenograft model. Thus, these studies set the stage for a possible clinical trial of this drug in cholangiocarcinoma patients.
Acknowledgments
Grant support: VA Merit Review Award (J.M. McDonald).
We thank Marion Spell, Center for AIDS Research, University of Alabama at Birmingham, for technical assistance with flow cytometry.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Sirica AE. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 2.Gores GJ. Cholangiocarcinoma: current concepts and insights. Hepatology. 2003;37:961–9. doi: 10.1053/jhep.2003.50200. [DOI] [PubMed] [Google Scholar]
- 3.Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33:1353–7. doi: 10.1053/jhep.2001.25087. [DOI] [PubMed] [Google Scholar]
- 4.O’Brian CA, Ioannides CG, Ward NE, Liskamp RM. Inhibition of protein kinase C and calmodulin by the geometric isomers cis- and trans-tamoxifen. Biopolymers. 1990;29:97–104. doi: 10.1002/bip.360290114. [DOI] [PubMed] [Google Scholar]
- 5.McCague R, Rowlands MG, Grimshaw R, Jarman M. Evidence that tamoxifen binds to calmodulin in a conformation different to that when binding to estrogen receptors, through structure-activity study on ring-fused analogues. Biochem Pharmacol. 1994;48:1355–61. doi: 10.1016/0006-2952(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 6.Vickers SM, Jhala NC, Ahn EY, et al. Tamoxifen (TMX)/Fas induced growth inhibition of human cholangiocarcinoma (HCC) by γ interferon (IFN-γ) Ann Surg. 2002;235:872–8. doi: 10.1097/00000658-200206000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan G, Vickers SM, Pickens A, et al. Apoptosis and tumorigenesis in human cholangiocarcinoma cells. Involvement of Fas/APO-1 (CD95) and calmodulin. Am J Pathol. 1999;155:193–203. doi: 10.1016/S0002-9440(10)65113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–13. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 9.Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25:270–5. doi: 10.1038/bjc.1971.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward HW. Anti-oestrogen therapy for breast cancer: a trial of tamoxifen at two dose levels. Br Med J. 1973;1:13–4. doi: 10.1136/bmj.1.5844.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001;6:469–77. doi: 10.1023/a:1012437607881. [DOI] [PubMed] [Google Scholar]
- 12.Perry RR, Kang Y, Greaves B. Effects of tamoxifen on growth and apoptosis of estrogen-dependent and -independent human breast cancer cells. Ann Surg Oncol. 1995;2:238–45. doi: 10.1007/BF02307030. [DOI] [PubMed] [Google Scholar]
- 13.Ahn EY, Pan G, Oh JH, Tytler EM, McDonald JM. The combination of calmodulin antagonists and interferon-γ induces apoptosis through caspase-dependent and -independent pathways in cholangiocarcinoma cells. Am J Pathol. 2003;163:2053–63. doi: 10.1016/s0002-9440(10)63563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chin D, Means AR. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000;10:322–8. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 15.Soderling TR, Stull JT. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem Rev. 2001;101:2341–52. doi: 10.1021/cr0002386. [DOI] [PubMed] [Google Scholar]
- 16.Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–40. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 17.Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res. 1999;31:577–96. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- 18.Soderling TR. CaM-kinases: modulators of synaptic plasticity. Curr Opin Neurobiol. 2000;10:375–80. doi: 10.1016/s0959-4388(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 19.Hoeflich KP, Ikura M. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 2002;108:739–42. doi: 10.1016/s0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 20.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–8. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 21.Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1:50–8. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- 22.Lagneaux L, Gillet N, Stamatopoulos B, et al. Val-proic acid induces apoptosis in chronic lymphocytic leukemia cells through activation of the death receptor pathway and potentiates TRAIL response. Exp Hematol. 2007;35:1527–37. doi: 10.1016/j.exphem.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 23.Longley DB, Wilson TR, McEwan M, et al. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene. 2006;25:838–48. doi: 10.1038/sj.onc.1209122. [DOI] [PubMed] [Google Scholar]
- 24.Valnet-Rabier MB, Challier B, Thiebault S, et al. c-Flip protein expression in Burkitt’s lymphomas is associated with a poor clinical outcome. Br J Haematol. 2005;128:767–73. doi: 10.1111/j.1365-2141.2005.05378.x. [DOI] [PubMed] [Google Scholar]
