

Abstract
The principle of microscopic reversibility states that at equilibrium the number of molecules entering a state by a given path must equal those exiting the state via the same path under identical conditions, or in structural terms, that the conformations along the two pathways are the same. There has been some indirect evidence indicating that protein folding is such a process, but there have been few conclusive findings. In this study, we performed molecular dynamics simulations of an ultra-fast unfolding and folding protein at its melting temperature in order to observe, on an atom-by-atom basis, the pathways the protein followed as it unfolded and folded within a continuous trajectory. In a total of 0.67 μs of simulation in water, we found 6 transient denaturing events near the melting temperature (323K and 330K) and an additional refolding event following a previously identified unfolding event at high-temperature (373K). In each case unfolding and refolding transition state ensembles were identified, and they agreed well with experiment based on comparison of S- and Φ-values. Based on several structural properties, these 13 transition state ensembles agreed very well with each other and with 4 previously identified transition states from high-temperature denaturing simulations. Thus, not only were the unfolding and refolding transition states part of the same ensemble, but in five of the seven cases, the pathway the protein took as it unfolded was nearly identical to the subsequent refolding pathway. These events provide compelling evidence that protein folding is a microscopically reversible process. In the other two cases, the folding and unfolding transition states were remarkably similar to each other, but the paths deviated.
In 1925, Richard C. Tolman coined the term “microscopic reversibility” in reference to chemical reactions:
In recent years increasing use has been made of a new postulate which perhaps cannot yet be stated in its final form, but which requires in a general way in the case of a system in thermodynamic equilibrium not only that the total number of molecules leaving a given state in unit time shall on the average equal the number arriving in that state in unit time, but also that the number leaving by any particular path shall on the average be equal to the number arriving by the reverse of that particular path, thus excluding any cyclical maintenance of the equilibrium state. The writer has ventured to name this postulate the principle of microscopic reversibility (1).
This description was recast into structural terms in 1967 by Frank H. Westheimer (2) and was adopted by the IUPAC in 1999:
In the case of SN2 reactions at tetrahedral centers implying a formation of the trigonal bipyramid transition state (or intermediate) structure, the original formulation of the principle was extended in the following way: if a molecule or reactant enters a trigonal bipyramid at an apical position, this (or another) molecule or reactant must likewise leave the trigonal bipyramid from an apical position (3).
The hypothesis that protein folding may, like chemical reactions, be microscopically reversible has since been offered. If this hypothesis is true, one would expect to observe identical transition states for folding and unfolding, and major events on the folding pathway would occur in reverse order in the unfolding pathway. Supporting evidence has been presented by Jackson et al. who showed through Φ-value analysis on chymotrypsin inhibitor 2 (CI21) that folding and unfolding transition states are the same, suggesting that the pathways are also the same (4). Additionally, molecular dynamics (MD) generated unfolding transition states (TS) of CI2 are in quantitative agreement with experimental data collected for both unfolding and refolding (5, 6). Furthermore, the unfolding and direct refolding pathways of CI2 were shown to be the same in a single continuous MD trajectory (7).
The latter MD study was done at the melting temperature (Tm) of the protein, at which point the folding and unfolding rates are equal and ΔG = 0. For these reasons, exchange between the folded and unfolded states is dependent on the energy barrier, ΔG‡ ≈ 2.3 kcal/mol (8), and unfolding and refolding may occur in a single trajectory on time scales tractable by MD. The protein, CI2, passed through three different states, native (N), nearly native (N′), and denatured (D), then returned to N′ over a time period of about 60 ns. When moving from N′ to D and back, unfolding and refolding transition states were identified, and they were the same. The Cα root mean square deviation (RMSD) to the native structure and internal contacts were analyzed to differentiate between the three different states. Day and Daggett (7) defined N′, an alternate, stable state for CI2 at elevated temperature. The protein passed through N′ before it moved through its TS to D. N′ was characterized by many near-native interactions but elongated contact distances. In particular, Trp 5, a fluorescence unfolding probe, was buried in both N and N′, but not in D, as would be expected if both N and N′ were native but D was not. D had a disrupted hydrophobic core and loss of secondary structure. This CI2 study showed direct unfolding and refolding in a single continuous trajectory by the same structural pathways for the first time. Consequently, this behavior needs to be demonstrated in another system to ensure it is reproducible, which we describe here.
The engrailed homeodomain (En-HD) of Drosophila melanogaster is a 61-residue three-helix bundle. It is ultra-fast folding (kF = 37,500 s-1 at 25°C and 51,000 s-1 around 42°C) and unfolding (kU = 1,100 s-1 at 25°C and 205,000 s-1 at 63°C), and its folding and unfolding pathways have been extensively characterized through combined experimental and MD studies (9-12). Folding for En-HD follows the framework model involving the docking of HI (residues 10-22), HII (28-38), and HIII (42-55) (13). These properties make En-HD especially well-suited for MD folding studies.
In this study, we performed MD simulations of En-HD near its Tm (52°C = 325K (12)) to compare unfolding and refolding under identical conditions. We analyzed 5 simulations, 3 at 323K and 2 at 330K. We compared them to 4 previously described thermal denaturation simulations, 2 each at 373K and 498K, and one native simulation at 298K. The first of the 373K simulations was found to contain a region of particular interest that had not been previously reported, so that simulation was also analyzed in detail. We identified and characterized 3 different states populated during unfolding and refolding: N, N′, and D. We also found 6 transient denaturing events in which En-HD partially unfolded and refolded in 3 of the 5 Tm simulations; from this 12 unfolding and refolding transition state ensembles (TSE) were identified. En-HD unfolded in the 373K/1 simulation, and a TSE in agreement with experiment was reported previously (9, 12, 13). Further investigation of this simulation showed that En-HD later refolded, so we also describe this high temperature refolding TSE. These 13 TSEs agree well with the 4 previously identified unfolding TSEs from the 4 high-temperature simulations. Besides defining TSEs, we analyzed the entire pathway En-HD followed as it unfolded and refolded. Five of the 7 refolding pathways were nearly identical to the unfolding pathways that preceded them. These 5 examples are further evidence that the ensembles of folding and unfolding pathways are one and the same, and that protein folding is a microscopically reversible process. However, in the other 2 cases, En-HD passed through remarkably similar unfolding and refolding transition states, but only a portion of the actual refolding pathway was similar to the unfolding pathway.
Methods
Molecular Dynamics Simulations
A total of 9 MD simulations are addressed in this paper at the following temperatures with simulation times in parentheses: 298K (100 ns), 323K (100 ns, 50 ns, 42 ns), 330K (100 ns, 100 ns), 373K (24 ns, 75 ns), 498K (20 ns, 60 ns), for a total of 0.67 μs. All 4 of the 373K and 498K simulations have been described previously (9, 12, 13), as have the first 2 323K and the 298K simulations (14).
Both of the 330K simulations were performed using our in-house molecular dynamics package, in lucem Molecular Mechanics (ilmm) (15) with the Levitt et al. force field (16) using previously described protocols (17). The crystal structure (PDB ID: 1ENH) was minimized for 1000 steps and solvated in a box of F3C water molecules (18) such that there was at least 12 Å between the protein and the edge of the periodic box. The density was set to 0.985 g/mol in agreement with the experimentally-determined liquid-vapor coexistence curve for this temperature (19). 1000 steps of steepest descent minimization were performed on the water alone followed by 1 ps of dynamics. Next, the water and the protein were independently minimized for an additional 500 steps. Production simulations were performed for 100 ns allowing all atoms to move with structures written out every 1 ps. Long-range interactions were truncated after 8 Å using a force-shifted nonbonded cutoff. Our force-shifted cutoff method at this distance is the most effective treatment of long-range interactions based on computational savings, energy conservation, and ability to reproduce experimental results (20). The 323K/3 simulation followed the same protocol, except there was only 8 Å of padding between the protein and the edge of the box, the protein was minimized for 200 steps before adding water, and the simulation was run for 42 ns.
