

Abstract
Picornaviruses have a 22–24 amino acid peptide, VPg, bound covalently at the 5’ end of their RNA, that is essential for replication. VPgs are uridylylated at a conserved Tyrosine to form VPgpU, the primer of RNA synthesis by the viral polymerase. This first complete structure for any uridylylated VPg, of poliovirus type 1 (PV1)-VPgpU, shows that conserved amino acids in VPg stabilize the bound UMP, with the uridine atoms involved in base pairing and chain elongation projected outward. Comparing this structure to PV1-VPg and partial structures of VPg/VPgpU from other picornaviruses suggests that enteroviral polymerases require a more stable VPg structure than does the distantly related aphthovirus, foot and mouth disease virus (FMDV). The glutamine residue at the C-terminus of PV1-VPgpU lies in back of the uridine base and may stabilize its position during chain elongation and/or contribute to base specificity. Under in vivo-like conditions with the authentic cre(2C) hairpin RNA and Mg++, 5-methylUTP cannot compete with UTP for VPg uridylyation in an in vitro uridylyation assay, but both nucleotides are equally incorporated by PV1-polymerase with Mn++ and a poly-A RNA template. This indicates the 5 position is recognized under in vivo conditions. The compact VPgpU structure docks within the active site cavity of the PV-polymerase, close to the position seen for the fragment of FMDV-VPgpU with its polymerase. This structure could aid in design of novel enterovirus inhibitors, and stabilization upon uridylylation may also be pertinent for post-translational uridylylation reactions that underlie other biological processes.
Keywords: uridylylation, post-translational modification, picornaviruses, disordered structures, Coxsackie virus B, polymerase priming mechanism, RNA editing, antiviral compounds, flexibility
Introduction
Picornaviruses are non-enveloped, +−strand RNA viruses that include many virulent and easily communicable human and animal pathogens. Although poliovirus (PV) has been nearly eradicated by vaccine campaigns, pockets of infectious virus persist in certain parts of the world. The World Health Organization has recently called for the development of new, safer vaccines and drugs for the treatment of poliomyelitis (Global Polio Eradication Initiative). Other enteroviruses also constitute public health problems, causing illnesses ranging from the common cold (Rhinoviruses) to dilated cardiomyopathies leading to heart transplants (primarily human enteroviruses such as Echovirus and Coxsackie virus B (CVB)[23, 30]). Picornaviruses spread rapidly, as outbreaks of hemorrhagic conjunctivitis caused by Coxsackie virus A[24, 40, 51] or Foot and Mouth Disease (FMDV)[11] in farm animals have illustrated, and there are few therapies.
One area that could be targeted for drug design is the unique priming mechanism used by picornaviral polymerases. The 5’ end of the viral RNA is bound to a 22–24 residue peptide, called VPg or 3B (the latter referring to its position in the P3 domain of the polyprotein)[32]. Free VPg and the primers of RNA synthesis, VPgpU or VPgpUpU (uridylyated at Tyr3) are found in PV-infected cells[12]. Uridylylation of VPg occurs independently of priming and initiation of RNA synthesis[44]. Mutations throughout VPg have been shown to prevent uridylylation and/or, when inserted into the genomic RNA, virus growth [49].
However, a molecular model of how VPgpU primes RNA synthesis is lacking. There are partial crystal structures of picornaviral VPgs complexed with their polymerases, one for the aphthovirus FMDV[18] and more recently, for the enterovirus Coxsackie virus B3 (CVB3)[25]. These differ from one another in both the structure of the VPg and its location on the polymerase.
Considering the evolutionary distance between the aphtho- and enteroviruses, differences in the structure and the mode of interaction of the VPgs with the RNA polymerases might be anticipated. The genome of FMDV (8400nt, with a 1300 n 5’ noncoding region) is longer than that of PV (7400 nt, 740 n in the 5’ noncoding region)[6]. The enteroviruses have a single copy of VPg in their genome, while FMDV strains encode and express 3 distinct copies of VPg (3B1, 3B2, 3B3), all of which can be found in vivo linked to viral RNA[29]; deletion of any of the 3 reduces RNA replication[17]. The VPgs of FMDV are longer (23–24 aa compared to 22 for the enteroviruses) and their amino acid sequences are only identical at 3 positions to those of the enteroviruses (Figure 1). In addition, the FMDV VPgs contain glutamic acid, which is not found in enterovirus VPgs.
Figure 1. Alignment of the sequence of PV-1 VPg (used in this study) with VPgs from other enteroviruses.
