

Abstract
Development of mesothelioma is linked mainly to asbestos exposure but the combined contributions of genetic and epigenetic alterations are unclear. We investigated the potential relationships between gene copy number (CN) alterations and DNA methylation profiles in a case series of pleural mesotheliomas (n=23). There were no instances of significantly correlated CN alteration and methylation at probed loci, whereas averaging loci over their associated genes revealed only two genes with significantly correlated CN and methylation alterations. In contrast to the lack of discrete correlations, the overall extent of tumor CN alteration was significantly associated with DNA methylation profile when comparing CN alteration extent among methylation profile classes. Further, there was evidence that this association was partially attributable to prevalent allele loss at the DNA methyltransferase gene DNMT1. Our findings define a strong association between global genetic and global epigenetic dysregulation in mesothelioma, rather than a discrete, local coordination of gene inactivation.
Keywords: Integrative genomics, DNA methylation, Copy number alteration, Mesothelioma
Introduction
Pleural mesothelioma is a rapidly fatal asbestos-associated malignancy with approximately 80% of pleural mesothelioma cases reporting a known history of exposure (1). Asbestos fibers have been shown to be both cytotoxic and clastogenic in vitro (2, 3), and interference with the mitotic machinery by asbestos can lead to abnormal chromosome segregation and hence deletion events (4). Although the number of phenotypically important point mutations in pleural mesotheliomas has been shown to be relatively low (5, 6), reports of extensive gene copy number (CN) alterations in this disease are numerous, and common regions of allele loss include 1p, 3p21, 6q, 9p21, 15q11–15, and 22q (7–14). There have also been several reports of promoter hypermethylation at tumor suppressor gene loci in mesothelioma (6, 15–21). Gene silencing by DNA methylation at CpG dinucleotides in promoter regions is a well-recognized mechanism of gene inactivation and known to contribute to tumorigenesis (22). We recently reported that increased asbestos exposure is significantly associated with an increased prevalence of gene promoter methylation at cell cycle control genes (23). Other recent work from our group confirmed the relationship between asbestos exposure and DNA methylation in an investigation of over 750 cancer-related genes, and defined the distinct profiles of coordinated DNA methylation in mesotheliomas relative to non-tumorigenic pleural tissues (24).
The spectrum of somatic alterations in mesothelioma and all human cancers includes both genetic and epigenetic events that act in concert to drive tumorigenesis and promote progression to a malignant phenotype. To date there are only a few examples of studies that aim to perform an integrative analysis of multiple types of somatic alterations. The Cancer Genome Atlas Research Network examined mutations, CN alterations, and DNA methylation in an integrative manner and highlighted alterations of core pathways in glioblastoma (25). Further, a subset of colorectal cancers is known to possess a DNA methylator phenotype, and an association between tumors harboring this phenotype and a lower degree of chromosomal aberrations strongly suggest that there are independent mechanisms of tumor progression in this disease (26). Although integrative genomics is still in its infancy, these types of approaches have tremendous potential to impact cancer biology and translational research as they may provide insight into more generalized mechanisms at work in carcinogenesis, and thus targets for therapeutic intervention which could have broader impact.
In pleural mesotheliomas, we investigated both genetic and epigenetic alterations using high-throughput, array based approaches and established and robust analytical strategies with the objective of gaining a more complete understanding of how genetic and epigenetic alterations may interact to drive tumorigenesis.
Materials and Methods
Study population
Mesotheliomas (n=158) and grossly non-tumorigenic parietal pleura (n=18) were obtained following surgical resection at Brigham and Women’s Hospital through the International Mesothelioma Program from a pilot study conducted in 2002 (n=70) and an incident case series beginning in 2005 (n=88) as previously described (27). All patients provided informed consent under the approval of the appropriate Institutional Review Boards. Clinical information, including histologic diagnosis was obtained from pathology reports. The study pathologist confirmed the histologic diagnoses and further assessed the percent tumor from resected specimens (mean >60%).
DNA extraction and methylation analysis
DNA from fresh frozen tissue and matched whole blood was isolated with QIAamp DNA mini kit (Qiagen, Valencia, CA) following the manufacturer’s protocol. Tumor DNA was modified with sodium bisulfite using the EZ DNA Methylation Kit (Zymo Research, Orange, CA). Illumina GoldenGate® methylation bead arrays interrogated 1505 CpG loci associated with 807 cancer-related genes processed at the UCSF Institute for Human Genetics, Genomics Core Facility using methods described in (28). The GoldenGate methylation data used in the analysis has been previously described (24). Methylation array data are publicly available via GEO (GSE20989).
