

Abstract
Many chronic inflammatory diseases are associated with increased risk of developing cancer. In the colon, strong support for a link between chronic inflammation and cancer extends, in part, from population-based studies of persons with inflammatory bowel disease (IBD). Patients with IBD are at increased risk of developing colorectal cancer (CRC). The general consensus is that IBD results from the combined effects of genetics and environment factors known to affect the immune system. Vitamin D, an important regulator of the immune system, has been linked to IBD. Despite the strong potential reported for 1,25-dihydroxyvitamin D (1,25-OH)2D), its strong effects on calcium metabolism limits its potential value. Recently, less active vitamin D metabolites, cholecalciferol and 25-hydroxyvitamin D (25(OH)D), have gained considerable attention as promising agents against IBD-related colon cancer. Yet, their anti-proliferative properties and mechanism of action remain to be better defined. We present several signaling pathways commonly regulated by vitamin D compounds and highlight their regulation on TLR4. The efficacy of 25(OH)D and 1α-hydroxyviatmin D5 are defined using the azoxymethane (AOM)/dextran sodium sulfate (DSS)-induced IBD-related colon carcinogenesis model. In summary, vitamin D supplementation may provide a cost-effective approach to reduce IBD related colon cancer.
Keywords: Vitamin D, inflammation, colon cancer
I. Introduction
Inflammation is a risk factor for many common malignancies including cancers of the lung, breast, and colon (1). The clearest link between inflammation and colon cancer is seen in patients with inflammatory bowel disease (IBD) (2). Colorectal cancer is one of the most serious complications of IBD, accounting for increased mortality in these disorders (2). The severity of inflammation correlates with the risk of colorectal cancer in patients with IBD (3). Epidemiological studies have reported that several environmental factors such as diet may play a pivotal role in mediating the occurrence, severity and neoplastic progression of this IBD (4). For example, vitamin deficiencies in general and vitamin D insufficiency in particular have been shown to occur in IBD patients. Furthermore, studies have shown that people living in areas that receive less sunlight have lower circulating 25(OH)D levels and higher prevalence rates of IBD (5). This link between low serum 25(OH)D levels and inflammation-associated colon cancer offers the possibility of identifying a mode to prevent cancer with vitamin D supplementation.
While the molecular mechanisms whereby chronic inflammation predisposes to cancer remain elusive, animal models have been instrumental in demonstrating a link between vitamin D and risk for inflammation-associated colon cancer. For example, Interleukin (IL)-10 knockout (KO) mice, which spontaneously develop symptoms resembling IBD, have been widely used to study the pathogenesis as well as the therapeutic and preventive strategies for IBD. Cantorna et al (6) investigated the role of vitamin D using IL-10 KO mice. For this, mice were fed diets deficient in vitamin D and compared to mice fed chow containing adequate levels vitamin D and/or diets supplemented with pharmacological concentrations of the active metabolite, 1,25(OH)2D. While the IL-10 KO mice fed the vitamin D sufficient chow experienced IBD symptoms beginning at 12 weeks of age, the mice receiving vitamin D deficient diets had accelerated symptoms with 60% mortality rates before week 9 of age. In contrast, pharmacological concentrations of 1,25(OH)2D alleviated the symptoms in IL-10 KO mice with pre-existing symptoms of IBD. In another study, the significance of the vitamin D receptor (VDR) in immune function was investigated using VDR KO, IL-10 and double KO (VDR/IL-10) mice (7). It was shown that colons from the double KO mice exhibit a more severe form of IBD accompanied with over-expression of numerous inflammatory cytokines.
