

Abstract
Background
OPN is a variably expressed secreted glycophosphoprotein that mediates hepatocellular cancer (HCC) growth and metastasis. MicroRNAs (miRNAs) may be responsible for variant OPN expression, interrupting translation by binding OPN messenger RNA (mRNA) 3′-untranslated regions (UTRs).
Methods
Microarray analysis identified microRNAs of interest. Plasmid luciferase reporter constructs with variable OPN 3′UTR mutations were transfected into two HCC cell lines to determine miRNA regulation of OPN expression. Western blot confirmed variable OPN expression in both cell lines. Invasion, adhesion, and migration evaluated metastatic behavior in Hep G2 and Hep 3B with modified microRNA and OPN expression.
Results
Hep 3B produces 36x microRNA 181a compared to Hep G2. Luciferase activity after transfection with miRNA 181a precursor was decreased in both cell lines (p<0.01); luciferase activity increased with miRNA 181a inhibitor transfection in both cell lines (p<0.01). Hep 3B transfected with mutated OPN 3′UTR increased luciferase activity 108% (p<0.01). Hep G2 transfected with miRNA precursor decreased OPN expression 5x (p<0.01) in Western blot. Hep 3B transfection with miRNA precursor increased OPN expression 3x (p<0.01) in Western blot. In vitro metastatic correlates increased in Hep 3B lines after transfection with siOPN and/or miRNA 181a inhibitor (p<0.01).
Conclusions
MiRNA 181a decreases OPN expression in HCC cell lines. This undescribed mechanism may confer metastatic characteristics to HCC.
Introduction
Hepatocellular carcinoma (HCC) is the third most common cancer worldwide and is a leading cause of cancer related death. HCC mortality is correlated with both recurrent disease and metastasis. Increased mortality and metastatic behavior of HCC subtypes have been linked to expression of osteopontin (OPN)1,2. OPN is a hydrophilic, phosphorylated glycoprotein that is naturally occurring in the extracellular milieu throughout the body. OPN has physiologic roles associated with bone remodeling, immune modulation, and inhibition of apoptosis. OPN also plays many roles in the pathologic sequence of tumor growth, migration, invasion, angiogenesis, and metastasis3,4. OPN is overproduced by carcinoma cell lines that have a proclivity for metastasis. OPN over expression has been associated with poor clinical outcome2. Few studies have investigated possible mechanisms responsible for differential OPN expression in HCC. In our studies we have used two distinct cell lines, Hep G2 and Hep 3B, with differential endogenous OPN expression. When compared to Hep G2, Hep 3B exhibits decreased OPN mRNA stability resulting in decreased OPN protein expression5. We hypothesized that differential OPN translational mRNA stability may be mediated by microRNA. MicroRNAs are oligonucleotides (~21 nt) that have been recently established as a regulatory mechanism of eukaryotic protein expression. They inhibit protein expression by preferentially binding with target mRNA 3′-UTRs. If sufficient miRNA homology exists, the target mRNA segment is degraded via the RNA-induced silencing complex (RISC) pathway. In this study, we demonstrate that differential OPN expression in Hep G2 and Hep 3B is mediated, in part, by microRNA 181a. We conclude that microRNA suppression may be one mechanism responsible for differential OPN expression in HCC.
Methods
Cell Culture
The HCC cell lines Hep G2 (ATCC# HB-8065) and Hep 3B (ATCC# HB-8064) obtained from the American Type Tissue Collection (ATCC, Manassas, Va) were maintained as monolayers in Minimum Essential Medium (MEM, Sigma-Aldrich, St. Louis, Mo) plus 10% fetal bovine serum at 37°C and 5% CO2.
Microarray analysis
Total RNA was isolated from Hep G2 and Hep 3B cultures using RNeasy Mini Kit (Quiagen, Germantown, MD). RNA isolates were sent to the Duke Microarray Facility (Durham, NC) to detect differing miRNA profiles using Ambion mirVana set2 + Invitrogen NCode Set V2 arrays. The samples were analyzed using Exiqon (Denmark) microRNA processing with microRNA reference panel, a multispecies array that encompasses all human viral miRNAs and 98% of human miRNAs from miRBase v.12.0 as well as 435 propietary human miRNAs from miRPlus™.