SNP 500K mapping array for copy number analysis
From the total study population, 23 tumors from the incident case series were chosen for copy number alteration profiling by hybridizing 5μl containing ≥ 50ng/μl of tumor or matched peripheral blood DNA to each of the two GeneChips® that comprise the Human Mapping 500K single-nucleotide polymorphism array set (Affymetrix, Santa Clara, CA) (29), following manufacturer protocols and standard operating procedures at the Harvard Partners Microarray Core servicing facility. Probe intensities at each locus were determined in the GCOS software and genotypes calls were generated using the Genotyping Analysis Software (Affymetrix) (30). Probe signals were normalized to their matched referent peripheral blood sample data using the Copy Number Analysis Tool v4.0.1 software (CNAT) (31) (Affymetrix) with median scaling and default tuning parameters, and copy number states were inferred by Hidden Markov Model analysis. SNP array data are publicly available via GEO (GSE21057), and the combined set of array data (methylation and SNP data) can be accessed via the GEO super-series GSE21058.
Statistical analysis
Illumina BeadStudio Methylation software was used for methylation dataset assembly. Fluorescent signalsfor methylated (Cy5) and unmethylated (Cy3) alleles give methylation level: β= (max(Cy5, 0))/(|Cy3| +|Cy5| + 100) with ~30 replicate bead measurements per locus. Detection P-values determined poor performing CpG loci (n=8), which were removed from analysis. X chromosome loci were also removed, leaving 1413 CpG loci associated with 773 genes. All 23 tumors analyzed for copy number alteration passed quality control.
Subsequent analyses were carried out in the R statistical software package (32) and were restricted to autosomal chromosomes. Kaplan-Meier survival strata and log-rank tests were used for univariate survival analyses and Cox proportional hazards models were used to control for potential confounders. For inference, data were clustered using a recursively partitioned mixture model (RPMM), and the number of classes was determined by recursively splitting the data via 2-class models, with Bayesian information criterion (BIC) used at each potential split to decide whether the split was to be maintained or abandoned as described in Houseman et al. (33) and employed in (24, 34).
GoldenGate CpG loci were matched to Affymetrix SNPs in the manner described in (35) (typically within 1kb). The local relationship between methylation and CN alterations at 1413 loci was tested by calculating the Pearson product-moment correlation coefficients, P-values were calculated via permutation test (5k permutations), and the qvalue package in R was used to correct for multiple comparisons (Q-values). To assess correlation at the gene (rather than locus) level, equivalent statistics were employed on CpGs matched to genes by chromosomal position, assigned promoter status if they were upstream of the TSS, and averaged by gene. Similarly, copy number calls were averaged together for all SNPs associated with a gene.
To investigate molecular alterations individually at each locus, two-sided, two-sample t-tests assuming unequal variance were used to compare methylation between the 23 tumors and 18 non-diseased tissues and a one-sample t-test was used with the normal log2 ratio assumed to be 0, for copy number alterations. Copy number log2 ratios were used in this analysis due to the possibility that discretization imposed by CNAT might bias associations towards zero. Mean alteration differences were considered statistically significant where P < 0.05. The association between DNA copy number and methylation class membership was tested using the mean value of |CNS-2|, where CNS is the copy number state of each of 500,446 loci, as defined by the CNAT. A permutation test with 5,000 iterations using the Kruskal-Wallis test statistic was performed.
Results
A representative series of mesotheliomas and their matched peripheral blood-derived DNA as referents (n=23) from the total study population were subjected to 500K SNP mapping arrays to assess copy number (CN) alteration (Supplementary Table 1). A summary of the copy number alterations identified is provided in Supplementary Table 2. Consistent with previous reports (36–38), we observed prevalent allele loss at 1p36 (35%), 1p21.3 (30%), 4q22 (30%), 4q31–32 (35%), 3p21.3 (44%), 6q25 (39%), 9p21 (39%), and 22q (44%). In addition, we observed prevalent gains at1q23 (35%), 5p (22%), 7p (22%), and 8q24 (22%). Across tumor samples, the overall prevalence of CN losses and CN gains varied widely, with a mean prevalence of CN loss events of 8% (Standard Deviation (SD) = 9%), and a mean prevalence of CN gains of 5% (SD = 6%, Supplementary Table 3). Tests for association between extent of CN alteration and age, gender, histology, and asbestos exposure burden were not statistically significant.