Consequently, a focus on the mechanism by which vitamin D mediates pro-inflammatory pathways (TLR4, cytokines) may provide insights into the prevention of colitis-associated cancer. Notwithstanding the numerous reports demonstrating the curative action of 1,25(OH)2D its potential usefulness as a chemopreventive agent are limited by its propensity to cause hypercalcemia. Recently, less active vitamin D metabolites such as dietary vitamin D3 (cholecalciferol) or 25(OH)D have gained considerable attention, in part, due the presence of 1α-hydroxylase. Yet reports on the anti-proliferative properties and mechanism of action of these vitamin D metabolites are limited. In the present study, the anti-proliferative actions and time-dependent effects on the expression of CYP27A1, CYP27B1, CYP24 and VDR mRNA levels were determined for several vitamin D metabolites (D3, 25(OH)D, 1,25(OH)2D and 1α-hydroxyvitamin D5 [1α(OH)D5] using the human colon cancer cell line HT29. Moreover, gene array analysis assisted to identify five common canonical pathways regulated by various vitamin D metabolites; The Toll-Like Receptor 4 (TLR4) pathway was the most statistically significant regulated. The common effects on inflammation associated signaling pathways by the vitamin D metabolites directed our interest in evaluating the efficacy of the less active vitamin D metabolites using the AOM/DSS-induced inflammation-associated CRC model.
II. Materials and Methods
Chemicals and supplies
All chemicals were purchased at the highest purity available. D3 (Cholecalciferol) was purchased Sigma-Aldrich (St. Louis, MO); 1,25(OH)2D was obtained from Cayman Chemical Co. whereas 25(OH)D and dextran sulfate sodium salt (MW 36,000-50,000) were purchased from MP Biomedicals, LLC.
Cell culture
Human colon cancer cell lines (Caco-2, HCT116 and HT-29) were obtained from American Type Culture Collection (Manassas, VA). Cell lines were maintained and cultured in either RPMI 1640 media (HT29) or MEM with Earle's salts (Caco-2, HCT116) (Life Technologies, Inc., Grand Island, NY) with 10% FBS, and 1% antibiotic-antimycotic solution at 37°C in a humidified atmosphere of 5% CO2.
Analysis of cell proliferation
Cells were seeded at a density of 5× 103 in 24-well cell culture plates and allowed to adhere for 24 h After incubation with or without 1,25(OH)2D (100 nM), 25(OH)D, D3, or 1α(OH)D5 (1 μM) for various time points, the study was terminated. Cells were trypsinized and cell number determined by Z1 Coulter Particle Counter (Beckman Coulter, Fullerton, CA).
RNA Extraction, qRT-PCR, and microarray analysis
HT29 cells were treated with ethanol control or 1,25(OH)2D (100 nM), 25(OH)D (1 μM), or 1α(OH)D5 (1 μM) for 24 h. Samples were processed as previously described (8). Next, the samples were hybridized to the Human Genome U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA). In GeneSpring v7.2 (Agilent Technologies, Santa Clara, CA) .cell files were preprocessed using Robust Multichip Average (RMA) and genes were normalized to the mean expression of the control sample. Canonical pathways were analyzed through the use of Ingenuity Pathway Analysis (IPA; Ingenuity Systems, www.ingenuity.com). For qRT-PCR, total RNA extraction and the RT reaction were performed as described previously (9). Briefly, qRT-PCR was performed with 2 μl diluted RT product in a MyiQ Real-time PCR Detection System (Bio-Rad, Hercules, CA) by using iQ™ SYBR Green PCR Supermix (Bio-Rad) according to manufacturer's guidelines. The PCR cycling conditions used were: (15 s at 95°C, 15 s at 60°C and 20 s at 72°C) for 40 cycles. Fold inductions were calculated using the formula 2-(ΔΔCt), where ΔΔCt is ΔCt(treatment) - ΔCt(control), ΔCt is Ct(target gene) − Ct(actin) and Ct is the cycle at which the threshold is crossed. PCR product quality was monitored using post-PCR melt curve analysis. Experiments were performed in triplicates for genes of interest and for actin controls.
Inflammation-associated Colon Carcinogenesis Model
To investigate the role of vitamin D compounds in inflammation-associated colon cancer, we used the AOM/DSS-induced carcinogenesis model. For this study, six week old female CF1 mice were purchased from Charles Rivers (Wilmington, MA) and randomized into one of five groups (control, D3, 25(OH)D, 1,25(OH)2D, or 1α(OH)D5). One week prior to carcinogen exposure, animals were placed on experimental diets. At week 1, mice received one injection of AOM (10mg/kg body weight). This was followed by administered 2.5% DSS in the drinking water for the duration of 7 days (week 2). The mice were continued on the diet until the time of termination (Week 16). Tumor incidence and multiplicity were determined at the time of sacrifice.