RT-PCR confirmation
Total RNA was prepared using TRIzol reagent. RNA was treated with DNase I (Promega,Madison,WI) at 37 °C for 30min, followed by the addition of 1 μL of stop solution at 65 °C for 15 minutes prior to analysis by qPCR. For qPCR analysis, 2 μg of DNase I-treated total RNA was used for reverse transcription to synthesize first strand cDNA using SuperScriptaseII RT (Invitrogen). Aliquots of the first strand cDNA were used in qPCR using SYBRGreen. The specific PCR primers for the OPN were designed using PrimerQuest software: Forward primer 5′-TGA ATG GTG CAT ACA AGG CCA TCC-3′, reverse primer 5′-TTCATAACTGTCCTTCCCACGGCT-3′. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the endogenous control: GAPDH forward: 5′-AGC CTC AAG ATC ATC AGC AAT GCC-3′; GAPDH reverse: 5′-TGT GGT CAT GAG TCC TTC CAC GAT-3′. OPN was normalized to a housekeeping gene, GAPDH; 2−(ΔΔCt) normalization6 was used to calculate the results.
3′-UTR luciferase plasmid constructs
For OPN 3′-UTR reporter vector constructs, OPN 3′-UTR fragments were generated by PCR using forward primer 5′-GCG CCT CTA GAA AAG GAG AAA AAA TAC AATT-3′and reverse primer 5′-GGC CGT CTA GAT TAC ATT CAA GAT AAA AGA-3′ (IDT). The Luc-OPN-3′-UTR fragment was generated by restriction enzyme Xba I digestion followed by insertion at a downstream site of luciferase containing the pGL3 promoter vector (Promega Co.). Mutant constructs, Luc-OPN-3′-UTR-M1, Luc-OPN-3′-UTR-M2, and Luc-OPN-3′-UTR-M3, were constructed using QuikChange® II site-directed mutagenesis kit (Stratagene, La Jolla, CA), with the wild type Luc-OPN-3′-UTR promoter vector as template. Consensus binding site mutations corresponded to OPN promoter 3′-UTR (Luc-OPN-3′-UTR-M1 (427–429); 5′-GTTGAATG-3′ to 5′-GTTAGCTG-3′, Luc-OPN-3′-UTR-M 2 (429–431); 5′-TTGAATGT-3′ to 5′-TTGACGAT-3′, Luc-OPN-3′-UTR-M3 (444–446); 5′-TGGTGGTG3′ to 5′-TGGCATTG-3′). Wild-type and mutational accuracy were verified using DNA sequencing.
Transient transfection assay
A confluent monolayer of Hep G2 and Hep 3B cells were transfected with 25 microgram OPN 3′UTR mutant construct pGL3 plasmid with luciferase reporter or wild-type OPN 3′UTR pGL3 control plasmid plus Renilla luciferase gene by Lipofection as per manufacturer’s protocol. After 48 h of transient transfection, cells were processed for luciferase activity analysis using a luminometer (TD-20/20; Tuner Designs, Sunnyvale, Calif) according to manufacturer’s instructions. The data represent mean-fold luminescence of the triplicates from 3 independent experiments.
Micro-RNA techniques
Hep G2 and Hep 3B cells were plated at a density of 5 × 105 cells/well in 6-well plates (Costar, Corning Inc., Corning, NY) in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen Co., Carlsbad, Calif) plus 10% fetal bovine serum 24 h before transfection as described previously. Cells were transfected with miRNA 181a precursor, miRNA 181a inhibitor, or null transfection (Santa Cruz Biotechnology) using electroporation (BTX, Holliston, MA ), as per manufacturer’s instructions. Transfected cells were harvested after 48 h, and OPN protein levels were quantified by Western blot analysis in triplicate assays.
Western blot for OPN
Cell culture media was harvested and cellular protein lysates obtained from the cell lines lysed with the buffer that contained 0.8% NaCl, 0.02% KCL, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholic acid, 0.144% NaH2PO4, and 0.024% KH2PO4 (pH 7.4) were separated by 12% SDS-PAGE. Transferred membranes were blocked and labeled with human monoclonal OPN primary antibody (1:1000 dilution; R&D Systems, Minneapolis,MN) for 3 h at room temperature followed by staining with horse radish peroxidase-conjugated secondary antibody for 1 h at room temperature. Bound peroxidase activity was detected by using Western Blot Luminol reagent as per manufacturer’s instructions (Santa Cruz Biotechnology, Santa Cruz, Calif).