Cα RMSD Matrix and 3D MDS
All-vs.-all Cα RMSD matrices were calculated to identify clusters of structures with similar conformations. Granularities were chosen to give 1000-5000 time points over the period of interest. The Cα RMSD between each structure and every other structure was computed, resulting in a matrix with 10002-50002 data points. Low Cα RMSD boxes on the diagonal represent a period of time during which the protein stayed in a particular conformation. When these boxes lie off the diagonal, they indicate conformations of similar structure visited discontinuously in time. As described previously (13), the “core” (residues 8-53) was usually used to calculate Cα RMSD, rather than the whole protein (residues 3-56). Since the fluctuations of the terminal residues are not indicative of the overall motion of the protein and introduce noise, the 5 residues at the N-terminus and 3 at the C-terminus were not included where specified.
Using the program R (21), multidimensional scaling (MDS) was performed to project the matrix down to 3 dimensions. This scaling results in a 3D plot in which each point represents a structure, and the distance between any two points is proportional to the Cα RMSD between the respective structures. The points are connected in order of time for the period of interest. As with the matrix, a series of points close together indicates that the structures are similar. Using this plot, TSEs were chosen as the last structures leaving the extended native cluster for an unfolding event (5) or the first structures upon returning to the native cluster for a refolding event. The TSE was defined as the point of cluster exit and previous 5 ps for unfolding (5) and as the cluster reentry and succeeding 5 ps for refolding.
HIII-core Distance Calculation
The distance between the closest backbone atoms in the C-terminus of HIII and the HI-HII scaffold in the crystal structure was chosen to represent the movement of HIII. The atoms chosen were the Cα of Phe 20 and the backbone carbonyl C of Lys 52, and the distance was measured at 10 ps granularity. Since the 373K/1 simulation was so short, a granularity of 1 ps was used to give consistent sampling.
Average Structures
Average structures were calculated using 100 ps granularity for the long N and N′ time-spans. For TS structures, all 6 structures in the TSE were included in the average. The Cα RMSD of the core residues (8-53) between the average structures was then calculated.
HIII-core Contacts
A contact for a pair of residues was defined based on whether any one of the heavy atoms in the first residue was below a set cutoff of any heavy atom in the second residue. This cutoff was defined as 5.4 Å for carbon-carbon distances and 4.6 Å for all other atom pairs. For Figure 4, the calculation was taken over the time period of interest with 1 ps granularity, and the percentage of structures in which the two specified residues were in contact was reported for the average measurements. For the whole-simulation graphs (Figure 1c), each of the contacts is listed as a separate horizontal line, and if the contact was present at the given time point along the x-axis, a cross (+) was plotted. 10 ps granularity was used.
Figure 4. HIII-core residue pairs fraction of time in contact for N, N′, and new TSEs in each simulation.
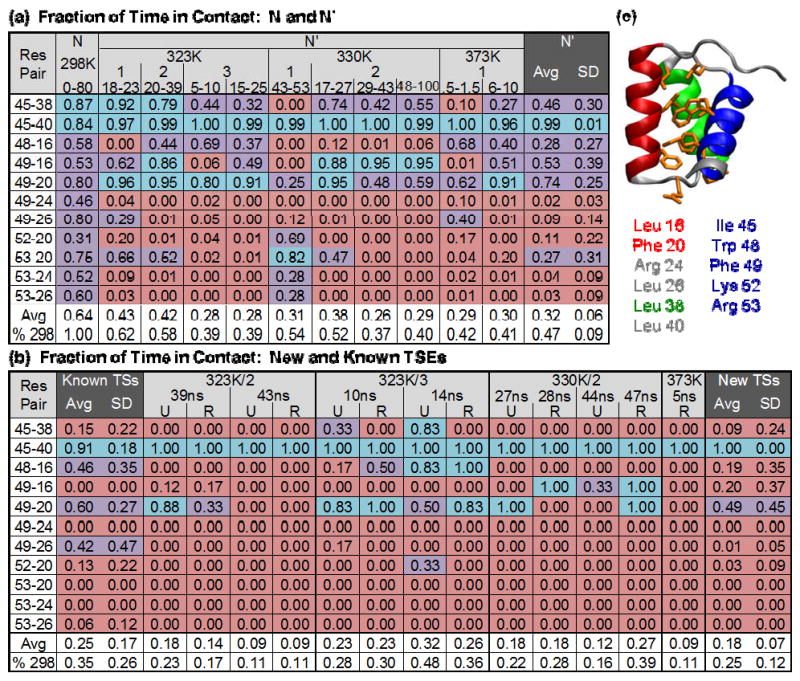
(a)-(b) are colored: < 25% pink, 25-75% purple, >75% blue. (a) Contacts for N in the 298K simulation and N′ in the 323, 330, and 373K simulations. The temperature of the simulation, simulation number, and time span in ns of the N′ state is given. (b) Contacts for new and previously identified TSEs. The simulation temperature and number is given (373K is 373K/1) as well as the ns during which the unfolding (U) or refolding (R) TSE occurred. The average fraction of time in contact was reported for each simulation as was the fraction of time in contact relative to the native (298K) simulation. Additionally, the average and standard deviation of the fraction of time in contact was calculated for each contact pair across all compared time spans. The average fraction of contacts is quite different for each of the states, N, N′, and TS, with a low standard deviation, and those contacts that are lost are lost fairly consistently. (c) Each of the selected contact residues is shown on the En-HD structure in orange.
Figure 1. General properties for each simulation.
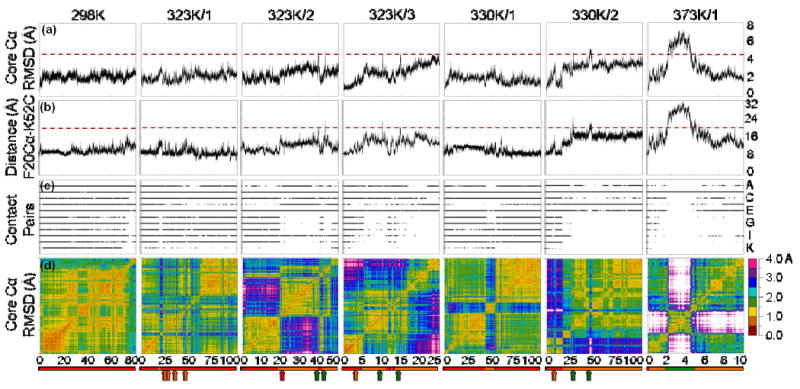
(a) Cα RMSD of the core (residues 8-53) calculated over time for each simulation relative to the 0 ns structure. Values above ∼4.5 Å are indicative of movement to D, as indicated by the dashed red line. (b) Distance between the Cα of Phe 20 and the backbone C of Lys 52 over time. N′ is characterized by values of ∼15 Å, while distances of greater than 20 Å (dashed red line) appear when the protein moves to D. (c) Contacts made between residue pairs. Alternate pairs are labeled on the right from top to bottom: (A) Ile 45 – Leu 38, (B) Ile 45 – Leu 40, (C) Trp 48 – Leu 16, (D) Phe 49 – Leu 16, (E) Phe 49 – Phe 20, (F) Phe 49 – Arg 24, (G) Phe 49 – Leu 26, (H) Lys 52 – Phe 20, (I) Arg 53 – Phe 20, (J) Arg 53 – Arg 24, (K) Arg 53 – Leu 26. Contacts 49-24, 49-26, 52-20, and 53-24 are characteristically present in N but not N′, while additional contacts are lost during D. (d) An all-vs.-all core Cα RMSD matrix. Low- core Cα RMSD squares on the diagonal represent a period of time with similar structures, and when they are off the diagonal, they indicate that the structures from the two corresponding time periods are similar. Below each matrix is a timeline depicting the different states the protein takes in each simulation: N (red), N′ (orange), and D (green). Arrows represent transiently occupied states.