Statistical Analyses
All data were analyzed using GraphPad Prism V3.0 (GraphPad Software, Inc., San Diego, CA). One-way analysis of variance (ANOVA) was used to determine significant differences between groups. If a significant difference (p <0.05) was observed, we used the Bonferroni t-test as a multiple comparison test.
III. Results
The growth inhibitory effects of D3, 25(OH)D, 1,25(OH)2D, and 1α(OH)D5 were compared using the human colon cancer cell line, HT-29. For these studies, HT29 cells were plated and treated with D3 (1 μM), 25(OH)D (0.5 or 1 μM), 1,25(OH)2D (0.1 μM), and 1α(OH)D5 (0.5 or 1 μM) for 96 hrs. As illustrated in Fig. 1A, compared to vehicle treated, all treatments significantly (p <0.01) inhibited the growth of HT-29 cells. Similar findings were demonstrated for in the Caco-2 cell line (data not shown). However, no significant inhibition was noted for the HCT116 cell line. The HCT116, express high baseline CYP24 mRNA levels (data not shown) and low VDR mRNA expression and therefore as expected they were not significantly inhibited by any of the vitamin D compounds.
Figure 1.
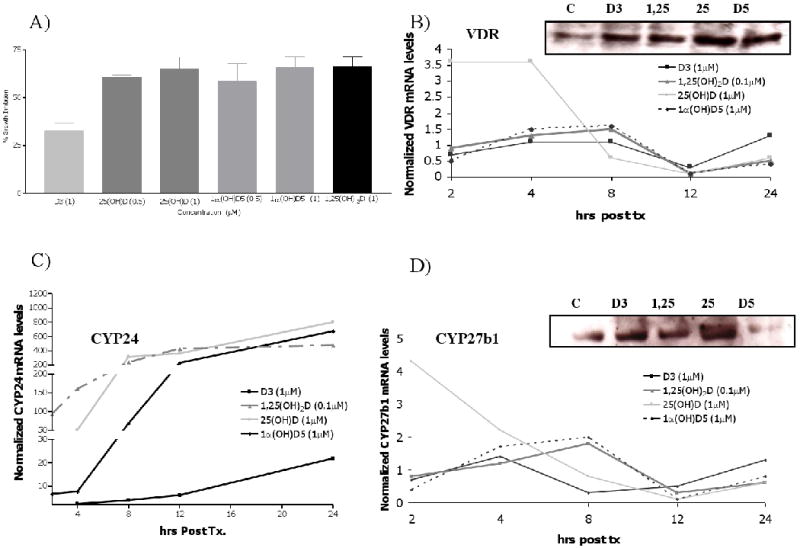
(a) Effects of vitamin D compounds on HT29 cell proliferation. Time dependent effects of vitamin D compounds on the mRNA expression for VDR (B), CYP24 (C), and CYP27b1 (D) in HT29 cells. Normalized values for vehicle-treated cells were set to 1 and fold induction by 1,25(OH)2D, 25(OH)D and 1α(OH)D5 are shown. RT-PCR experiments were conducted in triplicates and statistical analysis was determined an ANOVA (***p<0.001, ** p<0.01, ***p<0.05). Western blot analysis in HT-29 cells treated with [1] vehicle; [2] D3 (1 μM); [3] 1,25(OH)2D (0.1 μM); [4] 25(OH)D (1 μM); or [5] 1α(OH)D5 μM). Exposure to vitamin D metabolites resulted in a significant increase proteins levels for (B) VDR and 1α-hydroxylase (D) by 24 hr post treatment. Equal loading for each experiment is represented by separate actin bands.