- 25.Pawar PS, Micoli KJ, Ding H, et al. Calmodulin binding to cellular FLICE like inhibitory protein modulates Fas-induced signaling. Biochem J. 2008;412:459–68. doi: 10.1042/BJ20071507. [DOI] [PubMed] [Google Scholar]
- 26.Dong B, Valencia CA, Liu R. Ca(2+)/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J Biol Chem. 2007;282:25131–40. doi: 10.1074/jbc.M702123200. [DOI] [PubMed] [Google Scholar]
- 27.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 28.Franke TF, Yang SI, Chan TO, et al. The protein kinase encoded by the Akt protooncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–36. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 29.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–12. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 30.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 31.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 32.Ustundag Y, Bronk SF, Gores GJ. Proteasome inhibition-induces endoplasmic reticulum dysfunction and cell death of human cholangiocarcinoma cells. World J Gastroenterol. 2007;13:851–7. doi: 10.3748/wjg.v13.i6.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsumoto K, Nagahara T, Okano J, et al. The growth inhibition of hepatocellular and cholangiocellular carcinoma cells by gemcitabine and the roles of extracellular signal-regulated and checkpoint kinases. Oncol Rep. 2008;20:863–72. [PubMed] [Google Scholar]
- 34.Sacks DB, Porter SE, Ladenson JH, McDonald JM. Monoclonal antibody to calmodulin: development, characterization, and comparison with polyclonal anticalmodulin antibodies. Anal Biochem. 1991;194:369–77. doi: 10.1016/0003-2697(91)90243-m. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Xu J, Jhala N, et al. Fas-mediated apoptosis in cholangiocarcinoma cells is enhanced by 3,3′-diin-dolylmethane through inhibition of AKT signaling and FLICE-like inhibitory protein. Am J Pathol. 2006;169:1833–42. doi: 10.2353/ajpath.2006.060234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margolin N, Raybuck SA, Wilson KP, et al. Substrate and inhibitor specificity of interleukin-1 β-converting enzyme and related caspases. J Biol Chem. 1997;272:7223–8. doi: 10.1074/jbc.272.11.7223. [DOI] [PubMed] [Google Scholar]
- 37.Villa P, Kaufmann SH, Earnshaw WC. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22:388–93. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- 38.Que FG, Phan VA, Phan VH, et al. Cholangiocarcinomas express Fas ligand and disable the Fas receptor. Hepatology. 1999;30:1398–404. doi: 10.1002/hep.510300618. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Yue P, Schonthal AH, Khuri FR, Sun SY. Cellular FLICE-inhibitory protein down-regulation contributes to celecoxib-induced apoptosis in human lung cancer cells. Cancer Res. 2006;66:11115–9. doi: 10.1158/0008-5472.CAN-06-2471. [DOI] [PubMed] [Google Scholar]
- 40.Rogers KM, Thomas M, Galligan L, et al. Cellular FLICE-inhibitory protein regulates chemotherapy-induced apoptosis in breast cancer cells. Mol Cancer Ther. 2007;6:1544–51. doi: 10.1158/1535-7163.MCT-06-0673. [DOI] [PubMed] [Google Scholar]
- 41.Ullenhag GJ, Mukherjee A, Watson NF, et al. Overexpression of FLIPL is an independent marker of poor prognosis in colorectal cancer patients. Clin Cancer Res. 2007;13:5070–5. doi: 10.1158/1078-0432.CCR-06-2547. [DOI] [PubMed] [Google Scholar]
- 42.Ahn EY, Lim ST, Cook WJ, McDonald JM. Calmodulin binding to the Fas death domain. Regulation by Fas activation. J Biol Chem. 2004;279:5661–6. doi: 10.1074/jbc.M311040200. [DOI] [PubMed] [Google Scholar]
- 43.Mancino A, Mancino MG, Glaser SS, et al. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig Liver Dis. 2008 Apr 3; doi: 10.1016/j.dld.2008.02.015. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng A, Wang S, Yang D, Xiao R, Mattson MP. Calmodulin mediates brain-derived neurotrophic factor cell survival signaling upstream of Akt kinase in embryonic neocortical neurons. J Biol Chem. 2003;278:7591–9. doi: 10.1074/jbc.M207232200. [DOI] [PubMed] [Google Scholar]
- 45.Panka DJ, Mano T, Suhara T, Walsh K, Mier JW. Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells. J Biol Chem. 2001;276:6893–6. doi: 10.1074/jbc.C000569200. [DOI] [PubMed] [Google Scholar]
- 46.Schmitz KJ, Otterbach F, Callies R, et al. Prognostic relevance of activated Akt kinase in node-negative breast cancer: a clinicopathological study of 99 cases. Mod Pathol. 2004;17:15–21. doi: 10.1038/modpathol.3800002. [DOI] [PubMed] [Google Scholar]
- 47.Gupta AK, McKenna WG, Weber CN, et al. Local recurrence in head and neck cancer: relationship to radiation resistance and signal transduction. Clin Cancer Res. 2002;8:885–92. [PubMed] [Google Scholar]