

Abstract
Current models of oncogenesis incorporate the contributions that chronic inflammation and aging make to the patterns of tumor formation. These oncogenic pathways, involving leukocytes and fibroblasts are not readily applicable to brain tumors (glioma), and other mechanisms must account for microenvironmental influences on central nervous system tumorigenesis. Previous studies from our laboratories have employed Neurofibromatosis-1 (NF1) genetically-engineered mouse (GEM) models to understand the spatial restriction of glioma formation to the optic pathway of young children. Based on our initial findings, we hypothesize that brain region-specific differences in cAMP levels account for the pattern of NF1 gliomagenesis. To provide evidence that low levels of cAMP promote glioma formation in NF1, we generated foci of decreased cAMP, in brain regions where gliomas rarely form in children with NF1. Focal cAMP reduction was achieved by forced expression of phosphodiesterase 4A1 (PDE4A1) in the cortex of Nf1 GEM strains. Ectopic PDE4A1 expression produced hypercellular lesions with features of human NF1-associated glioma. Conversely, pharmacologic elevation of cAMP with the PDE4 inhibitor Rolipram dramatically inhibited optic glioma growth and tumor size in Nf1 GEM in vivo. Together, these results indicate that low levels of cAMP in a susceptible Nf1 mouse strain are sufficient to promote gliomagenesis, and justify the implementation of cAMP-based stroma-targeted therapies for glioma.
Keywords: NF1, glioma, astrocytoma, cAMP, PDE4, Rolipram, neurofibromin
Introduction
Carcinogenesis is a complex process frequently modeled as involving two phases, initiation and promotion (reviewed in (1)). During initiation, cancer cell progenitors sustain genetic alterations that result in loss of tumor suppressor and/or gain of oncogene function. Cells “initiated” in this manner possess a growth advantage through mutational activation of proliferation and/or survival pathways. These genetic changes are necessary, but not sufficient, for transformation. Susceptible tumor progenitors require additional signals like those derived from tissue stromal elements (e.g. endothelial cells, leukocytes and fibroblasts) to complete tumor promotion (reviewed in (2)). This multistage oncogenesis model, which also involves the effects of chronic inflammation and aging on stromal function, has been recently elucidated for several tumor types, including breast, gastric and hepatocellular carcinoma. These new insights have led to the identification of the essential constituent stromal cell types as well as many of the relevant pathways that mediate the critical interactions between supportive tumor stroma and receptive preneoplastic/neoplastic cells during tumor evolution.
Although the prevailing model of cancer initiation and promotion accounts for many events in the genesis of solid tumors, it does not fully explain the unique spatial and temporal pathogenesis of brain tumors, especially those in children. Tissue stroma and inflammation differ markedly in the brain. There are no brain fibroblasts, and immune system involvement in brain cancers appears to be minimal compared to that in peripheral tissues. Moreover, the stromal signals that drive tumorigenesis in the brain are unique and likely reflect the presence of spatially- and temporally-regulated brain-specific cues that sculpt the developing brain during embryogenesis and early fetal life.
Astrocytoma (glioma) formation in the common inherited disorder Neurofibromatosis-1 (NF1) is an excellent model system for elucidating the cellular and molecular mechanisms that underlie microenvironmental contributions to brain tumorigenesis. Similar to the initiating events in other cancers, complete loss of Nf1 protein (neurofibromin) function is necessary, but not sufficient, for NF1-associated gliomagenesis (3). In fact, gliomagenesis in NF1 displays such exquisite sensitivity to the genetics, age and region of the surrounding brain, that the necessary stroma-derived promoting factors can be localized in both place and time. Greater than 90 percent of astrocytomas in children with NF1 occur prior to age seven, and approximately 70 percent of these tumors involve the optic nerves and optic chiasm (4, 5). These observations indicate that the optic pathway of young children with NF1 is uniquely conditioned to support gliomagenesis.
A specialized function of the optic pathway in gliomagenesis is substantiated by studies in genetically-engineered mouse (GEM) models of NF1. Neither Nf1+/- mice nor mice with complete Nf1 loss in Glial Fibrillary Acidic Protein (GFAP)-expressing astroglial cells develop gliomas (3, 6). Only mice with both reduced Nf1 gene expression (Nf1+/- mice) in the microenvironment (“Tumor Microenvironment” mice) and complete loss of Nf1 gene expression in tumor progenitors (“Tumor Progenitor” mice) form gliomas. While the entire brain and spinal cord of these mice contains Nf1-/- glial cells within an Nf1+/- environment, tumors only form within the optic pathway, involving the prechiasmatic optic nerves and optic chiasm (“OPG” Mice) (7, 8). Together, these GEM strains highlight a complex set of requirements for gliomagenesis involving genetic events in tumor progenitors (complete loss of Nf1), genetic events in the microenvironment (reduced Nf1 expression; Nf1 heterozygosity), and region-specific signals (the optic pathway). Furthermore, these GEM strains afford a singular opportunity to investigate the mechanism(s) by which the tumor microenvironment and tumor progenitors interact to promote gliomagenesis.
Previously, we demonstrated a correlation between brain region-specific differences in cAMP levels and glioma formation (9), and hypothesized that low levels of cAMP constituted the region-specific condition necessary for NF1-associated gliomagenesis. In the current study, we experimentally test this hypothesis and demonstrate that focal cAMP suppression, together with global heterozygosity for neurofibromin and complete loss of neurofibromin in tumor progenitors, is sufficient to promote gliomagenesis in Nf1 genetically-engineered mice in vivo.
Materials and Methods
Animals
All animals were used in accordance with an established Animal Studies Protocol approved by the Washington University School of Medicine Animal Studies Committee. Wild type (Nf1flox/flox), Tumor Progenitor (Nf1flox/flox; GFAP-Cre), and OPG (Nf1flox/mut; GFAP-Cre) mice were generated as previously described (7)(Table 1).
Table 1.