In the last decade, evidence has been mounting which demonstrate the presence of the vitamin D metabolizing enzymes, 1α-hydroxylase, in a number of organs including the colon, lung, and breast. Reports have shown that these extra-renal organs are fully capable of metabolizing intermediate metabolites into to the active metabolite, 1α,25(OH)2D, via the local presence of 1α-hydroxylase (1-OHase; CYP27B1). Consequently, it has been suggested that raising blood levels of 25(OH)D may provide an adequate substrate for target tissues to produce their own local 1α,25(OH)2D. Likewise, the presence of 25-hydroxylase (25-OHase, CYP27A1) has been reported in human colon biopsies (10) and in prostate cancer cells (11, 12). Tokar et al (11) demonstrated the efficacy of cholecalciferol using human prostate cancer cells and demonstrate the presence of 25-hydroxylase in the cells. Lastly, target tissues have been shown to be fully capable of catabolizing 1,25(OH)2D by up-regulation of local production of 24-hydroxylase (24-OHase; CYP24). Accordingly, enzymatically active 24-OHase is expected to decrease the antiinflammatory properties of vitamin D and/or desensitize cancer cells to 1α,25(OH)2D. For this reason, we sought to investigate the time-dependent gene expression profile for VDR (Fig. 1B), CYP24 (Fig. 1C), and CYP27B1 (Fig. 1D). As illustrated in Fig. 1(B, C, and D), induction of steady state levels of VDR, CYP27B1, and CYP24 mRNA levels followed different expression patterns for each of the deltanoids. HT29 cells treated with 25(OH)D (1 μM) showed VDR and CYP27B1 mRNA levels significantly up-regulated 2 hrs post treatment; As expected, this increase was followed by a notable rise in CYP24 mRNA levels and decrease in CYP27B1 and VDR by 4 hrs. Exposure of HT29 cells with 1α(OH)D5 (1 μM) demonstrated a rise in VDR and CYP27B1 by 4 hrs, followed by a sharp rise in CYP24 and decreased CYP27B1 and VDR levels by 12 hr. Treatment with the active metabolite (0.1 μM) at 2hrs showed decreased VDR and CYP27B1 mRNA levels the highest rise in CYP24 expression. Lastly, these data demonstrate that D3 is capable of increasing CYP27B1 and VDR mRNA levels. Treatment with D3 significantly increased CYP27B1 (1.4 fold change) by 4hr; followed by an up-regulation of VDR mRNA levels at 8 hrs post treatment. Dietary vitamin D3, unlike the other deltanoids, showed a delay rise in CYP24 mRNA levels (24 hr). Since D3 appeared to be functionally effective at inducing CYP27B1 (Fig. 1D), we further investigated its effects on CYP27A1, the enzyme responsible for converting D3 to 25-hydroxycholecalciferol. We compared vehicle treated cells with those treated with 1,25(OH)2D (0.1 μM) and D3 (1.0 μM). These studies show that at 4 hr post treatment, CYP27A1 is significantly up-regulated in the D3 treated cells 3.09 ± 0.69 fold-change (p< 0.05), as compared to 1.5 ± 0.90 in the 1,25(OH)2D (p>0.05) treated cells. Given these results it is justifiable to further explore the use of D3 as a potential chemopreventive agent.
Building upon the results of our gene array studies, which facilitated the identification of five key signaling pathways (Fig. 2A) commonly regulated by vitamin D compounds, we sought to better elucidate the mediating effects of vitamin D on the most highly regulated pathway, that is, the TLR4 pathway. Using HT29 cells, we tested the effects of 25(OH)D (1μM), 1,25(OH)2D (0.1μM), and 1α(OH)D5 (1μM) on the selected genes by real-time-PCR (Fig. 2B) These studies confirm the previous findings showing the deltanoids significantly increase CD14 mRNA levels while decreasing the mRNA levels of TLR4, RAC1, RIP1, MYD88 and STAT1. Since both TLR4 and CD14 are believed to be early response genes, a second study was conducted to determine the time dependent effects on these two genes. Briefly, HT29 cells were treated with 25(OH)D (1μM), 1,25(OH)2D (0.1μM),D3 (1μM) for a period of 4, 8 or 24 hrs. These studies demonstrate that in comparison to vehicle, all treatment significantly up-regulated CD14 mRNA by 4 hrs. The mRNA levels for TLR4 mRNA were decreased following 8 hrs of treatment but continue attenuated at the 24 hr time point. Additional qRT-PCR studies were completed for MD-2, IRAK1, and IKKα. Figure 2C presents a schematic summary of the effects of vitamin D compounds on the TLR4 pathway. A decrease in activation by NF-κB was accompanied by a decreased expression of nuclear expression NF-κB (data not shown). Taken together these data provide support for additional investigations on the use of vitamin D as targeted therapy for inhibiting TLR4 especially in high risk populations (e.g., IBD patients with frequent relapses). In addition these data supports the use of the less active vitamin D metabolites [e.g., 25(OH)D and D3] for the prevention and treatment of IBD-related mucosal injury and CRC.