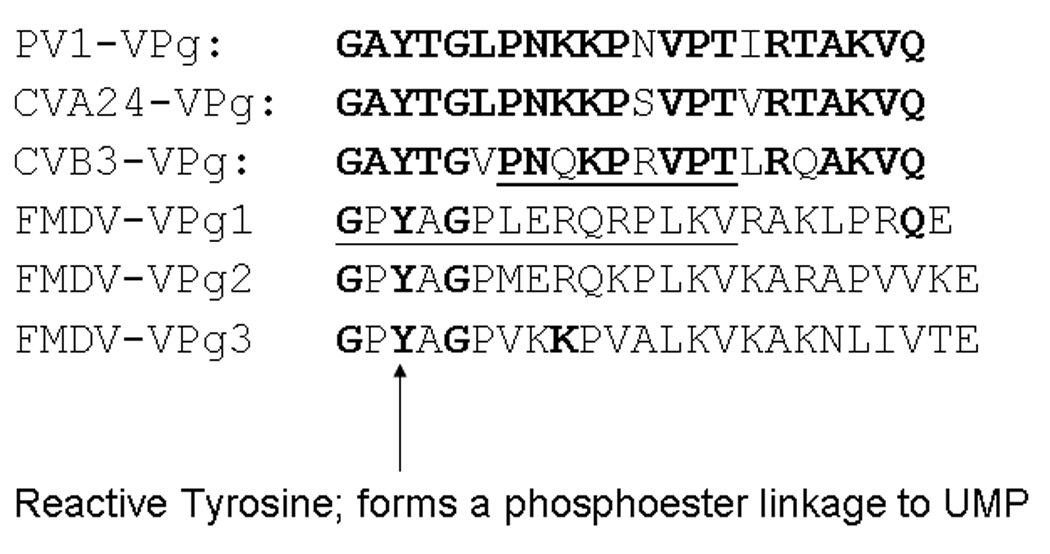
Coxsackie viruses A24 and B3, and the three VPgs of FMDV. The areas of the VPg visible in the crystal structures of CVB3 and FMDV-VPg1 (illustrated in Figure 4) in complex with their polymerases are underlined.
While VPgs have a flexible structure, the evidence summarized here suggests that enteroviral polymerases require a more structured primer than FMDV. Previously, we determined the first full-length structure of any picornavirus VPg, that of PV-1[55]. Here, we show the first full length structure of a uridylylated VPg, of chemically synthesized PV1-VPgpU. The most important experimental observations are that the conformation changed considerably upon uridylylation, became more stable, and could fit within the active site of the polymerase near the binding site of FMDV-VPgpU. This suggests that the role of VPgpU in initiating RNA synthesis is to reduce the mobility of the 5’ terminal-UMP moiety. Binding of UTP by positively charged amino acids and the C-terminal glutamine residue of VPg, which in the structure of the VPgpU binds the back face of the uridine base, could play an important role in base selectivity during priming. We show that under stringent in vitro assay conditions, 5-Methyluridine-5'-triphosphate (5-meUTP; thymidine as a ribonucleotide) cannot compete with UTP for uridylylation of VPg by the PV1-polymerase. Structural flexibility in the function of these VPgs is also discussed.
Materials and Methods
Synthetic PV1-VPgpU
(22 amino acids, uridylylated at the reactive Tyr3 (TYU3)) was prepared and purified essentially as described[31] via Fmoc-based solid phase peptide synthesis on Tentagel S resin. The uridylylated tyrosine residue was incorporated as a known pre-uridylylated Fmoc-amino acid derivative and coupled as previously described.(See Supporting information for additional details).
Sample preparation for NMR
Uridylylated PV1-VPgpU (3.7 mg) was dissolved in 0.5 ml water and extracted 3x with 1ml volumes of Diethyl ether (to remove residual acid from the purification). The sample was lyophilized and redissolved in 0.6 ml 10 mM sodium phosphate buffer pH 7.5; 0.06 ml of D20 was added (final concentration was thus about 2 mM). This sample was used for initial TOCSY and NOESY experiments, 1H-15N F1-coupled HSQC and natural abundance 1H-15N HMQC-J. For experiments in D2O (NOESY and natural abundance 1H-13C HMQC), the sample was lyophilized and re-dissolved in the original volume of 100% D2O.