Induced Tumor Frequency in Nf1 GEM.
| Nf1 GEM models | Induced Tumor Frequency | |||||||
|---|---|---|---|---|---|---|---|---|
| PDE4A1 |
PDE4A1-H229Q |
|||||||
| Experimental Group | Genotype | Strain | Description | tumor | no tumor | tumor | no tumor | P value |
| Wild-type mice | Nf1 flox/flox | Nf1 flox/flox | Wild-type mice. Do not develop spontaneous gliomas | 1 | 5 | 0 | 2 | n.s.* |
| Tumor Progenitor mice | Nf1flox/flox x GFAP-Cre | Nf1GFAPCKO | Wild-type mice lacking Nf1 expression in GFAP+ cells by E14.5. Do not develop spontaneous gliomas. | 1 | 6 | 0 | 7 | n.s. |
| OPG mice | Nf1flox/mut x GFAP-Cre | Nf1+/-GFAPCKO | Nf1+/- mice lacking Nf1 expression in GFAP+ cells by E14.5. 95% develop prechiasmatic/chiasmal optic gliomas by 10-12 weeks | 8 | 5 | 0 | 6 | 0.02 |
P values were determined by two-tailed Fisher's Exact Test comparing the frequency of tumor induction in response to PDE4A1 versus PDE4A1-H229Q injection.
Human Tumor and Brain Sections
Paraffin-embedded optic pathway glioma specimens from patients with NF1 (n=4) and normal human brain and optic chiasm autopsy specimens were retrieved from the archives of the Department of Pathology at the Washington University School of Medicine in accordance with an Institutional Review Board approved protocol for the use of human pathology specimens.
Chemicals, Reagents, and Antibodies
All chemicals were obtained from Sigma unless otherwise indicated. All tissue culture reagents and media were obtained from Invitrogen unless otherwise indicated. A construct containing mCherry cDNA was a gift of Dr. Roger Y. Tsien (University of California). Murine PDE4A1 (accession no. AJ297396) was provided by Dr. James Cherry (Boston University). Antibodies were obtained from Peprotech (CXCL12), Clontech (Dsred (mCherry)), Invitrogen (GFAP), Abcam (PDE4A & 58kD Golgi marker), Pharmingen (LCA), Wako (IBA-1), Cell Signaling (phosphorylated protein kinase A (pPKA) substrate) and Sigma (CNPase). Antibody directed against phosphorylated CXCR4 (pCXCR4) was developed in our lab and previously described (10). Antibody directed against Olig2 (DF-308) was a gift of Dr. Charles Stiles (Dana Farber Cancer Institute).
Lentiviruses
Full-length murine PDE4A1 or catalytically inactive PDE4A1-H229Q was cloned into a lentiviral packaging vector that also encodes mCherry fluorescent protein as previously described (11). A second packaging vector (FUW-FLG) encoding a fusion of firefly luciferase and enhanced green fluorescent protein driven by the human ubiquitin C promoter within an established lentiviral backbone (12) was also used as previously described (13, 14). Viral particles were produced from each packaging vector separately by the Viral Vectors Core Facility of The Hope Center for Neurological Diseases at Washington University School of Medicine.
Mutagenesis
The catalytically inactive PDE4A1-H229Q (15) was generated from full-length murine PDE4A1 using a Quikchange® mutagenesis kit (Stratagene) according to the manufacturer's instructions and the following mutagenesis primers:
Sense: 5’ – CAT GAC GTC GAC CAG CCT GGC GTC TCC AAC – 3’
Antisense: 5’ – GTT GGA GAC GCC AGG CTG GTC GAC GTC ATG – 3’.
Mutagenesis was confirmed by sequencing; expression and activity of catalytically active and inactive PDE4A1 constructs were determined first in Daoy medulloblastoma cells (American Type Culture Collection). Daoy cells were infected with lentivirus encoding either catalytically active or inactive PDE4A1 as previously described (11). Cells expressing PDE4A1 constructs were sorted and collected based on mCherry expression by high-speed fluorescence-activated cell sorting (MoFlo High-Performance Cell Sorter; DAKO). As described below, expression levels were measured by western blot, intracellular localization by immunofluorescence microscopy and effects on cAMP levels by ELISA.
Intracranial Viral Injections
Five to seven week old wild type (Nf1flox/flox), Tumor Progenitor (Nf1flox/flox; GFAP-Cre), and OPG (Nf1flox/mut; GFAP-Cre) mice were anesthetized [intraperitoneal ketamine (87 mg/kg)/xylazine (13 mg/kg); Phoenix Pharmaceuticals], the cranium was exposed, and a small hole was made 2 mm lateral and posterior to the bregma with a size 34 inverted cone burr (Dremel). Mice were positioned in a stereotactic frame (Stoelting) and 500,000 transducing units each of FUW-FLG and PDE4A1-Cherry or PDE4A1-H229Q viruses were injected through a 27-gauge needle over 2 min at 3 mm below the dura mater. The incision was closed with Vetbond (3M). A few hours after the injection and every 12 hours for 48 hours thereafter, all animals received subcutaneous injections of 0.5 mg/kg Buprenorphine hydrochloride.
Bioluminescence Imaging (BLI)
In vitro: Bioluminescence of Daoy cells expressing FUW-FLG and PDE4A1-Cherry or PDE4A1-H229Q constructs was evaluated at the Molecular Imaging Core facility at Washington University in St. Louis. Cells were grown in 96-well black-wall clear-bottom plates (Costar). In some experiments, cells were treated with either the PDE4 inhibitor Rolipram (0-20 µM) or vehicle for 18 hours. Cells were washed and exposed to a solution of 150 μg/ml luciferin for 10 minutes. Bioluminescence was measured using a charge-coupled device camera-based bioluminescence imaging system (IVIS 50, Caliper; exposure time 1-30 s, binning 8, field of view 12, f/stop 1, open filter). Data were expressed as total photon flux (photons/s) using LivingImage (Caliper) and IgorPro (Wavemetrics) image analysis software packages.
In vivo: Bioluminescence measurements were obtained 48 hours after viral injection and every two weeks thereafter for the subsequent six weeks. Hair was removed from mouse heads with chemical depilatory agents 24 hours prior to imaging. Images were obtained using the following parameters: IVIS 50, Caliper; exposure time 300 s, binning 8, field of view 12, f/stop 1, open filter and analyzed as previously described (14, 16).
Manganese Enhanced MRI
Manganese enhanced magnetic resonance images (MEMRI) of the optic nerve were acquired as previously described (17).