Figure 2.
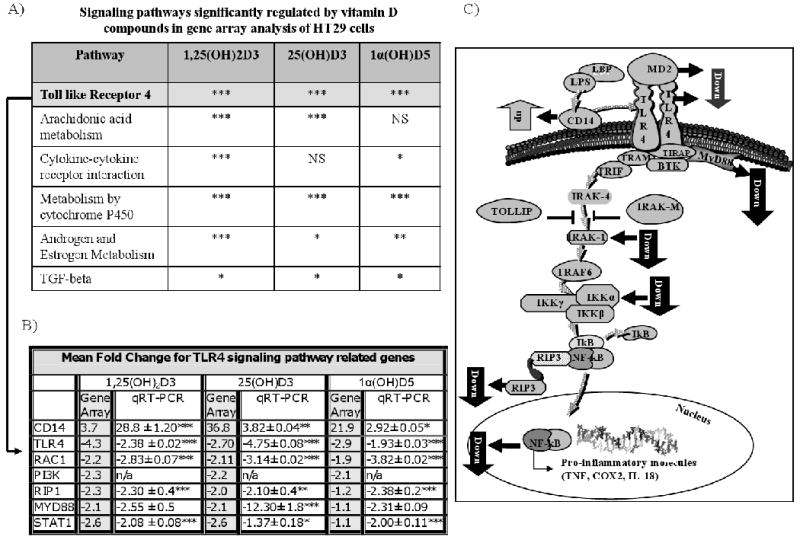
A) Gene array analysis established the five of six most highly significantly regulated pathways effectively activated and altered by 1,25(OH) 2D3 that were also common for 1α(OH)D5 and 25(OH)D. P-values for each of the corresponding pathways was determine for treatment group (*** p<0.001, ** p<0.01, * p<0.05); B) TLR4 pathway genes found to be regulated by vitamin D compounds and confirmed values via qRT-PCR. Vehicle-treated cells were set to 1 and mean fold induction by 1,25(OH) 2D, 25(OH)D and 1α(OH)D5 are shown. Statistical analysis was determined with an ANOVA (***p<0.001, ** p<0.01, ***p<0.05). D); C)schematic illustration of the effects that vitamin D has on the TLR4 pathway in HT29 cells.