Using prior data generated from interrogating DNA methylation in 158 mesotheliomas (24) we applied recursively partitioned mixture modeling (RPMM) to infer methylation profile classes wherein the 23 tumors profiled for CN alterations were included. This approach built classes of samples based on profiles of methylation with data from all measured autosomal loci using a mixture of beta distributions to recursively split the tumors into parsimoniously differentiated classes (39–41). Since methylation profile classes from the established model of all 158 mesotheliomas represent a more stable classification, we did not model the subset of 23 tumors with CN data separately. Instead, original class membership data were used in our analyses and among the seven RPMM methylation classes, the 23 tumors with CN data had a distribution of class membership that included all but methylation class five. Tumors in methylation class two were previously shown to be from patients with significantly lower asbestos burden than other classes, and relative to these class two cases, classes four and seven had significantly poorer prognosis (24).
Figure 1 directly compares copy number alterations and DNA methylation state in side-by-side, chromosome-specific plots of these alterations arranged by methylation class membership. Remaining chromosomes are illustrated in Supplementary Figure 1. Qualitatively, when focusing on single loci or specific genes, there was no obvious or consistent pattern of altered methylation when CN alterations were present. However, we next quantitatively evaluated whether – consistent with Knudson’s two hit paradigm of gene inactivation – coordinate dysregulation of CN state and methylation was present. No loci were found to have a significant correlation (Q < 0.05) between CN and methylation state across the examined tumors (Supplementary Table 4).
Figure 1.

Side by side plots of chromosome-specific mesothelioma copy number alteration and DNA methylation profiles ordered by RPMM methylation class. Left panel illustrates copy number alteration, SNP loci are matched to the nearest CpG locus and oriented coordinately along the chromosome, green=loss, red=gain. Right panel illustrates DNA methylation status at gene-loci listed, blue=methylated, yellow=unmethylated. A) Chromosome 1. B) Chromosome 6.
By averaging CN and methylation state within gene-specific regions we expanded this approach to a gene-level query. Only ITGB1 and FYN had significantly correlated (Q < 0.05) CN and methylation among 663 examined genes (Supplementary Table 4). However, Figure 2 shows that these significant correlations were not due to coordinate allele loss and hypermethylation. Other genes with highly correlated CN and methylation were also plotted. CDNKN2A had widespread methylation of one of its target CpGs, though this site is normally methylated in non-tumor pleura (Figure 2). Only two genes – TGFB2 and GDF10 – had any visually detectable coordinate methylation and CN loss and in each case only one tumor had both alterations (Figure 2). Though not among the genes in Supplementary Table 4, RASSF1A is commonly inactivated in mesothelioma and in our samples 7 tumors (30%) had allele loss, 2 (22%) were hypermethylated (Figure 2), and consistent with results above, only one tumor had coordinate methylation and CN loss.
Figure 2.
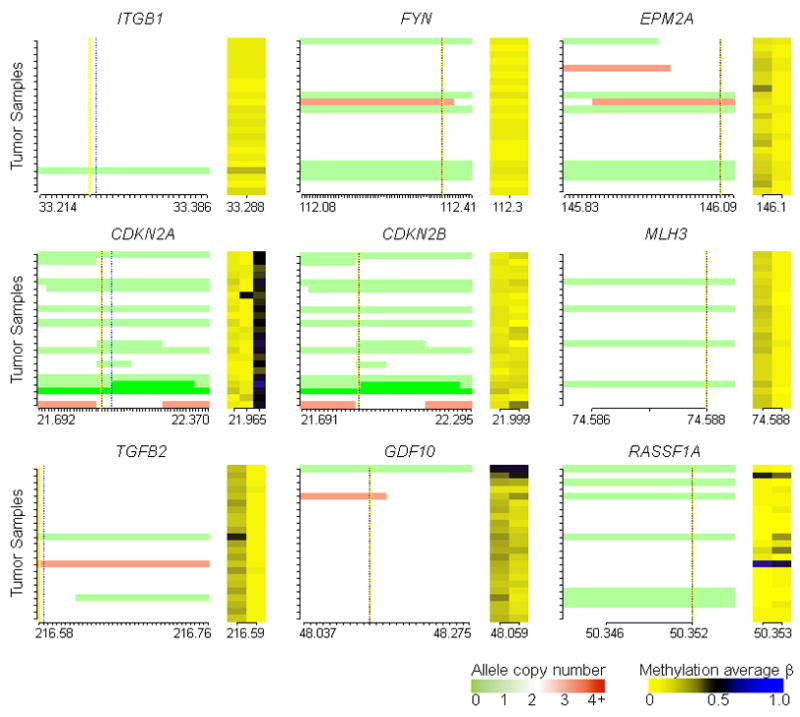
Gene-specific integrated copy number and methylation plots indicate a lack of coordinate copy number loss and hypermethylation events in mesotheliomas. Each ordinate tick mark represents a tumor sample, equivalently ordered across individual plots. In the left portion of the plot for each gene vertical yellow and dashed lines mark the positions of the TSS and CpG loci respectively, red represents amplified alleles, white represents two alleles, and green represents deleted alleles. The abscissa displays a tick mark for each SNP on the mapping array arranged 5′ to 3′ for each gene, and lists the first and last chromosomal position for SNP probes plotted (x10−6, not scaled to genomic distance) and the position of the CpG(s)(x10−6). The right portion of each plot shows methylation average β values, dark blue for fully methylated and bright yellow for unmethylated.