Preclinical Study Design
Treatment
Ten to 12 week old OPG mice with MRI-confirmed optic nerve gliomas were assigned to either the vehicle (water alone) or Rolipram (5 mg/kg/day in the drinking water (11)) treatment group (5 animals/group). After 1 or 4 weeks of treatment, all animals were deeply anesthetized and perfused with 4% paraformaldehyde, and optic nerves were isolated.
Response evaluation
Response evaluations included measurements of optic nerve volumes, tumor cell proliferation, and tumor cell apoptosis (18). Optic nerve volumes were assessed by measuring optic nerve diameters at the level of the optic chiasm (D0), as well as 200 (D200), 400 (D400) and 600 (D600) microns anterior to the chiasm. Total volume was calculated from the sum of each 200-micron segment volume (V1 = D0 to D200) as determined using the equation for a truncated cone:
Proliferation within normal and tumor-bearing optic nerve was determined as the number of Ki-67 positive cells per high power field (HPF). Three high power fields per specimen were evaluated. Apoptosis was measured using terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL assay, Roche Diagnostics) according to the manufacturer's instructions and evaluated in a similar manner to Ki-67 staining.
Tissue Sections, Immunohistochemistry and Immunofluorescence
Twenty micron thick tissue sections were cut from 4% paraformaldehyde-fixed, paraffin-embedded experimental brains (10). For evaluation of mCherry, GFAP, LCA, Olig2, CNPase, pPKA substrate and IBA-1 expression, slides underwent antigen retrieval with Target Retrieval Solution (DakoCytomation) according to the manufacturer's instructions. Primary antibody concentrations were as follows: Dsred (mCherry, 1:200), GFAP (2.5 ug/ml), PDE4A (1:300), LCA (1:50), IBA-1 (1:1000), 58 kD Golgi protein (1 ug/ml), Olig2 (1:20,000), CNPase (1:500) and pPKA substrate (1:100). With the exception of PDE4A, 58 kD Golgi protein, and PDE4A-GFAP costains, staining was detected using biotin-conjugated secondary antibodies augmented by streptavidin-horseradish peroxidase, and visualized using either 3,3'-diaminobenzidine or with the Vector VIP Peroxidase Substrate kit (DAKO). Sections were counterstained with either hematoxylin or methyl green (Olig2/CNPase). PDE4A, 58 kD Golgi protein, and PDE4A-GFAP staining was detected via immunofluorescence using the secondary AlexaFluor 555–conjugated and AlexaFluor 488–conjugated secondary antibodies at a concentration of 1:750 (Molecular Probes) for 90 minutes. Nuclei were counterstained with 4', 6-diamidino-2-phenylindole (DAPI). Parallel staining was performed on normal mouse and human brain and optic chiasm autopsy specimens. Control sections were processed identically in the absence of primary antibody.
cAMP measurement
cAMP was measured by competitive immunoassay using a Correlated Enzyme Immunoassay Kit (Assay Designs) according to the manufacturer's instructions and as previously described (9). Cyclic AMP values were normalized to protein for each sample individually.
Western blot analysis
Western blot analysis for PDE4A1 or PDE4A1-H229Q expression was performed as previously described (11). Briefly, protein extracts were obtained by lysing cells with lysis buffer [20 mmol/L Tris (pH 7.4), 137 mmol/L NaCl, 10% glycerol, and 1% Triton X-100] supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Calbiochem 524628). The proteins (25mg) were resolved with 10% Bis-Tris gels (Invitrogen) and transferred onto Hybond ECL nitrocellulose membrane (Amersham) according to standard protocols. Blots were probed with an antibody to PDE4A, followed by incubation with IRDye® conjugated secondary antibody (LI-COR). Actin expression served as a loading control. Blots were imaged with the Odyssey fluorescent scanning system (Li-Cor).
Statistical Analyses
Significance of induced tumor frequency was determined with two-tailed Fisher's exact test. Comparisons were made between the rate of tumor formation in response to PDE4A1 and PDE4A1-H229Q in each strain of mice. Differences in cAMP levels in Daoy cells and the effect of Rolipram on optic pathway glioma cell proliferation and apoptosis were evaluated by two-tailed t-test.
Results
Gliomagenesis in NF1 occurs with remarkable spatial and temporal specificity, predominantly affecting the optic nerves and chiasm of young children (19). This pattern of glioma formation indicates that a necessary interaction occurs between glial NF1 gene inactivation and signals present in the optic pathway microenvironment during NF1-associated gliomagenesis.
In prior studies involving human specimens and a genetically-engineered mouse model of NF1-associated optic glioma (“OPG” mice), we identified CXCL12 as a candidate stromal signal capable of stimulating Nf1-deficient glial cell growth. (9). Multiple brain-derived cells within optic pathway glioma express CXCL12 including endothelial cells (Figure 1A), entrapped axons and infiltrating microglia. The presence of a functional paracrine relationship between CXCL12-expressing stromal elements and tumor cells is demonstrated by the frequent presence of the CXCL12 receptor, CXCR4, in a CXCL12-induced phosphorylated form within bipolar pilocytic cells (Figure 1B, inset). The potential importance of stromal activation of CXCR4 lies in its ability to suppress intracellular levels of cAMP, which we found was necessary for the stimulation of Nf1-/- astroglial cell growth (9). Moreover, we found region-specific differences in cAMP levels in the mouse brain that correlated with the pattern of glioma formation in NF1. The optic pathway had the lowest, while the cortex had the highest cAMP levels. Based on these observations, we hypothesized that differences in cAMP levels underlie the distinct pattern of gliomagenesis in NF1.
Figure 1. Optic nerve gliomas in humans and mice.