To choose which contacts to report, we identified residue pairs in which one member of the pair was in HIII and the other was not. Of these, the only pairs that were selected were those that were in contact at least 25% of the time in the native (298K) simulation.
Calculation of S-values
The S-value is a semi-quantitative structure index that provides an overall measure of the secondary and tertiary structure for each residue of the protein (6). S-values agree well with experimental Φ-values for a variety of proteins (6, 9, 22-25). The S-value is the product of the extent of native secondary structure (S2°) and native and nonnative tertiary contacts (S3°) present in a given structure relative to the number of contacts in the crystal structure. A value of 1 for S corresponds to native-like extent of structure in the TS, while a value of 0 suggests the residue is unstructured. As previously described (9), S3° was used in place of S for residues Phe 8, Leu 26, and Leu 40. For these three residues, side-chain interactions were maintained despite disorder in the main chain. Consequently, the product of S2° and S3° did not accurately represent the degree of structure retention.
Protein- DNA Interactions
In order to generate a semi-quantitative measure of whether MD-generated En-HD conformers bind DNA, we measured distances between the DNA sugar-phosphate backbone and the HI-HII helical hairpin using the crystal structure of En-HD bound to DNA (PDB ID: 3HDD). The crystal structure contains two nearly identical En-HD structures (core Cα RMSD = 0.27 Å), so we selected the one bound to the ideal TAATTA sequence for all measurements (26). En-HD binds DNA primarily through residues in HIII (major groove) and the N-terminus (minor groove) (26, 27). Since the N-terminus becomes structured only upon binding DNA (26), its conformation during our simulations should have no bearing on whether free En-HD is structured enough to bind DNA. Using Profit (28), the MD structures were first aligned to the DNA-bound structure based on a least-squares fit of the Cα atoms in HIII, the DNA-binding helix. We selected a pairs of residues for the distance measurements, representative of one of two hydrogen bonds in the DNA-bound crystal structure that did not involve HIII or the N-terminus of En-HD. The atoms chosen for the measurement were the Cα of Tyr 25 from En-HD and backbone P of Thymine 28 from the DNA. For measurements over time, 10 ps granularity was used. A period of time beginning 1 ns after the TS and continuing for 1 ns was selected to represent D for all 4 high-temperature unfolding simulations. Since there was not a full nanosecond of denatured time for most of the 4 lower-temperature simulations, the most denatured structure based on 3D MDS of the Cα RMSD matrix was used.
Protein Images and Figures
All protein images were rendered using VMD (29), Cα RMSD matrices were made using R (21), and 3D MDS images were rendered with Chimera (30). Graphs were plotted and rendered in Gnuplot (31).
Results
A total of 10 MD simulations were performed at 5 different temperatures (298K, 323K, 330K, 373K, and 498K). We describe the major conformational states of En-HD in each of the 10 simulations: N, N′, TS, and D. When En-HD was in N, HIII was docked against the HI-HII scaffold. N′ was characterized by a slight movement of HIII towards the N-terminus without losing many contacts or solvating the hydrophobic core. When HIII moved out and away from the HI-HII scaffold, exposing the hydrophobic core, En-HD was deemed to be in D. The protein was not necessarily unfolded in D, but it was not native nor, by definition, biologically active. Whenever the protein moved from N′ to D or from D back to N′, a TS was identified. These four states will be discussed further in the context of each simulation.
Overview of Simulations
298K
En-HD remained folded in the native 298K simulation with a core Cα RMSD of 2.1 ± 0.3 Å (average ± 1 standard deviation) and an HIII-core distance (Phe20 Cα – Lys52 carbonyl C, see Methods) of 10.6 ± 1.4 Å for the first 80 ns of the 100 ns simulation (Figure 1a,b). During the final 20 ns of the simulation, the 9 C-terminal residues formed a π-helix, but HIII remained docked to the HI-HII scaffold. For this reason, the final 20 ns are not considered here.
323K/1
In this 100 ns simulation, En-HD stayed mostly in N but briefly moved to N′ from 18-23 ns. It transiently moved to N′ about 4 more times over the next 30 ns then stabilized in N for the remainder of the simulation (Figure 1). The protein did not populate D during this simulation, so there were no TSs identified.
323K/2
En-HD was in N for the first ∼20 ns. At 19 ns, HIII moved ∼10 Å towards the N-terminus, entering N′ (Figure 3a). It remained in N′ until 39 ns when there was a large jump in core Cα RMSD and HIII-core distance, reflecting the undocking of HIII and entrance into D. HIII moved far enough away from the core (20 Å) for it to lose 10 of its 11 native core contacts (Figure 1c) and for the hydrophobic core to be solvated. This altered position was only transient, however, and HIII moved back to its position in N′ a short 0.28 ns later. Over the next 1 ns, HIII moved back to its N position where it stayed for ∼3 ns. HIII then returned to its N′ position transiently before the protein once again entered D at 43 ns. This transition was marked by another jump in core Cα RMSD, HIII-core distance, and loss of contacts (Figure 1). After 0.16 ns, the protein returned to N′, where it remained for the duration of the 50 ns simulation. The structures of En-HD in all four of its states are shown in Figure 2, and these structures are representative of those seen in the other 6 simulations.
Figure 3. N and N′ structures and all-vs.-all core Cα RMSD matrix for 2 323K simulations.
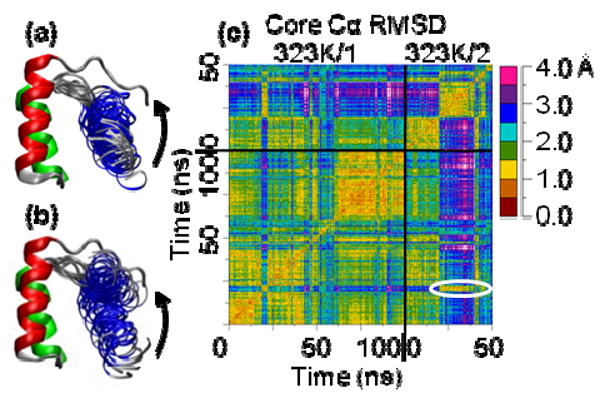
(a) N and N′ from the first 38 ns of the 323K/2 simulation taken every 0.5 ns for HIII. (b) N and N′ from the first 100 ns of the 323K/1 simulation with structures taken every 1 ns for HIII. HI-HII is from the 0 ns structure. HIII moves towards the N-terminus but not out, so the hydrophobic core is not solvated. (c) All-vs.-all core Cα RMSD matrix for the 323K/1 and 323K/2 simulations. 323K/1 and 323K/2 are in N′ for the longest time from 19-20 ns and 20-39 ns, respectively. The color of the circled, low core Cα RMSD box off the diagonal indicates that these two N′ states are as similar to each other as they are to themselves. Smaller boxes can be seen off the diagonal for the points in the 323K/1 simulation when En-HD moves to N′ transiently indicating these transient N′ states are the same as the longer two.