To assess whether global, rather than discrete trends for methylation and/or CN alteration might be observed, we plotted the magnitude and direction of CN and methylation alterations (log2 ratios versus a ratio of 0 for unaltered CN and tumor versus non-tumor methylation at each gene) to genes by their associated P-value (Figure 3A). The volcano plot of CN alterations was skewed to the left, indicating genome-wide trends for gene-level CN losses (15,790 genes). Similarly, the volcano plot of methylation alterations was also skewed to the left, indicating a genome-wide trend for loss of methylation relative to non-tumor pleural samples (663 genes). In order to determine whether CN and methylation alterations differ based on methylation profile, we split samples by the first partition of RPMM methylation classes. More specifically, the unsupervised nature of the RPMM model allows stratification of samples by branches of its associated dendrogram, since the model recursively partitions the methylation data, class membership can be retraced to the original parent methylation classes, the left branch and right branch class. Here, the “left branch” samples are those belonging to methylation classes 1 or 2 (n=10) the daughter classes of the initial left branch class while the “right branch” samples are those belonging to methylation classes 3 – 7 (n=13), the daughter classes of the initial right branch RPMM class. The volcano plots for CN alterations were strikingly different between methylation class branches: tumors in the left branch classes of the RPMM had far more loci with significantly altered CN compared to tumors in the right branch of RPMM (Figure 3B). Tumors in the left branch had a wider range of methylation alteration compared to tumors in the right branch, indicating that the trend toward an overall increase or decrease in the degree of methylation was associated with allele copy number loss. These results prompted further investigation of the global association between CN alterations and methylation state.
Figure 3.

Log significance plots for mean copy number and methylation alterations indicate global copy number losses, global hypomethyaltion, and differential copy number alteration by methyaltion class. Each region was compared to its expected normal value (normal tissue betas for methylation and log2=0 for CN alteration) and t-tests were performed. Negative log-transformed P-values (generated by tumor/normal t-tests) are plotted versus mean alteration of copy number or methylation. The space above the dotted line represents a significance level of P < 0.05. A) Promoter-associated copy number-altered genes (n=15,790, top) and methylation alterations (663 genes, bottom) are shown overall and B) stratified by RPMM methylation class structure (Left branch classes, n=10, Right branch classes, n=13) as indicated.
The evidence for a global relationship between CN alterations and methylation alterations could suggest that CN alterations of master epigenetic regulatory genes influence overall tumor methylation. Maintenance methyltransferase DNMT1 had CN loss in 7 tumors (30%, Supplementary Figure 1, Chromosome 19) and de novo methyltransferase DNMT3B had no CN alterations (Supplementary Figure 1, Chromosome 20). Plotting the average CpG methylation for tumors with DNMT1 allele loss versus tumors without loss, we observed a significant trend for increased methylation among tumors with no allele loss at DNMT1 compared to tumors with allele loss (Figure 4, P = 0.05). Further, DNMT1 allele loss was associated with significantly reduced patient survival in a Cox proportional hazards model controlling for age, gender and tumor histology (HR, 5.07; 95% C.I. 1.23 – 20.9, Supplementary Table 5). In similar investigations of alterations of critical DNA double strand break repair genes (e.g. ATM, XRCC4) prevalent CN alterations or methylation silencing events were not observed.