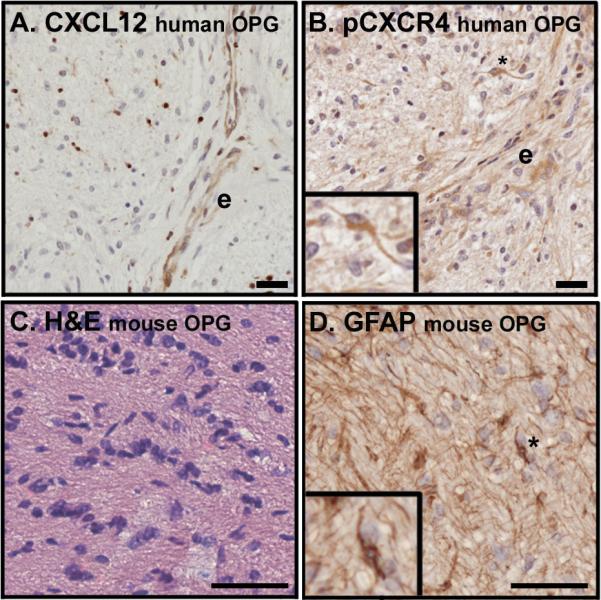
(A) An optic nerve glioma specimen from a patient with NF1 demonstrates mild hypercellularity and CXCL12 expression in the endothelium of tumor-associated blood vessels (e) as well as in scattered infiltrating cells. (B) Serial section from the tumor presented in panel A stained for the presence of a ligand-induced phosphorylated form of CXCR4 (pCXCR4) reveals a high level of receptor activation in proximity to the CXCL12-expressing endothelium. Phosphorylated CXCR4 is present in a pilocytic cell (inset). (C) A hypercellular lesion with nuclear atypia is evident within the optic nerve of an OPG mouse with hematoxylin and eosin stain. (D) The tumor pictured in panel C contains GFAP-expressing cells with the elongated bipolar morphology characteristic of pilocytic cells. In all cases expression appears brown. Scale bars for A and B equal 20 microns and for C and D equal 50 microns.
To evaluate this hypothesis in vivo, we sought to alter the pattern of brain region-specific cAMP levels to determine whether this would coordinately change the pattern of tumor formation in an established genetically-engineered mouse (GEM) model of NF1 OPG. Astrocytomas in Nf1 GEM models share many, but not all, of the features of human OPG. Gliomas within the optic nerves of OPG mice (Figure 1C) contain bipolar GFAP positive astrocytoma cells (Figure 1D) (20, 21) that also express the transcription factor Olig2 (Supplemental Figure 1A). Misexpression of Olig2 was recently shown to be highly correlated with pilocytic astrocytoma (22, 23). In addition, murine OPG exhibit microglial infiltration reminiscent of human tumors (Supplemental Figure 1B) (24-26). In contrast to human pilocytic astrocytomas, murine tumors do not contain Rosenthal fibers or eosinophilic granular bodies.
To generate foci of decreased cAMP, we ectopically expressed lentiviral-encoded cAMP specific phosphodiesterase-4A1 (PDE4A1) (27) via stereotactic injection into the cortex of OPG mice. PDE4A1 was chosen for these experiments based on our prior demonstration that increased PDE4A1 expression decreased intracellular cAMP levels and enhanced tumor growth in a xenograft model of malignant glioma in vivo (11). The cortex was selected as the site for PDE4A1 injection based on the following considerations that suggest it is usually not a permissive microenvironment for glioma formation in NF1: (1) Patients with NF1 rarely develop cortical tumors, (2) glioma formation in Nf1 mouse models does not occur in the cortex, and (3) the cortex exhibits the highest levels of cAMP (9).
Catalytically-active and inactive (PDE4A1-H229Q (15)) forms of PDE4A1 were cloned into a lentiviral packaging vector also encoding for mCherry fluorescent protein (11). In control studies, Daoy medulloblastoma cells were infected with lentivirus encoding PDE4A1 or PDE4A1-H229Q. While both forms of PDE4A1 were equally expressed (Supplemental Figure 2A) and localized to the Golgi apparatus (Supplemental Figure 2B), only catalytically-active PDE4A1 decreased intracellular cAMP levels (Supplemental Figure 2C). To mimic the low levels of cAMP normally present within the optic pathway, we co-injected PDE4A1/mCherry lentivirus with a second lentivirus encoding firefly luciferase (14) into the cortex of wild-type, Tumor Progenitor, and OPG mice. Bioluminescence imaging (BLI) of firefly luciferase expression was used to monitor transgene expression and served as a potential measure of neoplastic growth. In control studies, the expression of PDE4A1 or PDE4A1-H229Q in Daoy cells, or treatment with the pan-PDE4 inhibitor Rolipram had no effect on luciferase activity (Supplemental Figure 2D, E).
Since wild-type mice do not develop cortical gliomas, tumor induction following ectopic PDE4A1 expression in wild type mice (Nf1flox/flox) would indicate that cAMP suppression is sufficient for glioma initiation and promotion. Although Tumor Progenitor mice (Nf1flox/flox; GFAP-Cre) possess occasional foci of hyperproliferating astrocytes, they do not develop gliomas (3). PDE4A1-induced tumor formation in these mice would occur only if suppression of cAMP alone induced the combined tumor-promoting properties of an Nf1+/- microenvironment and the optic pathway. Finally, cortical glioma formation in OPG mice (Nf1flox/mut; GFAP-Cre) would suggest that cAMP suppression is sufficient, within the context of an Nf1+/- microenvironment to bring about the neoplastic growth of Nf1-/- tumor progenitors. PDE4A1-H229Q injections serve as controls for non-catalytic effects of lentiviral injection and PDE4A1 overexpression.
In 51 of 58 injected mice, we detected a bioluminescent signal, indicating successful viral transduction and transgene expression. Increased BLI on serial studies demonstrated replication of virally infected cells. The brain of each animal was subjected to an extensive blinded evaluation for the presence of neoplasia according to the algorithm outlined in Supplemental Figure 3.
Hematoxylin and eosin-stained sections from micro-injected cortices were examined for common features of neoplasia, including hypercellularity, cellular clustering, and nuclear atypia, as well as the specific features of low-grade glioma seen in the Nf1 OPG GEM model (Figure 1, Supplemental Figure 1). Twelve of 13 OPG mice injected with catalytically-active PDE4A1 lentivirus demonstrated increased BLI and expression of mCherry fluorescent protein/PDE4A at the injection site (Figure 2A-C). The diagnosis of an induced glioma was made when hypercellular lesions expressing mCherry fluorescent protein and PDE4 also contained GFAP-positive cells with the bipolar morphology characteristic of tumor cells (piloid cells) commonly found in the low-grade gliomas of NF1 patients (Figure 2D). Further indication of glioma was evident in increased Olig2 expression (Figure 3A). In each case, induced tumor sites exhibited greater numbers of Olig2 positive nuclei per high-powered field than did the same location in contralateral cortex. As a group, the induced tumors demonstrated significantly increased Olig2 expression compared to the uninvolved contralateral cortex (Figure 3B). Comparable histological findings were not observed in the contralateral cortex, nor are they observed in normal human or mouse cortex and optic nerve (Supplemental Figure 4).