Figure 2. Structures and order of population of the 4 states in the 323K/2 simulation.
Under each state is a set of representative structures from the course of the simulation overlaid and fit on HI-HII Cα atoms. HI (residues 8-20) is colored in red, HII (26-36) in green, and HIII (42-55) in blue. The time point in ns of each structure is listed below, and arrows connect them in the order they occurred. Structures within each state are similar, while each state is distinct. In N and N′, HIII forms a ∼15° angle with the HI-HII scaffold. When En-HD reaches the TS, the angle is ∼30°, and it becomes even wider in D.
323K/3
The first 5 ns of this simulation were spent mostly in N, after which En-HD stabilized in N′ for another 5 ns. There was a transient movement to D at 10 ns, indicated by a spike in core Cα RMSD and HIII-core distance. There was a transition back to N for 2 ns with a transient jump to N′ during that time. Then at 13 ns, there was a more stable transition to N′, interrupted only by a transient jump to D at 14 ns. En-HD remained in N′ until the end of the simulation. Only the first 25 ns of the 42 ns simulation are considered here in order to focus on the transitions of interest.
330K/1
En-HD was stable in N for the majority of this simulation, with the exception of 43-53 ns (Figure 1). During this time, HIII unwound slightly at the N-terminus causing it to move towards the N-terminus by a register, much like the movement previously described for N′. Because the movement was due to unwinding rather than a simple loop movement, N′ in this simulation had somewhat different properties (core Cα RMSD HIII-core and contact pattern) than N′ in the 323K simulations (Figure 1).
330K/2
This simulation began with 8 ns of N, moved to N′ for 1 ns, returned to N for 8 ns, then shifted to N′ again. At 27 ns, the protein moved from N′ to D for 0.33 ns, then returned to N′ at 28 ns. Again, at 44 ns, En-HD moved from N′ to D and stayed there until 47 ns when it returned to N′. It remained in N′ for the duration of the 100 ns simulation (Figure 1). Each of the three non-transient N′ states here agree very well with N′ in the 323K simulations as shown in Figure 2.
373K/1
En-HD moved from N to N′ within 0.5 ns, then proceeded on to D. An unfolding TS was previously identified at 1.720 ns (9, 12, 13), and we identified a new refolding TS shortly after 5 ns. After En-HD returned to N′, it remained there for the remainder of the 24 ns simulation. We will focus only on the first 10 ns of the simulation here. After refolding, N′ did not agree very well with N′ in the other simulations near the Tm, due to the fluctuating π-helical structure adopted by the C-terminus of HIII around 7 ns (data not shown). The temperature of this simulation was nearly 40K above the Tm of En-HD, so refolding is expected to occur only rarely and incompletely. We were lucky to observe such an event so that we can compare folding and unfolding within a continuous trajectory at high temperature.
Native' State at Elevated Temperature
Before HIII moved far away enough from the HI-HII scaffold for En-HD to become “denatured,” it occupied two distinct positions. The conformation of En-HD with HIII positioned most similarly to the crystal structure is N, and we define N′ as the state in which HIII slides ∼10 Å towards the N-terminus (Figure 3a,b).
In all three of the 323K simulations and the second 330K simulation, the same N′ structures were observed based on core Cα RMSD and HIII-core contact similarity (Figures 3c, 4a, and 5a). Based on the core Cα RMSD matrix of the first two 323K simulations (Figure 3c), the conformations from 18-23 ns and the subsequent transient deviations from N in 323K/1 were the same as those from 20-40 ns and 42-50 ns (with the exception of the unfolding events) in 323K/2. The core Cα RMSD between average structures of N′ in the 323K/1-3 and 330K/2 simulations also showed this similarity (Figure 5a). The 0.5-1.5 ns N′ in 373K/1 was in good agreement with N′ in 323K/1-3 and 330K/2 based on core Cα RMSD, but the 6-10 ns N′ was less similar due to disruption of HIII.
Figure 5. Matrix of core Cα RMSDs between average structures representative of N, N′, and TS.
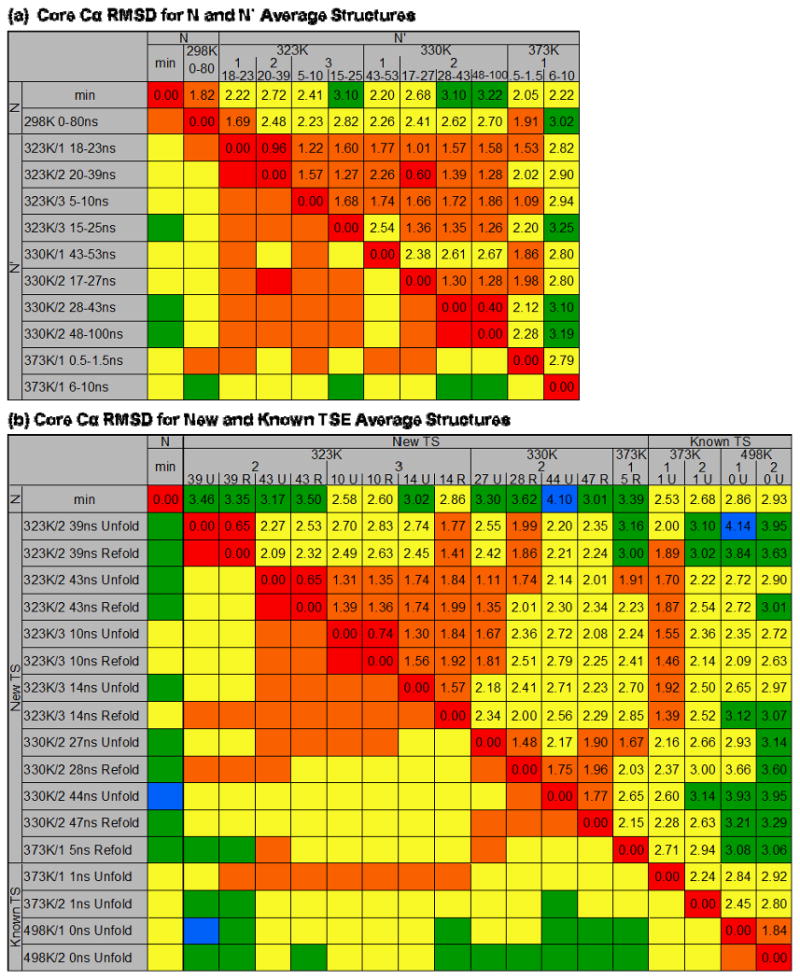
The RMSDs reported are for the core Cα residues of the average structure over the time periods indicated and are colored to visualize trends 0-1 Å (red), 1-2 Å (orange), 2-3 Å (yellow), 3-4 Å (green), 4-5 Å (blue). (a) Core Cα RMSDs for average N′ structures. The simulation temperature, number, and time span in ns of N′ is given. Excluding the later 373K/1 N′ structure, all of the N′ structures are within 3 Å core Cα RMSD of each other, and with the exception of 330K/1, all of the low-temperature N′ structures are within 2 Å. (b) Core Cα RMSD for the 13 new average TSE structures and 4 previously identified. The simulation temperature, number, and ns during which the unfolding (U) or refolding (R) TSE occurred is given. The average TSE structures are most similar to other TSE structures at the same temperatures, and all are about the same core Cα RMSD from N (∼2.5-3.5 Å).