Figure 4.

Tumors without DNMT1 allele loss have increased methylation relative to tumors with DNMT1 allele loss. Mean average β methylation values for tumors with allele loss at DNMT1 are plotted versus mean average β methylation for tumors without allele loss for each of the 1413 autosomal CpG loci. The line has an intercept of zero and a slope of one to help illustrate the relatively higher methylation values among tumors without DNMT1 allele loss.
In an effort to better visualize and test the global trends of association between methylation and CN alterations we plotted the CN alteration profiles of tumors based on RPMM methylation class membership (Figure 5). This plot illustrates the significant difference in the extent of CN alterations among methylation classes (Permutation test P < 0.02). Tumors in methylation classes two, six, and seven exhibit a greater extent of CN alteration than tumors in methylation classes one, three or four (Figure 5). The means and standard errors of class-specific percents of SNPs with CN alterations are also illustrated in Figure 5.
Figure 5.

Plot of mesothelioma copy number alteration profiles based on class membership from recursively partitioned mixture model (RPMM) of DNA methylation data indicates a global correlation between methylation class and extent of copy number alteration. The RPMM was generated with DNA methylation data from 158 tumors as we reported previously; the original RPMM contained seven classes, and the 23 tumors plotted here represent membership in six of the seven classes, where none of the 23 tumors were members of the original RPMM class 5. In the copy number profiles green indicates loss and red indicates gain. The right panel of the figure displays the mean (circle) and standard error (line) of methylation-class-specific percent of SNP loci with altered CN. Grey triangles in the middle portion of the figure indicate which samples have allele loss at DNMT1.
Discussion
The phenotype of any individual tumor arises as the result of a constellation of somatic alterations, and only recently has technology afforded the opportunity to profile and examine multiple types of alterations on a genome-wide scale. In an effort to further our understanding of the relationship between two forms of somatic alterations known to be important in the genesis of mesothelioma, we profiled CN alterations in a subset of tumors for which we had available DNA methylation profiling data. Investigating single loci and specific genes, we found no significant correlations between CN and methylation, indicating that two-hit gene inactivation is not commonly achieved by coordinate hypermethylation and allele loss in mesothelioma. However, when looking genome-wide, we found that the extent of CN alterations was significantly related to the DNA methylation profiles of these tumors, suggesting a strong link between genetic and epigenetic dysregulation in mesothelioma. The association between asbestos and mesothelioma carcinogenesis is well established, asbestos fibers are known to be clastogenic, and patient asbestos burden has been associated with methylation alterations in mesothelioma (3, 24). Combined with the known, low mutagenic action of asbestos (e.g. mesotheliomas almost never harbor TP53 mutations, and (5)), these tumors are an excellent candidate for integrative genomic profiling of copy number and epigenetic alterations.
It is possible that the observed global association between CN alteration and methylation could be influenced by a bias in methylation measurement at regions with CN alteration. If methylation measurements were biased by CN state, the bias could contribute to the classification of DNA methylation profiles modeled by RPMM. In fact, previous work has indicated certain microarray platforms will be impacted by just such a bias (42). However, we recently performed an extensive investigation including multiple tumor types and showed that the Illumina GoldenGate bead-array-based platform for methylation measurement is not subject to significant bias from CN alterations (35).
Despite generally extensive CN and methylation alterations, we observed very few instances of locally coordinate allele loss and methylation in these tumors. Although two genes had significant correlations between CN loss and methylation neither case had high enough methylation levels to be consistent with gene-silencing. On the other hand, among other genes with highly-ranked correlation between methylation and CN alteration there were two single instances of coordinate CN loss and hypermethylation, one at TGFB2 and one tumor at GDF10. The specific functions of TGFB2 are dependent upon cell type and presence of both TGF-β type II and type I receptors (43), and in mesothelioma it is thought that TGFB2 may play complex roles in growth and regulation of immune responses via cytokine production (44). Interestingly, the other target of coordinate methylation and CN loss was GDF10, which encodes bone morphogenic protein 3b (BMP3b); the BMPs are part of the transforming growth factor-β superfamily, and a low prevalence of GDF10 methylation has previously been shown in mesotheliomas (45).