Figure 2. Induced tumors occur at PDE4A injection sites.
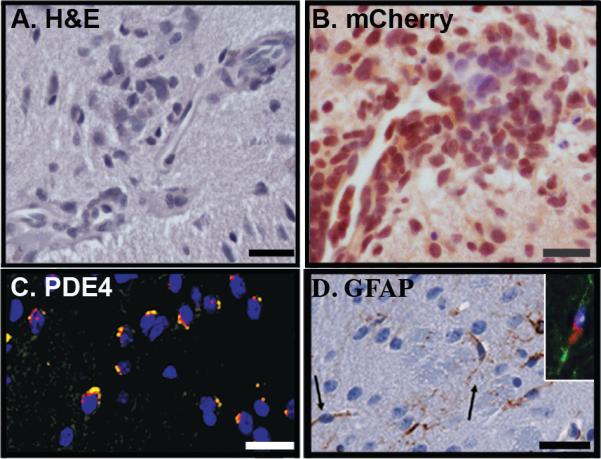
Tumors were identified in eight of 13 OPG mice injected with catalytically-active PDE4A1. The following representative images were taken of four such induced tumors: (A) Induced tumors had foci of cellular clusters and nuclear atypia evident on hematoxylin and eosin stained sections. Scale bar = 25 µm. (B) Injection sites were further identified as hypercellular, disorganized lesions expressing virally encoded mCherry fluorescent protein (brown, scale bar equals 25 µm), and (C) PDE4A in characteristic perinuclear localization (yellow). DAPI counterstains nuclei blue. Scale bar = 25 µm. (D) Glial Fibrillary Acidic protein (GFAP)-expressing cells (brown) display an elongated bipolar phenotype (arrows) characteristic of pilocytic cells. Scale bar = 50 µm. Inset: Co-labelling of a piloid cell with antibodies directed against GFAP (green) and PDE4 (red) support the role of PDE4A1 expression and cAMP suppression in the genesis of glioma.
Figure 3. Induced tumors exhibit increased Olig2 expression and evidence for decreased cAMP levels.
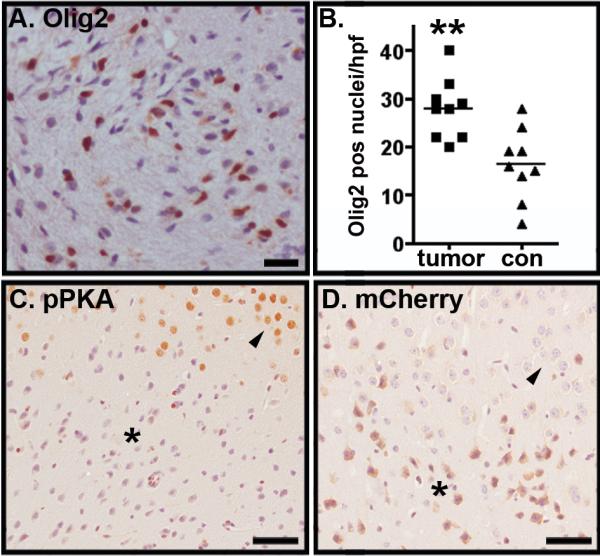
(A) Cells within induced tumors exhibited increased Olig2 expression. Scale bar = 25 μm. (B) The number of Olig2 positive nuclei per high-powered field in induced tumors was greater than in contralateral cortex. P<0.005 as determined by two-tailed t-test. (C) Immunolabeling of injection sites with antibody directed against the phosphorylated form of PKA substrates revealed a high level of staining in normal cortex (arrowhead) but low level of staining in tumor cells (asterisk). (D) Serial sections through the same tumor site pictured in C and labeled with antibody directed against mCherry fluorescent protein reveals the interface between tumor (asterisk) and cortex (arrowhead). Scale bars equal 50 microns.
Importantly, bipolar piloid cells expressed PDE4 in a peri-nuclear distribution characteristic of PDE4A1 localization (Figure 2D inset), suggesting a role for PDE4A1 expression in gliomagenesis. The mechanistic significance of this finding was further underscored by our finding that induced tumors had lower levels of cAMP. Since the induced tumors are small and preclude direct cAMP measurements, cAMP levels were inferred from an immunohistochemical analysis using a surrogate marker, the intensity of Protein Kinase A (PKA) substrate phosphorylation. Tumor sites exhibited decreased labeling with phosphoPKA (pPKA) substrate antibody, indicating decreased PKA activity (Figure 3C,D) and suggestive of decreased levels of cAMP. These findings are consistent with a relationship between lower levels of cAMP and gliomagenesis in NF1.
In total, 41 of the 51 mice with increased BLI had injection sites with evidence for exogenous PDE4A1 or PDE4A1-H229Q expression and were deemed evaluable for tumor formation. Eight of the 13 OPG mice with increased BLI following PDE4A1 injection were considered to have induced cortical gliomas (Table 1). This was a statistically significant rate of induced tumor formation, as determined by Fisher's Exact Test (P=0.02). In contrast, only 1 of 7 Tumor Progenitor mice and 1 of 6 wild-type mice had evidence of induced glioma. None of the 15 control injections of PDE4A1-H229Q resulted in a tumor. The frequency of tumor formation in control mice was not significantly significant.
While PDE4A (not shown) and mCherry positive cells (Figure 4A) were observed at injection sites in mice without tumors, these differed from induced tumors by the absence of abnormal astrocyte morphologies (Figure 4B) and their lack of increased Olig2 expression (Figure 4C). Disruption in tissue architecture at these injection sites was limited to inflammation, as evidenced by the presence of LCA-positive cells (Figure 4D). LCA-positive inflammatory cells were only a minor component of the cellular constituents in the induced tumors (data not shown). Together with our prior studies, the pathological features of PDE4A1-induced cortical tumors in OPG mice demonstrate that low cAMP levels are sufficient to promote glioma formation when Nf1-/- glioma progenitors arise within the context of an Nf1+/- microenvironment.
Figure 4. Histological features of non-tumor reactions.