An in vivo study was conducted to investigate the efficacy of vitamin D in IBD-related colon cancer. For these experiments, we chose to use the AOM/DSS-induced carcinogenesis model. The AOM/DSS-induced inflammation CRC model was selected because it facilitates the study of onset, severity and recovery from mucosal injury (a subset of mice were sacrificed 7days after DSS) and serves to assess the effects of vitamin D on tumor incidence. Figure 3A serves to illustrate the experimental protocol employed, describes the experimental diets, and the effects of the experimental diets on animal weights. Briefly, the experimental diets were initiated at day 0. Next, animals received one injection of AOM (10mg/kg body weight), on week 1. DSS (2.5%) was provided to the mice in the drinking water (week 2-3). At week 9, the animals receiving 1,25(OH)2D (1.25 μg/kg diet) were found to have significantly lower weights than the control animals; To determine whether lowering the concentration of 1,25(OH)2D would results in recovery of weights, the dose was reduced to 0.625 μg/kg diet. However as shown in Fig. 3A, the 1,25(OH)2D group continued to have decreased weight as compared to control (p<0.001), 25(OH)D (p<0.01), D3 (p<0.05) or 1α(OH)D5 (p<0.05) groups. Given the dramatic weight los in the 1,25(OH)2D group, this group was not included in the final analysis. At the end of the study, the animals in the control (AOM+DSS) group were found to have an 84% incidence of tumors. The distribution of the tumors was greatest in the distal colon, with the fewest tumors found in the proximal colon. The mean (±SE) number of tumors adjusted for length of colon for control was 0.24 ± 0.07 compared to 0.13 ± 0.03, 0.10 ± 0.04, 0.13 ± 0.03, and 0.13 ± 0.08 for D3, 1α(OH)D5, 25(OH)D, and 1,25(OH)2D, respectively. The greatest inhibition of tumor incidence was noted in the group receiving 1α(OH)D5 (56.2%) (p< 0.01). Mice in the D3 and 25(OH)D diet groups showed a reduction in tumor incidence of 42.4 (p=0.07) and 45.8% (p< 0.05), respectively (Fig.3 B). These data suggest that less active vitamin D metabolites or synthetic analogs (e.g., 1α(OH)D5), may be beneficial against inflammation-induced colon carcinogenesis.
Figure 3.

A) Overview of azoxymethane/dextran sodium sulfate (AOM/DSS)-induced inflammation-associated colon carcinogenesis studies. For this, CF1 mice were started on experimental diets on Day 0. At week 1, mice received one injection of AOM (10 mg/kg body weight). From week 2 to 3, animals were administered 2.5% DSS in the drinking water. Mice were sacrificed on week 16 and tumor incidence and multiplicity were assessed; Weekly mean body weight for each of the groups is presented. B) Mean (± SE) number of tumors/colon, % inhibition in tumor incidence and respective statistical significance is shown.
IV. Discussion
The risk for UC-associated CRC is increased at least 2-fold compared to the normal population. The risk for CRC is much higher among IBD patients with frequent relapses. Despite all the advances in our understanding of the pathophysiology of IBD, the etiology remains unknown. The general consensus is that IBD results from the combined effects of genetics and environment factors (e.g., diet) known to affect the immune system. Vitamin D, an important regulator of the immune system, has been linked to several auto-immune diseases including IBD. The results generated from the gene array analysis highlight the control that vitamin D compounds have on key members of the TLR4 pathway. Recently, it was shown that patients with IBD exhibit high TLR4 expression in the intestinal mucosa. Moreover, an increased expression of TLR4 has been shown to be associated with increased expression of other pro-inflammatory markers including cyclooxygenase-2 (Cox-2). These findings have increased the interest in generating agents capable of blocking the TLR4 pathway. Currently, TLR4 antagonist are being tested in clinical trials for sepsis, but enthusiasm for their use is dampened since distribution of TLR4 signaling pathway may bring about unwarranted side effects. Further studies are warranted to better elucidate the safety of vitamin D alone or in combination with other agents as a mode to safely modulate the TLR4 pathway and ultimately the occurrence of IBD-related colon cancer.
Previously the role of oral administration of D3 has been shown to be effective at regulating the calcium/phosphorus and vitamin D Homeostasis in animals (13). Moreover, the usefulness of D3 is being evaluated in patients diagnosed with sporadic colorectal cancer. For example, an ongoing trial is investigating the use of D3 in combination with calcium for tertiary prevention of colon tumors in surgically treated patients. In the present study, D3 was found to significantly up-regulated the vitamin D metabolizing enzymes, CYP27A1 (25-hydroxylase) and CYP27B1 (1-OHase) in colon cells. Our current findings demonstrate that D3 is also transcriptionally active in increasing the expression of CYP27A1 in colon cancer cells. This is in agreement with previous findings in prostate cancer cells (11,12). Therefore, we conclude that the conventional pathway of D3 activation (i.e., via liver and kidney) may not be mandatory for colonic epithelium. Moreover, the rapid activation of CYP27A1 and CYP27B1 following D3 treatment was not found to be accompanied by a sharp rise in CYP24. As shown in Figure 1c, D3 has a significantly delayed effect on CYP24. Cumulatively these data reiterate the potential use of D3 as an effective oral chemopreventive agent. Additionally it appears that unlike the activation of D3 in kidneys, 1,25(OH) 2D synthesized in the colon will most likely should have minimal impact on blood calcium and phosphorus levels. In summary, future studies are warranted to confirm whether local production of the active metabolite (from dietary vitamin D3 or 25(OH)D) in the colon epithelium may provide a wider and safer therapeutic window, more efficient and cost effective way for chemoprevention of inflammation induced-colon cancer.