Invasion/adhesion/migration assay
For adhesion assay, 1 × 105 cells/well were seeded in triplicates on 96-well plates pre-coated with fibronectin (10 mg/mL) for 2 h at 37°C. Nonadherent cells were aspirated, and the bound cells were washed in 1× phosphate-buffered solution (PBS) and fixed with 3.7% paraformaldehyde for 10 min followed by staining with 0.4% crystal violet for 10 min at room temperature. After several washes in 1× PBS, 30% acetic acid was added to release the dye and was assayed on a microplate reader at 590 nm (Molecular Devices Co., Sunnyvale, Calif). For migration and invasion assays, cells were seeded on 8-mm pored, 12-well trans-wells in triplicates (1 × 105/well) and incubated for 24 h at 37°C and 5% CO2. The upper chamber was coated with matrigel (0.2 mg/mL) 24 h before seeding of cells in Minimum Essential Medium (MEM, Sigma-Aldrich, St. Louis, Mo) plus 1% FBS culture medium for the contained invasion assay. The lower chamber contained Minimum Essential Medium (MEM, Sigma-Aldrich, St. Louis, Mo) plus 10% FBS. The migrated cells were fixed for 10 min in 1× PBS with 3.7% paraformaldehyde followed by 3 washes in 1× PBS after the nonmigrated cells on the top surface of the filters were removed with cotton swabs. The filters were stained with 0.4% crystal violet for 10 min at room temperature. Migrated cells were counted under microscopy.
Data presentation
Results are expressed as mean ± standard deviation (SD). Analysis was performed using Student’s T test. Values of p<0.05 were considered significant.
Results
OPN Expression in Hep G2 and Hep 3B Cell Lines
Expression of OPN protein and mRNA was determined in the two HCC cell lines. OPN protein expression was 12-fold greater in the cellular lysate and culture media (secreted protein) of Hep G2 cells compared to Hep 3B (p<0.01) in which OPN expression was virtually undetectable. (Figure 1A) We have previously demonstrated that OPN mRNA expression was significantly greater in Hep G2 cells compared to Hep 3B cells. In addition, OPN promoter activity and transcription were not significantly different between the two cell lines. (Data not shown.) To assess OPN mRNA degradation, Hep G2 and Hep 3B cells were cultured in the medium with or without actinomycin D (20 μg/ml). At several time points after incubation (0, 4, 8, 16, 24, and 36 h), total RNA was extracted and subjected to Northern blot analysis to determine expression and quantification of OPN mRNA. Half-life of OPN mRNA in Hep G2 cells was 21 ± 1 h whereas the half-life of OPN mRNA in Hep 3B cells was only 3 ± 1 h (p<0.01) (Figure 1B). These data suggest that the difference in OPN protein expression between the HCC cell lines may be, in part, due to accelerated OPN mRNA degradation in Hep 3B cells compared to Hep G2 cells5.
Figure 1.

Determination of differential OPN expression and microRNA transcription in 2 HCC cell lines. Figure 1a. OPN and β-actin expression by Western blot analysis in Hep G2 and 3B cell lysates and culture medium. Figure 1b. Normalized percent OPN mRNA expression in the presence of actinomycin D (20μg/mL) after 0, 4, 8, 16, 24, and 36 h incubation. Figure 1c. 2−(ΔΔCt) normalized real time PCR transcription of miRNA candidates mi96 and miRNA 181a in 2 HCC cell lines. Figure 1d. 2 −(ΔΔCt) normalized real time PCR transcription of miRNA 181a in 2 HCC cell lines.