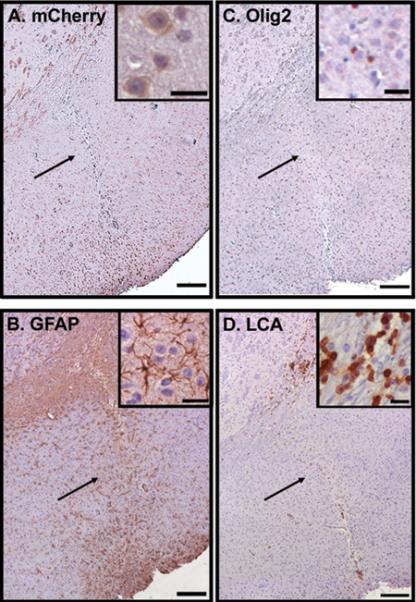
In 31 cases, no induced tumor was identified in GEM brains injected with PDE4A1 or PDE4A1-H229Q. Pictured is the sequential assessment of an injection site from a single animal with positive BLI but negative tumor evaluation. (A) The injection site, which had been identified by Hematoxylin and Eosin stain and PDE4A expression (not shown), demonstrated expression of mCherry fluorescent protein. (B) There are numerous GFAP positive cells at the injection site, but they display a stellate morphology typical of reactive (nonneoplastic) astrocytes. (C) There is a lack of increased Olig2 staining. (D) Also evident at this injection site is an inflammatory response characterized by numerous LCA-positive leukocytes. In all cases expression appears brown. Scale bars for large images are 200 µm. Inset scale bars = 25 µm.
To determine whether restoration of normal cAMP levels would attenuate optic glioma growth in vivo, we treated OPG mice for one or four weeks with Rolipram or vehicle following the detection of an optic glioma by manganese-enhanced MRI (Figure 5A). We previously showed that PDE4 inhibition with Rolipram blocked in vivo malignant glioma growth through elevation of cAMP levels (11, 14). Using this preclinical paradigm, four weeks of Rolipram resulted in tumor regression as measured by a restoration of optic nerve volumes to near normal values (Figure 5B, C). Moreover, Rolipram treatment suppressed tumor cell proliferation (Figure 5D), but had no effect on tumor cell apoptosis as measured by TUNEL staining (data not shown). These observations demonstrate that low levels of cAMP are necessary for the maintenance of optic glioma proliferation in vivo and further support the critical role of cAMP signaling in gliomagenesis and continued tumor growth.
Figure 5. Targeted inhibition of PDE4 attenuates tumor growth in OPG mice.
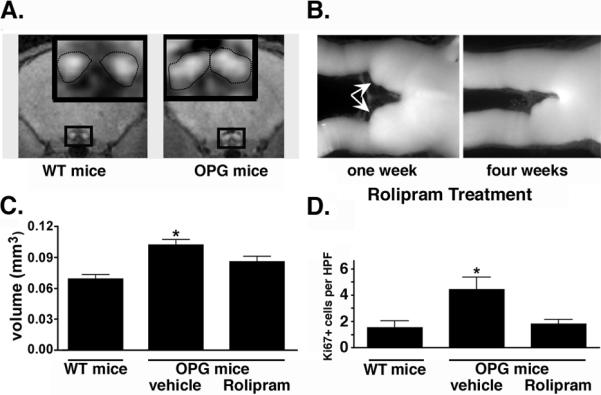
(A) Optic nerve gliomas were identified by enlarged optic nerves (white areas within the boxed region and demarcated by dotted line in inset) in OPG mice by MRI and assigned to treatment (Rolipram) or control (Vehicle) groups. (B) Optic nerve volumes in animals treated for 1 or 4 weeks with Rolipram were compared. The arrowheads indicate thickened optic nerves characteristic of these tumors. After 4 weeks of Rolipram treatment, optic nerves returned to a normal diameter. (C) Quantitation of optic nerve volumes indicates that there is a statistically significant increase in optic nerve volumes in untreated mice compared to wildtype mice (WT, without tumors) and that Rolipram abrogates this difference in volume (n = 5 mice/group). (D) The anti-tumor effect of Rolipram was associated with inhibition of tumor cell proliferation as determined by Ki67 staining and the numbers of Ki67 positive cells per high power field (HPF). N = 5 mice/group. * = p<0.05 for difference between OPG and WT mice as determined by two tailed t test.
Discussion
Clinical observations and experimental data indicate that cancers frequently arise within specific tissue contexts ((2) for review). For example, gastrointestinal cancers are often associated with chronic inflammation and senescent fibroblasts can promote the transformation of preneoplastic epithelial cells (28). Prevailing models of how inflammation and aging contribute to tumor promotion focus on the actions of fibroblast and leukocyte-derived cytokines and growth factors. However, this model is not directly applicable to brain tumors, as there are no fibroblasts within the CNS, and chronic CNS inflammation involving leukocytes is uncommon. Moreover, pediatric brain tumors develop in the absence of the effects of aging. Nevertheless, the unique spatial and temporal distribution of brain tumors (gliomas) in children with NF1 highlights the significant role that microenvironmental factors can play in brain tumor formation.
Based on the natural history of patients with NF1 as well as Nf1 GEM models (7, 29), we and others have proposed that the milieu of the normally growing nervous system functions similarly to aging or inflamed peripheral tissues by providing the necessary cellular elements and growth-regulatory factors for neoplastic transformation. Thus, while complete loss of neurofibromin function in glial progenitors is a requisite initiating step in transformation; the formation of NF1-associated mouse gliomas also requires a permissive Nf1 heterozygous microenvironment and region specific effectors. It is the confluence of all three of these elements: homozygous Nf1 gene inactivation in tumor progenitors, heterozygous Nf1 gene inactivation in the tumor surround, and region-specific effectors, that results in NF1-associated glioma formation in the optic nerves and chiasm of young NF1 heterozygous children and mice.
Several recent studies suggest that Nf1 heterozygosity creates a permissive microenvironment by altering the function of specific cell types that can be co-opted to promote tumorigenesis. In the central nervous system, we demonstrated that Nf1 heterozygosity alters microglia function, resulting in increased proliferation and motility, and elevated production of growth-promoting molecules such as CXCL12 and hyaluronidase (9, 25). Similarly, peripheral nerve tumor (neurofibroma) development requires Nf1+/- mast cells, which are released from the bone marrow in response to c-kit receptor activation (30).