Acknowledgments
Supported by NIH Grants K01 CA103861, RO1 CA121157 and R01 CA122299
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121:2381–6. doi: 10.1002/ijc.23192. [DOI] [PubMed] [Google Scholar]
- 2.Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009;29:2727–37. [PubMed] [Google Scholar]
- 3.Ahmadi A, Polyak S, Draganov PV. Colorectal cancer surveillance in inflammatory bowel disease: the search continues. World J Gastroenterol. 2009;15:61–6. doi: 10.3748/wjg.15.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantorna MT. Vitamin D and its role in immunology: multiple sclerosis, and inflammatory bowel disease. Prog Biophys Mol Biol. 2006;92:60–4. doi: 10.1016/j.pbiomolbio.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 5.Jahnsen J, Falch JA, Mowinckel P, Aadland E. Vitamin D status, parathyroid hormone and bone mineral density in patients with inflammatory bowel disease. Scand J Gastroenterol. 2002;37:192–9. doi: 10.1080/003655202753416876. [DOI] [PubMed] [Google Scholar]
- 6.Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents an ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr. 2000;130:2648–2652. doi: 10.1093/jn/130.11.2648. [DOI] [PubMed] [Google Scholar]
- 7.Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mole Endocrinol. 2003;17:2386–2392. doi: 10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- 8.Murillo G, Peng X, Torres KEO, Mehta RG. Deguelin inhibits growth of breast cancer cells by modulating the expression of key members of the Wnt signaling pathway. Cancer Prevention Res. 2009;2:942–50. doi: 10.1158/1940-6207.CAPR-08-0232. [DOI] [PubMed] [Google Scholar]
- 9.Peng X, Maruo T, Cao Y, Punj V, Mehta R, Das Gupta TK, Christov K. A novel RARbeta isoform directed by a distinct promoter P3 and mediated by retinoic acid in breast cancer cells. Cancer Res. 2004;64:8911–8. doi: 10.1158/0008-5472.CAN-04-1810. [DOI] [PubMed] [Google Scholar]
- 10.Matusiak D, Benya RV. CYP27A1 and CYP24 expression as a function of malignant transformation in the colon. J Histochem Cytochem. 2007;55:1257–64. doi: 10.1369/jhc.7A7286.2007. [DOI] [PubMed] [Google Scholar]
- 11.Tokar EJ, Webber MM. Chemoprevention of prostate cancer by cholecalciferol (vitamin D3): 25-hydroxylase (CYP27A1) in human prostate epithelial cells. Clin Exp Metastasis. 2005;22:265–73. doi: 10.1007/s10585-005-8394-y. [DOI] [PubMed] [Google Scholar]
- 12.Tokar EJ, Webber MM. Cholecalciferol (vitamin D3) inhibits growth and invasion by up-regulating nuclear receptors and 25-hydroxylase (CYP27A1) in human prostate cancer cells. Clin Exp Metastasis. 2005;22:275–84. doi: 10.1007/s10585-005-8393-z. [DOI] [PubMed] [Google Scholar]
- 13.Vieth R, Milojevic S, Peltekova V. Improved cholecalciferol nutrition in rats is noncalcemic, suppresses parathyroid hormone and increases responsiveness to 1,25-dihydroxycholecalciferol. J Nutr. 2000;130:578–584. doi: 10.1093/jn/130.3.578. [DOI] [PubMed] [Google Scholar]