Differential MicroRNA Expression in Hep G2 and Hep 3B Cell Lines
As microRNAs play a critical role in differential protein expression, microarray analysis of differential microRNA expression in Hep G2 and Hep 3B was performed. This demonstrated increased levels of miRNA 33b and 514 in Hep G2 and increased levels of miRNA 181a and 96 in Hep 3B.(p<0.0001) The OPN 3′-UTR sequence (548 nt) was then sequenced in both cell ines and found to be identical. Given the inhibitory role of miRNAs in protein expression, this sequence was analyzed using Targetscan and miRBase Target and found to have potential miRNA 181a and 96 binding sites. Quantitative RT-PCR was performed for miRNA 181a and 96.(Figure 1c and 1d) There was no significant difference in the levels of miRNA 96 between Hep G2 and Hep 3B. However, there was a 36 ± 5 fold increase of miRNA 181a in Hep 3B over Hep G2, determined using the 2 −(ΔΔCt) technique. Subsequent studies focused on miRNA 181a.
MicroRNA 181a Targeting of OPN 3′-UTR
The OPN 3′-UTR segment was cloned into the pGL3 vector with the SV40 promoter followed by transient transfection and assayed for luciferase activity; luciferase acts as a surrogate for OPN in this setting.(Figure 2a) In selected settings, miRNA 181a precursor or miRNA 181a inhibitor was added. The data demonstrate increased baseline luciferase activity in Hep G2, mirroring the relative excess of OPN in Hep G2 cells. In the presence of miRNA 181a precursor, luciferase expression was decreased in Hep G2 and Hep 3B by 52% and 28%, respectively (p<0.01 vs reporter alone for Hep G2 and Hep 3B). Conversely, addition of the miRNA 181a inhibitor increased luciferase activity 15% in Hep G2 (p<0.01 vs reporter) and 32% in Hep 3B (p<0.01 vs WT reporter). Site-directed mutagenesis of the 3 putative miRNA 181a binding sites in OPN 3′-UTR was then performed and the luciferase reporters, M1, M2 and M3, constructed. These were then transfected into Hep G2 and Hep 3B (Figure 2b). In both Hep G2 and Hep 3B, neither the M1 nor M2 exhibited significantly different luciferase expression when compared to the WT reporter control. However, the M3 construct was found to increase luciferase in Hep 3B by 108% (p<0.01 vs WT reporter) and 15% in Hep G2 (p=NS vs WT reporter). The lack of effect in Hep G2 may be secondary its relatively low baseline levels of miRNA 181a. When the M3 construct was transfected into Hep G2 and Hep 3B in the presence of the miRNA 181a precursor or miRNA 181a inhibitor, there was not a significant change in luciferase expression when compared to the M3 construct alone (data not shown). These results indicate that miRNA 181a alters OPN 3′-UTR dependent reporter expression and it may interact with OPN 3′-UTR sequence nt 444–446.
Figure 2.
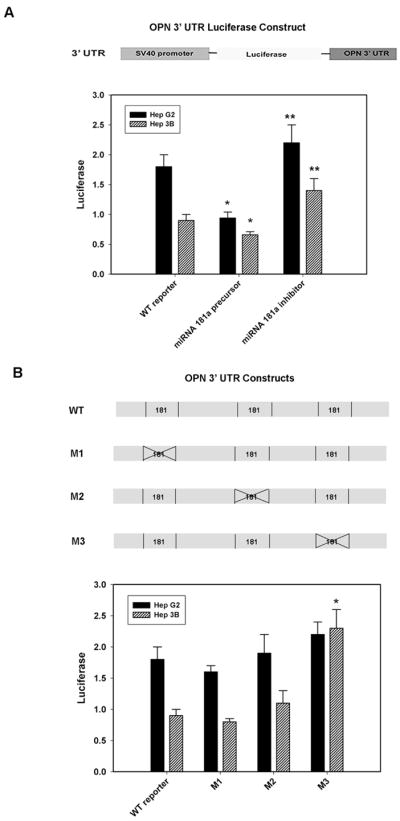