Despite a lack of locally coordinated CN and methylation alterations, we observed strong evidence for an association between genome-wide extent of CN alteration and methylation alterations. In fact, a global propensity for both CN and methylation losses was observed. The trend for methylation losses in these tumors was previously reported in the total study population (n=158): relative to non-tumor pleura, among 1413 autosomal CpG loci nearly 1000 CpGs had significantly altered methylation between non-tumor pleura and mesotheliomas (24), and over 75% were losses of methylation. Hence, one potential explanation for the lack of coordinate CN loss and hypermethylation is a low prevalence of hypermethylation events in these tumors. In the context of a propensity for methylation loss, further investigation of the global coordination between CN alterations and methylation lead us to the methyltransferases themselves. Not only was there prevalent CN loss of the maintenance methyltransferase, but tumors without DNMT1 loss had higher average methylation across all CpGs than those with loss. It is reasonable to suggest that loss of DNMT1 may also result in hypomethylation of repeat elements and thereby further contribute to genomic instability and increased CN alterations, though additional investigations are necessary. Furthermore, we found that loss of DNMT1 was associated with a significantly increased risk of death. We have previously reported associations between methylation profile class membership and survival in mesothelioma (46). It is possible that DNMT1 loss is also associated with methylation class membership, (potentially those classes with significantly poorer survival) although additional investigation is necessary. Nonetheless, when studying human tumors it may not be possible to estimate the timing of critical events that contribute to dysregulation of genetic and/or epigenetic maintenance.
We have shown that fulfillment of the two-hit paradigm is generally not achieved by coordinate methylation and CN alteration mechanisms in mesothelioma, but that there is a strong association between CN alteration and methylation state in these tumors. Initial exploration of the cause(s) behind overall CN and methylation associations indicate promise for the investigation of master regulatory genes (such as the DNA methyltransferases) in additional studies with integrative genomic approaches. Other forms of gene inactivation mechanisms not measured here (e.g. miRNA/mRNA alterations, gene mutation) may still act coordinately with methylation or CN alteration to fulfill the two hit paradigm at a gene-specific level, may also be globally associated with one another, and should be investigated in future studies. Certainly, further studies including higher-resolution methylation arrays, additional forms of somatic alteration, and/or different tumor types with distinct etiologies are all warranted. In fact, in solid tumors of the head and neck, our group has now reported correlation between overall CN alteration extent and methylation profiles in the absence of gene-specific coordinate inactivation events, strong evidence that our findings may extend to many other forms of cancer (47). Collectively, these data further demonstrate the feasibility and promising utility of integrative genomics approaches for the advancement of cancer biology and translational research.
Supplementary Material
Acknowledgments
Financial Support: R01CA126939, R01CA100679, International Mesothelioma Program at Brigham and Women’s Hospital (Research grant), Mesothelioma Applied Research Foundation (Research grant).
Footnotes
Conflicts of Interest: None
References
- 1.Robinson BW, Lake RA. Advances in malignant mesothelioma. The New England journal of medicine. 2005;353:1591–603. doi: 10.1056/NEJMra050152. [DOI] [PubMed] [Google Scholar]
- 2.Jaurand MC. Mechanisms of fiber-induced genotoxicity. Environmental health perspectives. 1997;105 (Suppl 5):1073–84. doi: 10.1289/ehp.97105s51073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelsey KT, Yano E, Liber HL, Little JB. The in vitro genetic effects of fibrous erionite and crocidolite asbestos. British journal of cancer. 1986;54:107–14. doi: 10.1038/bjc.1986.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yegles M, Saint-Etienne L, Renier A, Janson X, Jaurand MC. Induction of metaphase and anaphase/telophase abnormalities by asbestos fibers in rat pleural mesothelial cells in vitro. Am J Respir Cell Mol Biol. 1993;9:186–91. doi: 10.1165/ajrcmb/9.2.186. [DOI] [PubMed] [Google Scholar]