The fraction of time the HIII-core contacts were made was remarkably similar for all 10 non-transient N′ conformations, with the exception of 330K/1 (Figure 4a). The average standard deviation for contact time over all 11 residue pairs was 19%, and if the 330K/1 simulation was excluded, it dropped to 15% (Figure 4). Contact vs. time plots are useful to probe which pairs are in contact over the course of a simulation (Figure 1c). Based on this, contacts Phe 49 – Arg 24 (F), Phe 49 – Leu 26 (G), Lys 52 – Phe 20 (H), and Arg 53 – Arg 24 (K) were lost when En-HD moved from N to N′ and were mostly regained if it moved back to N from N′.
Properties of the Transition State Ensembles
A total of 13 new unfolding and refolding TSEs were identified in 4 simulations at 323K, 330K, and 373K. The TSEs all had low core Cα RMSD to each other (2.1 ± 1.0 Å), particularly the Tm TSEs (1.7 ± 0.9 Å, Figure 5b). The Tm TSEs were less similar to the high-temperature TSEs, with lower core Cα RMSDs to the 373K TSEs than the 498K TSEs, but most were 2-4 Å core Cα RMSD between any two high temperature and Tm TSE average structures. The core Cα RMSD to the native state was 3.1 ± 0.4 Å over all 17 TSEs, and the lowest core Cα RMSDs were observed between the exit and reentry TSEs at 39 and 43ns in the 323K/2 simulation (0.65 Å in both cases).
The HIII-core contacts agreed very well between the new and previously identified TSEs (Figure 4b). The Ile 45 – Leu 40 contact was consistently sustained in all of the TSEs, likely due to the residues' positions in the HII-HIII loop and at the N-terminal end of HIII, respectively. Where there were dissimilarities in contacts made between the known and new TSEs, it was usually the case that there were less contacts made in the new, lower temperature TSEs.
There was a pattern of gain and loss of HIII-core contacts preceding and following the TSEs across the simulations. There were 6 residues involved in contacts characteristically lost in N′: Phe 20, Arg 24, Leu 26, Phe 49, Lys 52, and Arg 53; and aside from the 4 pairs that lost contact in N′, these 6 residues were also involved in 3 more contacts: Phe 49 – Phe 20, Arg 53 – Phe 20, and Arg 53 – Leu 26. Of these 3 pairs, the 53-26 contact was lost early in all 6 simulations at elevated temperature (Figure 1c). In the case of the 12 new TSEs in the 323K and 330K simulations, the 49-20 contact was lost within 1 ns before or immediately after the exit TS, and it was reformed within 0.1 ns following the reentry TSs. The 53-20 contact was also lost during in the same time (with one exception: 323K/3 10 ns), and it was regained within 1 ns of all 4 reentry TSs in the 323K simulations, but it never reformed after the first reentry TS in the 330K/1 simulation. In the 373K/1 simulation the 49-20 pair was lost 0.5 ns after the exit TS and regained 0.1 ns after the reentry TS, and the 53-20 pair was lost 1.5 ns before the exit TS and regained 1 ns after reentry.
S-values, which quantify local structure in TSEs, were calculated for the 13 TSEs and compared with experimentally determined Φ-values. The S- and Φ-values agree very well for 10 of the 13 new TSEs, with linear correlation coefficients ranging from R = 0.71-0.86 (Figure 6). The two 330K/2 44-47 ns TSEs did not agree well with experiment (S vs. Φ, R = 0.10 and 0.03). Loss of secondary structure in the termini of HIII explains some of the disagreement. For example, Ala 43 from the N-terminal end of HIII completely lost its secondary structure, giving it an S-value of 0 rather than its Φ-value of 1.05. The 373K/1 5 ns reentry TS had a slightly lower correlation coefficient (R = 0.60), with the largest disagreements seen for residues Leu 26, Leu 38, and Gly 39 which are located at the ends of HII. The secondary structure was as expected for these residues (turn, helix, helix; respectively), so the discrepancy is due to altered packing.
Figure 6. S-values for the 13 new TSEs.

The correlation between calculated S-values and experimentally determined Φ-values is quite good for the first 10 TSEs. The S- and Φ-values do not agree as well for the 330K/2 44-47ns or 373K/1 TSEs, which is due to loss of secondary structure or altered packing. For the 3 residues noted by *, only S3° was reported (see Methods).
Protein- DNA Interactions
When En-HD can no longer bind DNA, it loses its biological activity and is therefore denatured, although it may not be unfolded. Consequently, it is informative to determine whether the structures we identified as N, N′, and D can bind DNA. In an effort to quantify En-HD's DNA binding ability, we fit En-HD to DNA based on the Cα atoms of the major binding helix (HIII) and assessed to what extent the other binding interactions could be formed. Tyr 25 is not in HIII or the unstructured N-terminus, and it makes a hydrogen bond to the DNA backbone (26, 27). Since we forced HIII to bind, this residue was selected for distance calculations. Across the simulations, the distances for Tyr 25 – Thymine 28 show a general trend of being longer for D than N, but the distance is also longer in N′ than N. Structures from the 323K/2 simulation and the Tyr 25 – Thymine 28 distance are given in Figure 9 and are representative of what we saw in the other simulations.
Figure 9. Structures from 323K/2 fit to DNA.
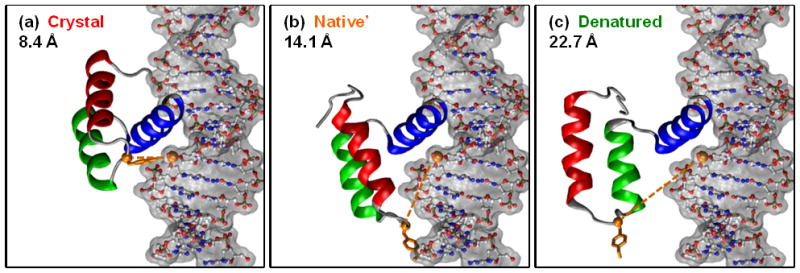
En-HD colored with Tyr 25 in orange sticks. En-HD's Tyr 25 Cα and the DNA's Thymine 28 phosphate are shown in orange van der Waals spheres, and the distance between these two atoms is given and marked with an orange dashed line. (a) The crystal structure of En-HD bound to DNA (PDB ID: 3HDD) (b) N′ at 30 ns in the 323K/2 simulation. (c) The most denatured state (D) from the 43 ns N′→D→N′ transition, 43.195 ns. The structures show that En-HD moved slightly away from the DNA in N′ compared to N, and much farther in D.