Luciferase activity after transient transfection in 2 HCC cell lines. Figure 2a. Schematic of OPN 3′ UTR luciferase construct in pGL3 vector. Luciferase activity after OPN 3′ UTR luciferase reporter transfection with wild type miRNA 181a precursor or miRNA 181a inhibitor in 2 HCC cell lines. Hep G2 and Hep 3B luciferase activity decreased by 52% and 28% respectively after transfection with miRNA 181a precursor (*p< 0.01 vs. WT) Hep G2 and Hep 3B increased luciferase activity 15% and 32% respectively with miRNA 181a inhibitor transfection (** p< 0.01 vs WT). Figure 2b. Depiction of three candidate miRNA 181a sites of interaction with sequential mutants interrupting each sequence as predicted by algorithmic software. Luciferase activity after transfection in 2 HCC cell lines with variable OPN 3′UTR luciferase reporter: wild type, mutant 1 (M1), mutant 2 (M2), or mutant 3 (M3). Transfection of M1 or M2 did not alter luciferase activity in either cell line. M3 transfection increased luciferase activity in Hep G2 by 15% (p=NS vs WT) and in Hep 3B by 108% (*p < 0.01 vs WT reporter)
MicroRNA 181a and OPN Dependent Function
To determine the effect of miRNA 181a on OPN protein expression, Western blot analysis was performed in Hep G2 and Hep 3B. (Figure 3) OPN protein in Hep G2 + miRNA 181a precursor was decreased 5-fold (p<0.01 vs Hep G2), while that of Hep 3B + miRNA 181a inhibitor was increased 3-fold (p<0.01 vs Hep 3B). In vitro adhesion and migration/invasion assays were performed with Hep G2 and Hep 3B cells in the absence and presence of miRNA 181a precursor or miRNA 181a inhibitor. (Figure 4) Wild-type Hep G2 typically exhibits 3 fold greater adhesion/migration/invasion than wild-type Hep 3B. (Data not shown.) In Hep G2, addition of miRNA 181a precursor decreased adhesion and migration/invasion by 57% and 48% (p<0.01 vs Hep G2 alone); miRNA 181a inhibitor did not significantly alter adhesion or migration/invasion. In Hep 3B, addition of miRNA 181a precursor did not alter adhesion and migration/invasion; miRNA 181a inhibitor increased adhesion and migration/invasion by 60% and 550% (p<0.01 vs Hep 3B alone). Overall, miRNA 181a accounts for a significant amount of the relative baseline differences between Hep G2 and Hep 3B in this context. To corroborate the specific effect of miRNA 181a on OPN regulated functions, the above experiments were repeated in the presence of siRNA to OPN (siOPN). In this instance, addition of miRNA 181a precursor or miRNA 181a inhibitor to Hep G2 and Hep 3B in the presence of siOPN resulted in a significant decrease in adhesion and migration/invasion in both WT cells. (Figure 4) In total, our data indicates that miRNA 181a regulates OPN expression and OPN dependent in vitro correlates of metastatic behavior in the HCC cell lines, Hep G2 and Hep 3B.
Figure 3.
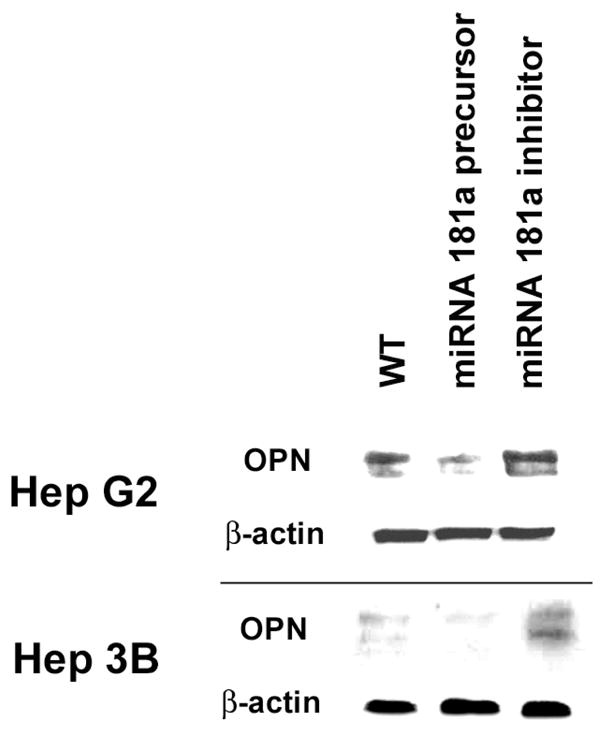
Western blot analysis of OPN and housekeeping gene β-actin in control and after transient transfection with miRNA 181a precursor or miRNA 181a inhibitor in 2 HCC cell lines. MiRNA 181a precursor transfection into Hep G2 decreased OPN expression by 5-fold (p< 0.01). MiRNA 181a inhibitor transfection into Hep 3B cell line augmented OPN expression by 3-fold (p< 0.01).