- 5.Sugarbaker DJ, Richards WG, Gordon GJ, et al. Transcriptome sequencing of malignant pleural mesothelioma tumors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3521–6. doi: 10.1073/pnas.0712399105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirao T, Bueno R, Chen CJ, Gordon GJ, Heilig E, Kelsey KT. Alterations of the p16(INK4) locus in human malignant mesothelial tumors. Carcinogenesis. 2002;23:1127–30. doi: 10.1093/carcin/23.7.1127. [DOI] [PubMed] [Google Scholar]
- 7.Xio S, Li D, Vijg J, Sugarbaker DJ, Corson JM, Fletcher JA. Codeletion of p15 and p16 in primary malignant mesothelioma. Oncogene. 1995;11:511–5. [PubMed] [Google Scholar]
- 8.Balsara BR, Bell DW, Sonoda G, et al. Comparative genomic hybridization and loss of heterozygosity analyses identify a common region of deletion at 15q11.1–15 in human malignant mesothelioma. Cancer research. 1999;59:450–4. [PubMed] [Google Scholar]
- 9.Huncharek M. Genetic factors in the aetiology of malignant mesothelioma. Eur J Cancer. 1995;31A:1741–7. doi: 10.1016/0959-8049(95)00365-p. [DOI] [PubMed] [Google Scholar]
- 10.Whitaker D. The cytology of malignant mesothelioma. Cytopathology. 2000;11:139–51. doi: 10.1046/j.1365-2303.2000.00247.x. [DOI] [PubMed] [Google Scholar]
- 11.Murthy SS, Testa JR. Asbestos, chromosomal deletions, and tumor suppressor gene alterations in human malignant mesothelioma. J Cell Physiol. 1999;180:150–7. doi: 10.1002/(SICI)1097-4652(199908)180:2<150::AID-JCP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 12.Lu YY, Jhanwar SC, Cheng JQ, Testa JR. Deletion mapping of the short arm of chromosome 3 in human malignant mesothelioma. Genes Chromosomes Cancer. 1994;9:76–80. doi: 10.1002/gcc.2870090114. [DOI] [PubMed] [Google Scholar]
- 13.Bell DW, Jhanwar SC, Testa JR. Multiple regions of allelic loss from chromosome arm 6q in malignant mesothelioma. Cancer research. 1997;57:4057–62. [PubMed] [Google Scholar]
- 14.Bjorkqvist AM, Tammilehto L, Anttila S, Mattson K, Knuutila S. Recurrent DNA copy number changes in 1q, 4q, 6q, 9p, 13q, 14q and 22q detected by comparative genomic hybridization in malignant mesothelioma. British journal of cancer. 1997;75:523–7. doi: 10.1038/bjc.1997.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He B, Lee AY, Dadfarmay S, et al. Secreted frizzled-related protein 4 is silenced by hypermethylation and induces apoptosis in beta-catenin-deficient human mesothelioma cells. Cancer research. 2005;65:743–8. [PubMed] [Google Scholar]
- 16.Lee AY, He B, You L, et al. Expression of the secreted frizzled-related protein gene family is downregulated in human mesothelioma. Oncogene. 2004;23:6672–6. doi: 10.1038/sj.onc.1207881. [DOI] [PubMed] [Google Scholar]
- 17.Murthy SS, Shen T, De Rienzo A, et al. Expression of GPC3, an X-linked recessive overgrowth gene, is silenced in malignant mesothelioma. Oncogene. 2000;19:410–6. doi: 10.1038/sj.onc.1203322. [DOI] [PubMed] [Google Scholar]
- 18.Ohta Y, Shridhar V, Kalemkerian GP, Bright RK, Watanabe Y, Pass HI. Thrombospondin-1 expression and clinical implications in malignant pleural mesothelioma. Cancer. 1999;85:2570–6. [PubMed] [Google Scholar]
- 19.Shigematsu H, Suzuki M, Takahashi T, et al. Aberrant methylation of HIN-1 (high in normal-1) is a frequent event in many human malignancies. International journal of cancer. 2005;113:600–4. doi: 10.1002/ijc.20622. [DOI] [PubMed] [Google Scholar]
- 20.Shivapurkar N, Toyooka S, Toyooka KO, et al. Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types. International journal of cancer. 2004;109:786–92. doi: 10.1002/ijc.20041. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki M, Toyooka S, Shivapurkar N, et al. Aberrant methylation profile of human malignant mesotheliomas and its relationship to SV40 infection. Oncogene. 2005;24:1302–8. doi: 10.1038/sj.onc.1208263. [DOI] [PubMed] [Google Scholar]
- 22.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–16. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 23.Christensen BC, Godleski JJ, Marsit CJ, et al. Asbestos exposure predicts cell cycle control gene promoter methylation in pleural mesothelioma. Carcinogenesis. 2008;29:1555–9. doi: 10.1093/carcin/bgn059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christensen BC, Houseman EA, Godleski JJ, et al. Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer research. 2009;69:227–34. doi: 10.1158/0008-5472.CAN-08-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng YW, Pincas H, Bacolod MD, et al. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin Cancer Res. 2008;14:6005–13. doi: 10.1158/1078-0432.CCR-08-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen BC, Marsit CJ, Houseman EA, et al. Differentiation of Lung Adenocarcinoma, Pleural Mesothelioma, and Nonmalignant Pulmonary Tissues Using DNA Methylation Profiles. Cancer research. 2009;69:6315–21. doi: 10.1158/0008-5472.CAN-09-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bibikova M, Lin Z, Zhou L, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome research. 2006;16:383–93. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bignell GR, Huang J, Greshock J, et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004;14:287–95. doi: 10.1101/gr.2012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu WM, Di X, Yang G, et al. Algorithms for large-scale genotyping microarrays. Bioinformatics. 2003;19:2397–403. doi: 10.1093/bioinformatics/btg332. [DOI] [PubMed] [Google Scholar]
- 31.Huang J, Wei W, Zhang J, et al. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics. 2004;1:287–99. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.R Development CT. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2007. [Google Scholar]
- 33.Houseman EA, Christensen BC, Yeh RF, et al. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics. 2008;9:365. doi: 10.1186/1471-2105-9-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marsit CJ, Christensen BC, Houseman EA, et al. Epigenetic Profiling Reveals Etiologically Distinct Patterns of DNA Methylation in Head and Neck Squamous Cell Carcinoma. Carcinogenesis. 2009 doi: 10.1093/carcin/bgp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houseman EA, Christensen BC, Karagas MR, et al. Copy number variation has little impact on bead-array-based measures of DNA methylation. Bioinformatics. 2009;25:1999–2005. doi: 10.1093/bioinformatics/btp364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krismann M, Muller KM, Jaworska M, Johnen G. Molecular cytogenetic differences between histological subtypes of malignant mesotheliomas: DNA cytometry and comparative genomic hybridization of 90 cases. The Journal of pathology. 2002;197:363–71. doi: 10.1002/path.1128. [DOI] [PubMed] [Google Scholar]
- 37.Taniguchi T, Karnan S, Fukui T, et al. Genomic profiling of malignant pleural mesothelioma with array-based comparative genomic hybridization shows frequent non-random chromosomal alteration regions including JUN amplification on 1p32. Cancer Sci. 2007;98:438–46. doi: 10.1111/j.1349-7006.2006.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindholm PM, Salmenkivi K, Vauhkonen H, et al. Gene copy number analysis in malignant pleural mesothelioma using oligonucleotide array CGH. Cytogenetic and genome research. 2007;119:46–52. doi: 10.1159/000109618. [DOI] [PubMed] [Google Scholar]
- 39.Shen L, Toyota M, Kondo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18654–9. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siegmund KD, Connor CM, Campan M, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegmund KD, Laird PW, Laird-Offringa IA. A comparison of cluster analysis methods using DNA methylation data. Bioinformatics (Oxford, England) 2004;20:1896–904. doi: 10.1093/bioinformatics/bth176. [DOI] [PubMed] [Google Scholar]
- 42.Pfister S, Schlaeger C, Mendrzyk F, et al. Array-based profiling of reference-independent methylation status (aPRIMES) identifies frequent promoter methylation and consecutive downregulation of ZIC2 in pediatric medulloblastoma. Nucleic acids research. 2007;35:e51. doi: 10.1093/nar/gkm094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marzo AL, Fitzpatrick DR, Robinson BW, Scott B. Antisense oligonucleotides specific for transforming growth factor beta2 inhibit the growth of malignant mesothelioma both in vitro and in vivo. Cancer research. 1997;57:3200–7. [PubMed] [Google Scholar]
- 44.Valle MT, Porta C, Megiovanni AM, et al. Transforming growth factor-beta released by PPD-presenting malignant mesothelioma cells inhibits interferon-gamma synthesis by an anti-PPD CD4+ T-cell clone. Int J Mol Med. 2003;11:161–7. [PubMed] [Google Scholar]
- 45.Kimura K, Toyooka S, Tsukuda K, et al. The aberrant promoter methylation of BMP3b and BMP6 in malignant pleural mesotheliomas. Oncol Rep. 2008;20:1265–8. [PubMed] [Google Scholar]
- 46.Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poage GP, Christensen BC, Houseman EA, et al. Genetic and epigenetic somatic alterations in head and neck squamous cell carcinomas are globally coordinated but not locally targeted. PLoS ONE. 2010:e0009651. doi: 10.1371/journal.pone.0009651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.