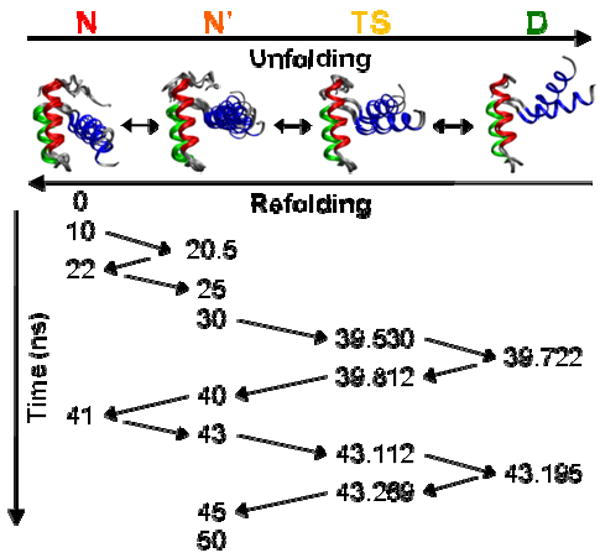
Unfolding and Refolding Pathways
There were 7 unfolding and refolding events identified in 4 simulations during the following times: 39 and 43 ns in 323K/2, 10 and 14 ns in 323K/3, 27-28 and 44-47 ns in 330K/2, and 1-5 ns in 373K/1. Comparison of the structures indicates that the protein moved through the same conformations when it refolded as when it unfolded for a given unfolding/refolding event. This path retracing can be seen as an “X” on the core Cα RMSD matrix and as overlaid paths in the 3D projection of the matrix (Figure 7). As the protein moved from its most denatured point back to N′, a line perpendicular to the diagonal of the matrix is apparent. This line represents a series of conformations that is very similar to the series of conformations the protein passed through just previously, but in reverse order. In the 3D projection, this same phenomenon is seen as overlapping points along the path from N′ to the most denatured point, then back to N′. This evidence was present to different extents for each of the 7 unfolding/refolding events, but it is the most striking for 323K/2 39 and 43ns, 323K/3 10ns, 330K/2 27-28ns, and 373K 1-5ns.
Figure 7. Comparison of 323K/2 43 ns unfolding and refolding TSEs and pathway.
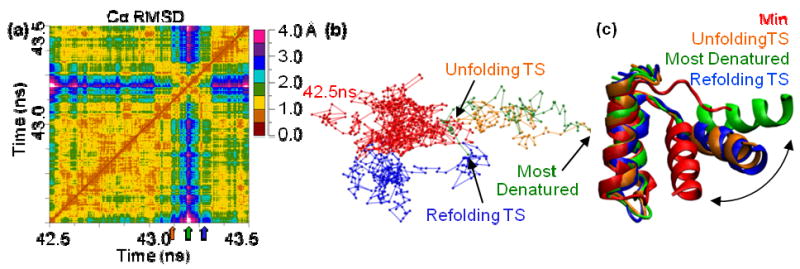
(a) An all-vs.-all Cα RMSD matrix showing the unfolding TS (43.112 ns, orange arrow), most denatured state (43.195 ns, green arrow), and the refolding TS (43.269 ns, blue arrow). The visible “X” is evidence that the conformations the protein takes as it leaves N′ to go to D are the same as those it takes when it returns to D, but in the reverse order. (b) The 3D MDS projection of the matrix from (a) in which each of the points represents a structure, and the distance between any two points is proportional to the Cα RMSD between the respective structures. The colors denote different periods in time: 42.5 ns to the unfolding TS (red), unfolding TS to the most denatured conformation (orange), most denatured to the refolding TS (green), and refolding to 43.5 ns (blue). That the paths that the protein followed as it moved from N′ to D and back are overlaid in the 3D projection indicates the conformations the protein took were very similar and in reverse order. (c) Structures of the unfolding TS (orange), refolding TS (blue), most denatured conformation (green), and the starting minimized structure (red). The structures were fit based on the Cα atoms of HI-HII. The 2 TS structures are nearly identical, while they are distinct from both N and D.
While N′ is not in the denatured ensemble, it is distinct from N, and thus there must exist a low-energy pathway to move between the two states. As with the unfolding and refolding pathways, there was an “X” on the core Cα RMSD matrix when En-HD transiently moved from N′ to N and back. In the 323K/3 simulation, there were 2 transient N→N′→N movements. There was an “X” visible on the core Cα RMSD matrix around both N→N′→N transitions, and there was a third “X” off the diagonal, around the intersection of the times corresponding to both of the individual N→N′→N transitions (Figure 8). This third “X” indicates that not only were the N→N′ and N′→N pathways very similar for a single transition, but also that the two N′→N→N′ transitions were almost equally as similar.
Figure 8. All-vs.-all core Cα RMSD matrix of 2 transient N→N′→N transitions from 323K/3.
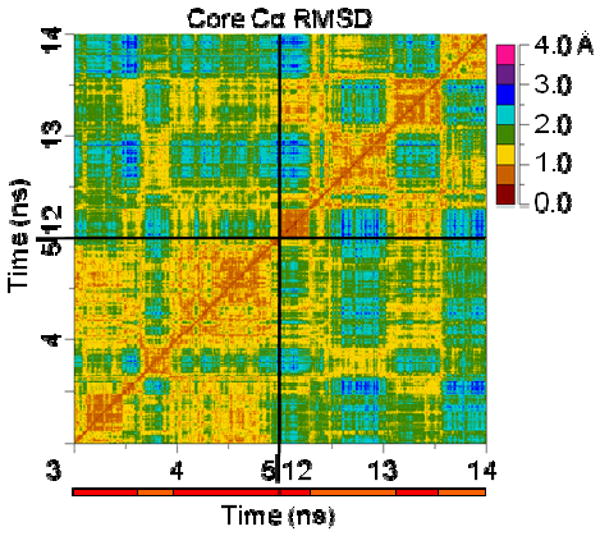
Much like for the N′→D→N′ transition in the 323K/2 43 ns TS, an “X” is visible on the core Cα RMSD matrix for both N→N′→N transitions. In this case, there is a third “X” that is apparent off the diagonal at the intersection of the times at which the other two “X”s occur. This suggests that not only are the N↔N′ paths very similar, but also the N→N′→N path from the first transition is remarkably similar to that from the second.
Discussion
Three different conformational states were populated by En-HD in our 7 simulations: N, N′, and D. The protein's state was determined based on a combination of measurements: core Cα RMSD, HIII-core distance, and HIII-core contact pattern (Figure 1). Measuring the core Cα RMSD to the minimized crystal structure was the foremost method in determining En-HD's state. The core Cα RMSD generally fluctuated between 1-3 Å when the protein was in N and 2-4 Å for N′ (Figures 1a and 5). Values over 4.5 Å usually indicated a departure from N′ to D. The Phe 20 Cα – Lys 52 C distance, representative of the distance between HIII and the HI-HII scaffold, was also a good indicator of state. When the protein was in N, the distance fluctuated around 10 ± 2 Å, while an increase to 14 ± 2 Å indicated movement to N′ (Figure 1b). Distances over 20 Å occurred when En-HD moved to D. Movement between states was more clearly discerned based on the HIII-core distance than on core Cα RMSD. There was nearly always a clean jump in distance between the different states, which suggests that N and N′ are distinct states despite both being “native.” The pattern of contacts between HIII and the rest of the protein also helped discriminate different states. The contact pairs selected for analysis were deliberately chosen to be good representatives of N. Jumps in core Cα RMSD to 2-4 Å and HIII-core distance to ∼14 Å, which were characteristic of N′, coincided with loss of 4 contacts pairs: Phe 49 – Arg 24, Phe 49 – Leu 26, Lys 52 – Phe 20, and Arg 53 – Arg 24 (Figure 1c).