Figure 4.

In vitro correlates of metastatic phenotype after transfection in 2 HCC cell lines. Hep G2 increased adhesion (Figure 4a) and invasion/migration (Figure 4b) after transfection with miRNA 181a precursor by 57% and 48% respectively compared to null transfection in Hep G2. Hep G2 transfection with siOPN and either miRNA 181a precursor or miRNA 181a inhibitor significantly decreased both adhesion and invasion/migration (*p< 0.01). Hep 3B transfection with miRNA 181a precursor increased adhesion (Figure 4a) and invasion/migration (Figure 4b)60% and 550% respectively compared to null transfected Hep 3B cell lines. Hep 3B transfection with siOPN and either miRNA 181a precursor or miRNA 181a inhibitor significantly decreased both adhesion and invasion/migration as well (**p< 0.01).
Discussion
MicroRNA’s are known to modulate protein expression, most commonly acting to degrade target mRNA thereby limiting protein expression. In the two cell lines investigated HepG2 readily produces OPN while Hep3B has limited OPN expression. Previous studies document that while both cell lines effectively translate OPN mRNA, Hep3B OPN mRNA is unstable and has a significantly shortened half-life when compared to HepG2 OPN mRNA5. Our proposed mechanism of Hep3B mRNA instability is microRNA regulated OPN expression. Spotted array microchip analysis confirmed differential microRNA profiles. MiRNA 181a is readily produced in Hep 3B, with only limited transcription in Hep G2. Real time PCR confirms the vast differences in miRNA181a expression between the two cell lines: 36-fold increase in Hep3B versus HepG2. To confirm miRNA 181a regulation of OPN expression, both cell lines were transfected with precursor miRNA 181a and inhibitor miRNA 181a. As expected, squelching miRNA181a in Hep 3B lines with inhibitor miRNA 181a increased OPN expression, as confirmed by Western blot analysis. Sites of miRNA 181a interference were predicted by algorithmic software Targetscan and miRBase Target determination of potential miRNA 181a interaction within the OPN 3′-UTR. Three potential sites of homology were identified in the OPN 3′-UTR. Creating mutated OPN 3′-UTR plasmids with luciferase reporter at each potential interaction site, we tested each candidate by transfecting both cell lines with each mutant plasmid. Ultimately, mutation at the OPN 3′-UTR nt 444–446 (5′-TGGTGGTG3′ to 5′-TGGCATTG-3′), or M3, transfected into Hep 3B showed increased luciferase activity when compared to wild type,. This is presumed to be secondary to inhibited miRNA 181a binding to the OPN 3′-UTR which in vivo, we propose, targets the mRNA strand for degradation or OPN expression quiescence. OPN expression following M3 transfection in Hep G2 cell line was not different when compared to WT, as Hep G2 does not express appreciable levels of miRNA 181a and thus mutation of the binding site is not of significant consequence. Transfection of miRNA181a inhibitor into Hep 3B cell lines directly suppressed miRNA 181a and increased OPN protein expression in Western blot analysis. Manipulation of OPN translation in HCC cell lines modified correlates of metastatic behavior. Suppressing OPN expression in Hep G2 diminished adhesion and migration/invasion. Inhibiting miRNA 181a in Hep 3B cell lines augmented adhesion and migration/invasion. Interestingly, siOPN had a more profound effect on the Hep 3B cells, which produce little OPN. These siOPN experiments suggest that there may be additional mechanisms for OPN regulation, or that the siOPN reduces adhesion and migration/invasion through other mechanisms.