Region-specific effects could also include region-specific differences in cell types that participate in gliomagenesis (31) or region-specific differences in growth-regulatory signals. Previously, we suggested CXCL12 as a candidate optic pathway-derived glioma-promoting factor and demonstrated that complete loss of neurofibromin function alters CXCR4 signaling, such that CXCL12 induces significant sustained suppression of cAMP levels. This abnormal CXCL12-mediated cAMP suppression creates an inappropriate survival response in Nf1-/- astrocytes. Neither CXCL12-induced cAMP suppression nor the resultant enhanced survival response is observed in wild-type astrocytes (9). Collectively, these data provide an attractive model for tumorigenesis, in which complete loss of neurofibromin function not only initiates oncogenesis, but also secondarily contributes to tumor promotion by dysregulating CXCR4-cAMP signaling. In this fashion, loss of neurofibromin results in the acquisition of two hallmark features of malignant transformation - hyperproliferation and increased survival - but only when neurofibromin loss occurs in a CXCL12-rich microenvironment, such as the optic pathway of young children and mice.
The current study provides novel in vivo support for the hypothesis that suppression of cAMP constitutes an important region-specific signal for gliomagenesis in NF1. The creation of cortical foci in which cAMP levels were suppressed rendered the cortex similar to the optic pathway and thus, susceptible to glioma. Whether CXCL12 is the primary regulator of cAMP effects during spontaneous gliomagenesis in NF1 remains unknown. Further, the critical cell type(s) through which PDE4A1 effects gliomagenesis are not yet identified.
Lentiviral expression of PDE4A1 could have reduced cAMP levels in Nf1-/- glial cells, and/or other cells in the Nf1+/- cortex, including neurons, microglia and endothelial cells. However, while cAMP might regulate tumor-promoting functions in these multiple cell types, growth-regulatory effects of cAMP in tumor cells are well described. For example, increased cAMP levels induced p38 phosphorylation and apoptosis in acute promyelocytic leukemia cells (32) and enhanced radiation-induced apoptosis of lung cancer cells through upregulation of Bak (33). Similarly, decreased cAMP inhibited the function and expression of the pro-apoptotic Bcl-2 family member Bim (34, 35). Consistent with cAMP suppression functioning within glioma progenitors, our previous studies indicated that suppression of cAMP within Nf1-/- astroglial cells endowed them with a survival advantage (9). Moreover, cAMP promotes astrocyte differentiation (36). Thus, suppression of cAMP levels in Nf1-/- tumor progenitors might inhibit differentiation and maintain a progenitor-like state and, in concert with tumor-promoting genetic and microenvironmental changes, facilitate tumorigenesis. Current work is focused on identifying in which cells cAMP suppression is required for gliomagenesis and whether cAMP dysregulation in Nf1 preneoplastic cells modulates apoptosis and/or proliferation.
Important issues also raised by these studies include the identity of the initiated tumor progenitor cell, the downstream mediators of cAMP signaling in tumor promotion and how best to target the cAMP pathway in the treatment of gliomas. Previous studies reported that both differentiated astrocytes and neural stem cells could serve as glioma cell-of-origin in a model of gliomagenesis dependent upon loss of Ink4a/Arf and gain of EGF receptor activation (37). In contrast, recent work on dermal neurofibromas in a GEM model of NF1 suggested that only tissue stem cells could function as tumor progenitors (38). The cortical injections in this study did not target any specific cell population. Thus, the PDE4A1-induced gliomas could have developed from transformed differentiated astrocytes or neural stem cells. The known ability of stem cells to extensively migrate throughout the brain to sites of pathology suggests this latter possibility is a tenable hypothesis (39). Future studies will be required to identify the cell of origin of low-grade gliomas in Nf1 GEM.
Finally, as underscored by Rolipram-mediated attenuation of optic glioma growth in OPG mice (this study) and by reduced Nf1 GEM neurofibroma size following Imatinib-mediated c-kit pathway inhibition (30), components of the stromal signaling pathway represent potential targets for future anti-neoplastic therapy. These exciting results firmly establish that the experimental tractability of Nf1 GEM model systems offer unprecedented opportunities to identify the cellular and molecular bases for stromal control of tumor formation. In addition, this spontaneous brain tumor model is an excellent platform for the development of stroma-directed chemotherapeutic strategies. Further, it was recently demonstrated that 23% of high-grade glioblastoma tumors harbor inactivating NF1 gene mutations (40). Thus, insights gained from the NF1 model will be informative with regard to stromal contributions to and stroma-directed therapy for other brain tumors in children and adults.
Supplementary Material
Acknowledgements
The authors thank JT Forys and Mahil Rao for help in generating PDE4A1 constructs and lentiviral vectors. This work was supported by Pilot Study Funding from the Molecular Imaging Center at Washington University School of Medicine and the NIH P50 CA94056 (D. Piwnica-Worms), NIH Neuroscience Blueprint Core Grant P30 NS057105 to Washington University (Viral Vectors Core), NCI UO1CA84314 (D.H. Gutmann), NCI/NIH RO1CA118389 (J.B. Rubin) and The Children's Tumor Foundation (J.B. Rubin). The authors have no conflicting financial interests.