To estimate the likelihood that the 3 different En-HD conformations binds DNA, we fit structures from our MD simulations onto the DNA-bound crystal structure and took a distance measurement that might discern between native (N or N′) and D. Representative structures from the 323K/2 simulation and distances are given in Figure 9. Even though both the N′ and D structures were distinct from the crystal structure, N′ conformations could more easily move back to N and be in a position to bind the DNA (and thus be biologically active) than those in D. For N′→N movement to occur, HI and HII would have to slide along the DNA and HIII so that they could dock against HIII as in N. This movement was what we saw for all N→N′ transitions in our simulations without DNA. For a D structure to move to N, it would have to expel the water from its solvated hydrophobic core before HI and HII could dock back onto HIII and the DNA. Overall, D appears to be too distorted to function properly, and N′ falls somewhere between N and D. While N′ may be able to recover and clamp down on the DNA, it is also possible that it is too distorted and therefore inactive.
Having defined three different states for En-HD in 7 independent simulations, it is interesting to consider the variations within a state and how En-HD passes between them. N′ was observed in all 6 of the elevated temperature simulations, but it was more similar in 5 of them (323K/1-3, 330K/2, and 373K/1) than it was in 330K/1 based on core Cα RMSD and HIII-core contacts (Figures 4a and 5a). Also, the first period of N′ in the 373K/1 simulation matched the first 4 simulations, while the second period was ambiguous. In all cases, HIII moved a register towards the N-terminus, away from the HI-HII turn, but N′ in 330K/1 and the end of 373K/1 was somewhat different from N′ in the other 4 simulations and in the beginning of 373K/1, despite having the same overall topology. This difference suggests that there may be multiple, subtly different N′ states.
In the 4 simulations where there was N′↔D movement, a total of 13 new TSEs were identified. The TSEs within one temperature were most similar, and the best agreement was between the unfolding and refolding TSEs for a single transient unfolding/refolding event (Figure 5b). In these cases, only a small portion of D was sampled so it was likely that the protein would find a refolding path very similar to its unfolding path from the ensemble of paths available.
Based on the 11 HIII-core contact pairs selected for analysis, there was good agreement among all 17 TSEs (13 new and 4 previously identified, Figures 1c and 4b). In almost all of the cases where there was disagreement between the new and previously identified TSEs, it was the case that there were more contacts present in the high-temperature TSEs, which suggests they were more native-like. It is expected that high-temperature TSEs will more closely resemble N than TSEs at the protein's Tm due to Hammond effects. This phenomenon causes the structure of the TSE to become more native-like upon destabilization, in this case by heat (22, 32, 33).
Not only were the 13 new TSEs consistent with those previously identified, but 10 of them were in good agreement with experiment based on comparing calculated S-values to experimental Φ-values (R = 0.71-0.86, Figure 6). The correlation was somewhat lower (R = 0.60) for the 373K/1 refolding TSE and significantly lower (R = 0.10 and 0.03) for the 330K/2 44-47ns TSEs. The 330K/2 44-47ns unfolding/refolding event followed a cyclical path as it folded and unfolded, yet its unfolding and refolding TSEs were very similar to each other (average structure core Cα RMSD = 1.77 Å, Figure 5b). The lack of agreement in this case may be illustrating discrepancies between single-molecule behavior versus bulk measurements. That is, our aberrant TSE pair may be extreme members of the much broader ensemble probed experimentally.
The 13 new TSEs generally agreed well with each other, the 4 previously identified TSEs from high-temperature unfolding simulations, and experiment. The unfolding and refolding TSEs were equally similar, which is evidence that all 17 TSEs come from the same global ensemble of transition states for En-HD folding. Further, our data suggest that the ensemble of paths for unfolding and refolding is also the same, which supports our long-standing contention that protein folding is a microscopically reversible process.
The symmetrical order of contact pair loss and gain upon unfolding and refolding is further evidence that protein folding is microscopically reversible. The 6 residues involved in the 4 contact pairs that were characteristically lost in N′ made 3 additional contacts (Figures 1c and 4). One pair was lost early in the simulations, and the other two, Phe 49 – Phe 20 and Arg 53 – Phe 20, were usually lost just before the unfolding TS and regained right after the refolding TS in the 7 unfolding/refolding events. These 6 residues make up the half of the HIII-core contacts closest to the C-terminus (Figure 4c). Arg 24, Leu 26, and Lys 52 lost all of their HIII-core contacts in N′, but Phe 20, Phe 49, and Arg 53 maintained 2 of the original 7 contact pairs. It was not until right after these 2 contact pairs were lost that En-HD reached its TS and became denatured, and they reformed right after reentering N′ from D in most cases. Thus, loss and gain of these 6 hydrophobic core contacts are critical steps on the N↔N′ and N′↔D pathways, based on our 323 and 330K simulations. This is evidence for microscopic reversibility in protein folding because there is a consistent order of loss of contacts in unfolding that is repeated in reverse order upon refolding. However, the pattern of loss by these 6 contact pairs was not consistently repeated in the 4 high-temperature unfolding simulations previously run at 373 and 498K (data not shown).
The all-vs.-all core Cα RMSD matrix and its 3D projection are arguably the best ways to observe the similarity of MD structures over time. Indeed, there was a visible “X” on the core Cα RMSD matrix for 5 of the 7 unfolding/refolding events. Similarly, the structures from the unfolding TS to the most denatured point back to the refolding TS overlay on the 3D projection of the core Cα RMSD matrix for these 5 unfolding/refolding events (Figure 7). Both the “X” and the overlaid paths indicate that the conformations En-HD moved through from the unfolding TS to the most denatured point had low core Cα RMSD to the conformations En-HD took as it moved back to the refolding TS, but in reverse order.
“X”s were also visible for transient movements from N to N′. In 323K/3, not only was the N→N′ path similar to the N′→N path, but both N→N′→N movements followed highly similar paths. There is an “X” on the diagonal of the all-vs.-all core Cα RMSD matrix for each N→N′→N movement, and there is also an off-diagonal “X” at the intersection of the times corresponding to each of the N→N′→N events. While these four N↔N′ pathways are not complete folding or unfolding pathways, they are transitions between discrete states along the full folding pathway.
En-HD never moved from N to D without first passing through N′ each of the 7 times it unfolded. In fact, in the 323K/2 and 323K/3 simulations, it moved back to N between the two unfolding/refolding events, passing through N′ on the way. Based on our simulations, the N↔N′ paths were part of the same ensemble as were the N′↔D paths, and N′ is a necessary step between N and D. Together, these findings are evidence that the entire N→N′→D and D→N′→N pathways are mirror images of the same process, and thus protein folding is a microscopically reversible process.
We identified and characterized four distinct states of En-HD: N, N′, TS, and D which were consistent across simulations at 298, 323, 330, and 373K. Core Cα RMSD, HIII-core contacts, HIII-core distance, and predicted DNA binding ability were used to discriminate between the states and place them on the folding pathway. We identified 7 transient denaturing events in 6 simulations and identified 13 new unfolding and refolding TSEs. The 13 new TSEs agreed well with 4 previously identified TSEs based on core Cα RMSD and HIII-core contacts as well as with experimental data based on Φ- and S-values. In 5 of the 7 transient denaturing events, the unfolding pathway was nearly identical to the refolding pathway. We also found two N↔N′ transitions that followed the same pathway in the folding and unfolding directions for both transitions. These phenomena are evidence that the ensemble of folding and unfolding pathways are one and the same and that protein folding can be a microscopically reversible process.
Acknowledgments
We thank Darwin Alonso, Amanda Jonsson, and Dustin Schaeffer for helpful discussions and technical assistance.