The role of miRNA 181a has been studied in various malignancies, but little has been accomplished in delineating the molecular mechanism. In gene expression and microchip analyses of pathological states, miRNA 181a is associated with improved survival in some hematalogic malignacies, such as chronic lymphocytic leukemia and acute myelogenous leukemia7. MiRNA 181a has been shown to modulate T cell maturation as well as oocyte nuclear rearrangement. MiRNA 181a expression has been linked to improved survival and decreased recurrence in gliomas8. Showing poor outcome in patients with glioma and down-regulated miRNA 181a, it is proposed that miRNA 181a is a powerful inhibitor of oncogenesis and tumor growth. In vitro transfection of three glioma cell lines with precursor miRNA 181a inhibited invasion and migration while promoting apoptosis, although the underlying molecular mechanism is yet undescribed. Another study of HCC metastasis strongly linked miRNA181a with improved survival while clearly identifying sixteen prometastatic microRNA’s9, from which miRNA 181a was excluded. In contrast, a comprehensive investigation of miRNA 181a in HCC cell lines has identified a prometastatis phenotype presumed to be initiated by the miRNA181a family. It is proposed that miRNA 181a is of importance in the development of epithelial cell adhesion molecule+/alpha-fetoprotein+ (EpCAM+/AFP+) HCC, which is associated with increased metastasis and poor survival10. The miRNA 181a locus is identified, but a mechanism is not described. While an association is noted between miRNA 181a transcription of this particularly malignant tumor cell line and poor outcome, there is no proposed causal relationship linking the two.
MiRNA 181a plays a role in gene expression in a host of pathological states, shown to both mediate and inhibit malignancy, but little has been accomplished in proposing a mechanism. OPN expression has been reported to be indirectly inhibited by miRNA 146. This mechanism describes increased miRNA 146 in the presence of BRMS-1, a mediator of suppressed metastasis in breast cancer cell lines11. BRMS-1, in addition to promoting miRNA 146 expression, also inhibits prometastasic markers EGFR, uPA, and OPN. MicroRNA dependent OPN expression is yet to be definitively identified.
OPN has been clearly shown to bestow a prometastatic phenotype to cancer cell lines. Reviews of OPN function demonstratively link expression to metastatic behavior, including but not limited to adhesion, invasion, migration, neovascularization, and diminished anti-tumor host immune response12. MiRNA 181a regulation of OPN expression provides a novel mechanism of suppressing metastasis in cancer cell lines. Further investigation of this pathway and potential manipulation of OPN expression may provide a new therapeutic avenue for particularly aggressive OPN expressing malignancies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gotoh M, Sakamoto M, Kanetaka K, et al. Overexpression of osteopontin in hepatocellular carcinoma. Pathol Int. 2002;52:19–24. doi: 10.1046/j.1440-1827.2002.01316.x. [DOI] [PubMed] [Google Scholar]
- 2.Pan HW, Ou YH, Peng SY, et al. Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer. 2003;98:119–127. doi: 10.1002/cncr.11487. [DOI] [PubMed] [Google Scholar]
- 3.Dai J, Peng L, Fan K, et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene. 2009;28:3412–3422. doi: 10.1038/onc.2009.189. [DOI] [PubMed] [Google Scholar]
- 4.Oates AJ, Barraclough R, Rudland PS. The role of osteopontin in tumorigenesis and metastasis. Invasion Metastasis. 1997;17:1–15. [PubMed] [Google Scholar]
- 5.Emani S, Zhang J, Guo L, et al. RNA stability regulates differential expression of the metastasis protein, osteopontin, in hepatocellular cancer. Surgery. 2008;143:803–812. doi: 10.1016/j.surg.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 7.Marcucci G, Radmacher MD, Maharry K, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1919–1928. doi: 10.1056/NEJMoa074256. [DOI] [PubMed] [Google Scholar]
- 8.Shi L, Cheng Z, Zhang J, et al. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008;1236:185–193. doi: 10.1016/j.brainres.2008.07.085. [DOI] [PubMed] [Google Scholar]
- 9.Budhu A, Jia HL, Forgues M, et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897–907. doi: 10.1002/hep.22160. [DOI] [PubMed] [Google Scholar]
- 10.Ji J, Yamashita T, Budhu A, et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50:472–480. doi: 10.1002/hep.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurst DR, Edmonds MD, Scott GK, et al. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009;69:1279–1283. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wai PY, Kuo PC. Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev. 2008;27:103–118. doi: 10.1007/s10555-007-9104-9. [DOI] [PubMed] [Google Scholar]