References
- 1.Laconi E, Doratiotto S, Vineis P. The microenvironments of multistage carcinogenesis. Semin Cancer Biol. 2008;18:322–9. doi: 10.1016/j.semcancer.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 2.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–50. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 3.Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–13. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63–6. doi: 10.1016/s0022-3476(94)70122-9. [DOI] [PubMed] [Google Scholar]
- 5.Guillamo JS, Creange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain. 2003;126:152–60. doi: 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- 6.Kluwe L, Hagel C, Tatagiba M, et al. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol. 2001;60:917–20. doi: 10.1093/jnen/60.9.917. [DOI] [PubMed] [Google Scholar]
- 7.Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–7. [PubMed] [Google Scholar]
- 8.Zhu Y, Harada T, Liu L, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132:5577–88. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warrington NM, Woerner BM, Daginakatte GC, et al. Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res. 2007;67:8588–95. doi: 10.1158/0008-5472.CAN-06-2220. [DOI] [PubMed] [Google Scholar]
- 10.Woerner BM, Warrington NM, Kung AL, Perry A, Rubin JB. Widespread CXCR4 activation in astrocytomas revealed by phospho-CXCR4-specific antibodies. Cancer Res. 2005;65:11392–9. doi: 10.1158/0008-5472.CAN-05-0847. [DOI] [PubMed] [Google Scholar]
- 11.Goldhoff P, Warrington NM, Limbrick DD, Jr., et al. Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes brain tumor regression. Clin Cancer Res. 2008;14:7717–25. doi: 10.1158/1078-0432.CCR-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 13.Smith MC, Luker KE, Garbow JR, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–12. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 14.Yang L, Jackson E, Woerner BM, Perry A, Piwnica-Worms D, Rubin JB. Blocking CXCR4-Mediated Cyclic AMP Suppression Inhibits Brain Tumor Growth In vivo. Cancer Res. 2007;67:651–8. doi: 10.1158/0008-5472.CAN-06-2762. [DOI] [PubMed] [Google Scholar]
- 15.Jacobitz S, Ryan MD, McLaughlin MM, Livi GP, DeWolf WE., Jr. Torphy TJ. Role of conserved histidines in catalytic activity and inhibitor binding of human recombinant phosphodiesterase 4A. Mol Pharmacol. 1997;51:999–1006. doi: 10.1124/mol.51.6.999. [DOI] [PubMed] [Google Scholar]
- 16.Gross S, Piwnica-Worms D. Real-time imaging of ligand-induced IKK activation in intact cells and in living mice. Nat Methods. 2005;2:607–14. doi: 10.1038/nmeth779. [DOI] [PubMed] [Google Scholar]
- 17.Banerjee D, Hegedus B, Gutmann DH, Garbow JR. Detection and measurement of neurofibromatosis-1 mouse optic glioma in vivo. Neuroimage. 2007;35:1434–7. doi: 10.1016/j.neuroimage.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hegedus B, Banerjee D, Yeh TH, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68:1520–8. doi: 10.1158/0008-5472.CAN-07-5916. [DOI] [PubMed] [Google Scholar]
- 19.Listernick R, Charrow J, Gutmann DH. Intracranial gliomas in neurofibromatosis type 1. Am J Med Genet. 1999;89:38–44. doi: 10.1002/(sici)1096-8628(19990326)89:1<38::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 20.Kleihues P, Burger PC, Collins VP, Newcomb EW, Ohgaki H, Cavenee WK. Glioblastoma. In: Kleihues P, Cavenee WK, editors. World Health Organization Classification of Tumours. 3rd ed. International Agency for Research on Cancer Press; Lyon: 2000. pp. 29–39. [Google Scholar]
- 21.Scheithauer BW, Hawkins C, Tihan T, VandenBerg SR, Burger PC. Pilocytic Astrocytoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. World Health Organization Classification of Tumours. 4th ed. International Agency for Research on Cancer Press; Lyon: 2007. pp. 14–21. [Google Scholar]
- 22.Bouvier C, Bartoli C, Aguirre-Cruz L, et al. Shared oligodendrocyte lineage gene expression in gliomas and oligodendrocyte progenitor cells. J Neurosurg. 2003;99:344–50. doi: 10.3171/jns.2003.99.2.0344. [DOI] [PubMed] [Google Scholar]
- 23.Takei H, Yogeswaren ST, Wong KK, et al. Expression of oligodendroglial differentiation markers in pilocytic astrocytomas identifies two clinical subsets and shows a significant correlation with proliferation index and progression free survival. J Neurooncol. 2008;86:183–90. doi: 10.1007/s11060-007-9455-7. [DOI] [PubMed] [Google Scholar]
- 24.Badie B, Schartner J. Role of microglia in glioma biology. Microsc Res Tech. 2001;54:106–13. doi: 10.1002/jemt.1125. [DOI] [PubMed] [Google Scholar]
- 25.Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16:1098–112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- 26.Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40:252–9. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- 27.Huston E, Houslay TM, Baillie GS, Houslay MD. cAMP phosphodiesterase-4A1 (PDE4A1) has provided the paradigm for the intracellular targeting of phosphodiesterases, a process that underpins compartmentalized cAMP signalling. Biochem Soc Trans. 2006;34:504–9. doi: 10.1042/BST0340504. [DOI] [PubMed] [Google Scholar]
- 28.Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–96. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–2. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang FC, Ingram DA, Chen S, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/-- and c-kit-dependent bone marrow. Cell. 2008;135:437–48. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeh TH, Lee da Y, Gianino SM, Gutmann DH. Microarray analyses reveal regional astrocyte heterogeneity with implications for neurofibromatosis type 1 (NF1)-regulated glial proliferation. Glia. 2009;57:1239–49. doi: 10.1002/glia.20845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahn YH, Jung JM, Hong SH. 8-Chloro-cyclic AMP-induced growth inhibition and apoptosis is mediated by p38 mitogen-activated protein kinase activation in HL60 cells. Cancer Res. 2005;65:4896–901. doi: 10.1158/0008-5472.CAN-04-3122. [DOI] [PubMed] [Google Scholar]
- 33.Choi YJ, Kim SY, Oh JM, Juhnn YS. Stimulatory heterotrimeric G protein augments gamma ray-induced apoptosis by up-regulation of Bak expression via CREB and AP-1 in H1299 human lung cancer cells. Exp Mol Med. 2009 doi: 10.3858/emm.2009.41.8.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marani M, Hancock D, Lopes R, Tenev T, Downward J, Lemoine NR. Role of Bim in the survival pathway induced by Raf in epithelial cells. Oncogene. 2004;23:2431–41. doi: 10.1038/sj.onc.1207364. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Insel PA. The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem. 2004;279:20858–65. doi: 10.1074/jbc.M310643200. [DOI] [PubMed] [Google Scholar]
- 36.McManus MF, Chen LC, Vallejo I, Vallejo M. Astroglial differentiation of cortical precursor cells triggered by activation of the cAMP-dependent signaling pathway. J Neurosci. 1999;19:9004–15. doi: 10.1523/JNEUROSCI.19-20-09004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bachoo RM, Maher EA, Ligon KL, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–77. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 38.Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–63. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aboody KS, Brown A, Rainov NG, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97:12846–51. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.