Footnotes
This research was supported by the National Institutes of Health Grant GM50789 (to V.D.).
Abbreviations: CI2, chymotrypsin inhibitor 2; MD, molecular dynamics; TS, transition state; Tm, melting temperature; ΔG‡, energy barrier / activation energy; N, native; N′, nearly native; D, denatured; RMSD, root mean square deviation; En-HD, engrailed homeodomain; TSE, transition state ensemble; ilmm, in lucem molecular mechanics; MDS, multidimensional scaling.
References
- 1.Tolman RC. The Principle of Microscopic Reversibility. Proc Natl Acad Sci U S A. 1925;11:436–439. doi: 10.1073/pnas.11.7.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Westheimer FH. Pseudo-rotation in the hydrolysis of phosphate esters. Accounts of Chemical Research. 1968;1:70–78. [Google Scholar]
- 3.Minkin VI. Glossary of Terms Used in Theoretical Organic Chemistry. Pure Appl Chem. 1999;71:1919–1981. [Google Scholar]
- 4.Jackson SE, elMasry N, Fersht AR. Structure of the hydrophobic core in the transition state for folding of chymotrypsin inhibitor 2: a critical test of the protein engineering method of analysis. Biochemistry. 1993;32:11270–11278. doi: 10.1021/bi00093a002. [DOI] [PubMed] [Google Scholar]
- 5.Li A, Daggett V. Characterization of the transition state of protein unfolding by use of molecular dynamics: chymotrypsin inhibitor 2. Proc Natl Acad Sci U S A. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daggett V, Li A, Itzhaki LS, Otzen DE, Fersht AR. Structure of the transition state for folding of a protein derived from experiment and simulation. J Mol Biol. 1996;257:430–440. doi: 10.1006/jmbi.1996.0173. [DOI] [PubMed] [Google Scholar]
- 7.Day R, Daggett V. Direct observation of microscopic reversibility in single-molecule protein folding. J Mol Biol. 2007;366:677–686. doi: 10.1016/j.jmb.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 9.Gianni S, Guydosh NR, Khan F, Caldas TD, Mayor U, White GW, DeMarco ML, Daggett V, Fersht AR. Unifying features in protein-folding mechanisms. Proc Natl Acad Sci U S A. 2003;100:13286–13291. doi: 10.1073/pnas.1835776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayor U, Grossmann JG, Foster NW, Freund SM, Fersht AR. The denatured state of Engrailed Homeodomain under denaturing and native conditions. J Mol Biol. 2003;333:977–991. doi: 10.1016/j.jmb.2003.08.062. [DOI] [PubMed] [Google Scholar]
- 11.Mayor U, Guydosh NR, Johnson CM, Grossmann JG, Sato S, Jas GS, Freund SM, Alonso DOV, Daggett V, Fersht AR. The complete folding pathway of a protein from nanoseconds to microseconds. Nature. 2003;421:863–867. doi: 10.1038/nature01428. [DOI] [PubMed] [Google Scholar]
- 12.Mayor U, Johnson CM, Daggett V, Fersht AR. Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation. Proc Natl Acad Sci U S A. 2000;97:13518–13522. doi: 10.1073/pnas.250473497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeMarco ML, Alonso DOV, Daggett V. Diffusing and colliding: the atomic level folding/unfolding pathway of a small helical protein. J Mol Biol. 2004;341:1109–1124. doi: 10.1016/j.jmb.2004.06.074. [DOI] [PubMed] [Google Scholar]
- 14.Beck DA, Daggett V. A One-Dimensional Reaction Coordinate for Identification of Transition States from Explicit Solvent Pfold-Like Calculations. Biophys J. 2007;93:3382–3391. doi: 10.1529/biophysj.106.100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck DAC, Alonso DOV, Daggett V. ilmm, in lucem Molecular Mechanics. University of Washington; Seattle: 2000-2008. [Google Scholar]
- 16.Levitt M, Hirshberg M, Sharon R, Daggett V. Potential-Energy Function and Parameters for Simulations of the Molecular-Dynamics of Proteins and Nucleic-Acids in Solution. Computer Physics Communications. 1995;91:215–231. [Google Scholar]
- 17.Beck DAC, Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. Journal of Physical Chemistry B. 1997;101:5051–5061. [Google Scholar]
- 19.Haar L, Gallagher JS, Kell GS. NBS/NRC Steam Tables: Thermodynamic and Transport Properties and Computer Programs for Vapor and Liquid States of Water in SI Units. Hemisphere; Washington, DC: 1984. [Google Scholar]
- 20.Beck DA, Armen RS, Daggett V. Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochemistry. 2005;44:609–616. doi: 10.1021/bi0486381. [DOI] [PubMed] [Google Scholar]
- 21.Team, R. C. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2004. [Google Scholar]
- 22.Daggett V, Li A, Fersht AR. Combined molecular dynamics and phi-value analysis of structure-reactivity relationships in the transition state and unfolding pathway of barnase: Structural basis of Hammond and anti-Hammond effects. J Am Chem Soc. 1998;120:12740–12754. [Google Scholar]
- 23.Li A, Daggett V. Molecular dynamics simulation of the unfolding of barnase: characterization of the major intermediate. J Mol Biol. 1998;275:677–694. doi: 10.1006/jmbi.1997.1484. [DOI] [PubMed] [Google Scholar]
- 24.Fulton KF, Main ER, Daggett V, Jackson SE. Mapping the interactions present in the transition state for unfolding/folding of FKBP12. J Mol Biol. 1999;291:445–461. doi: 10.1006/jmbi.1999.2942. [DOI] [PubMed] [Google Scholar]
- 25.Day R, Daggett V. Sensitivity of the folding/unfolding transition state ensemble of chymotrypsin inhibitor 2 to changes in temperature and solvent. Protein Sci. 2005;14:1242–1252. doi: 10.1110/ps.041226005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraenkel E, Rould MA, Chambers KA, Pabo CO. Engrailed homeodomain-DNA complex at 2.2 Å resolution: a detailed view of the interface and comparison with other engrailed structures. J Mol Biol. 1998;284:351–361. doi: 10.1006/jmbi.1998.2147. [DOI] [PubMed] [Google Scholar]
- 27.Kissinger CR, Liu BS, Martin-Blanco E, Kornberg TB, Pabo CO. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 Å resolution: a framework for understanding homeodomain-DNA interactions. Cell. 1990;63:579–590. doi: 10.1016/0092-8674(90)90453-l. [DOI] [PubMed] [Google Scholar]
- 28.Martin ACR. ProFit: Protein Least Squares Fitting. University College London; London, England: 1992-2001. [Google Scholar]
- 29.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 30.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 31.Williams T, Kelley C. Gnuplot. 1986-1993 1998 2004 www.gnuplot.info.
- 32.Matthews JM, Fersht AR. Exploring the energy surface of protein folding by structure-reactivity relationships and engineered proteins: observation of Hammond behavior for the gross structure of the transition state and anti-Hammond behavior for structural elements for unfolding/folding of barnase. Biochemistry. 1995;34:6805–6814. doi: 10.1021/bi00020a027. [DOI] [PubMed] [Google Scholar]
- 33.Day R, Bennion BJ, Ham S, Daggett V. Increasing temperature accelerates protein unfolding without changing the pathway of unfolding. J Mol Biol. 2002;322:189–203. doi: 10.1016/s0022-2836(02)00672-1. [DOI] [PubMed] [Google Scholar]