NMR experiments
All spectra (TOCSY, NOESY, 1H-15N F1-coupled HSQC and natural abundance 1H-15N HSQC-J and 1H-13C HMQC) were recorded on Varian NMR system 750 MHz spectrometer equipped with Z-axis gradients. For water suppression, a water-flip-back principle [26, 60]together with WATERGATE using the 3–9–19 composite pulse train[57] was implemented. During the NOE mixing time, the water magnetization goes immediately back to +z through radiation dumping [60]. At the end of the mixing time, a water-selective EBURP pulse[22] is applied to keep the water-magnetization at +z during the detection. The implementation of the water-flip-back principle improves the sensitivity for amide protons cross peak detection. All spectra were collected at 5°C. Data were processed with NMRPipe.
Chemical shift assignment
The 1H, 15N and 13C shifts were obtained from TOCSY (using NOESY connectivities to resolve the redundant Ala(2), Asn (2), Lys (3), Pro(3), Thr (3), and Val (2)) residues. Additional shifts, especially for the UMP and peptide side chains, were obtained from natural abundance 1H-15N F1-coupled HSQC and natural abundance 1H-13CHMQC-J cross peak data. 13C-HMQC cross peak data was especially useful for determining the chemical shift assignment for UMP (base and ribose). The 15N and 13C chemical shift values for the uridine were compared to average values for each position from the Biological Magnetic Resonance Bank (BMRB).
Dihedral angle constraints
Coupling constants were calculated after [63], according to the equation: JHNHa = 0.5(Δv1/2) − 0.5(Δv1/2(min)) + 4.0Hz for TOCSY data, where the smallest peak width at one half the height (Δv1/2(min) for our data is 16 Hz.
Angular constraints for secondary structure was done by assuming that all with JHNHa <5 Hz would have α character, those >8.5 would have β-character [62] and appropriate constraints derived. By these criteria, Y3-G5 had β character (J>8.5 Hz), and L6, K10, A19-Q22 were α (so J<5); N12, T15, I16, R17, T18 were Intermediate/RC. (See Supplementary data table)
Structure calculation with NOAH/DIAMOD
The NOAH/DIAMOD[46] suite was used to automatically assign the NOESY cross-peaks and calculate bundles of structures[20, 65]. At the start of each cycle after the first, a statistical analysis was performed to test the consistency of the current assignments with the bundle of structures. Based on this analysis assignments were kept or rejected. These cycles of assignments, generation of structures and re-evaluation of the assignments was repeated until the bundle of structures converged. Previous studies demonstrated that the 3D structures and automated assignments generated with these programs are in excellent agreement with results from conventional manual assignments [41, 64]. To add coordinates for uridylylated Tyrosine (TYU) to the DIAMOD library, atom entries for Tyr and UMP were combined, so that the hydroxyl of the Tyr was replaced by that of the 5’ phosphate-OH of UMP. The resulting phosphate ester bond was made rotatable, and the bonding distance at the junction was corrected.
Molecular docking
The ZDOCK program[8, 33] was used to dock VPgpU on the crystal structure of the poliovirus polymerase (PDB file1RA6[61]). ZDOCK uses a fast Fourier transform method to evaluate possible binding modes for a ligand on a given receptor protein. The scoring algorithm takes into account shape complementarity, desolvation energy, and the electrostatic potentials of the molecular surfaces.
VPg uridylylation assays
Assay 1 measures the synthesis of VPgpU and VPgpUpU on a poly(A) template [49]. The reaction mixture (20 µl) contained 50 mM HEPES, pH 7.5, 8% glycerol, 0.5 µg of poly(A), 0.5 mM manganese(II) acetate, 1 µg of purified PV 3Dpol, 2 µg of synthetic VPg, 1 µCi of 32P-UTP (3000 Ci/mmole), Perkin Elmer), and 10 µM unlabeled UTP. The reaction mixtures were incubated at 34°C for 1 hr and the reaction was stopped by the addition of 5 µl of loading buffer. The samples were analyzed by a Tris-Tricine SDS-polyacrylamide gel (13.5% acrylamide) electrophoresis (Bio-Rad) and the uridylylated VPg products were quantitated with a Phosphorimager (Storm 860; Molecular Dynamics).
Assay 2 is similar to Assay 1 except that the poly(A) template is replaced by 0.5 µg of cre(2C) RNA template, 0.5 mM manganese (II) acetate is replaced by 3.5 mM magnesium acetate, and 0.5 µM purified PV 3CDpro is added to the reaction mixture.[48]. 5-Methyluridine-5'-triphosphate (5me-UTP) was obtained from Trilink Biotechnologies.
Growth of poliovirus (PV1M) in the absence and presence of 5-methyluridine
HeLa R19 cells were grown in 35 mm dishes in Dulbecco's Modified Medium, supplemented with 5% bovine calf serum, and were infected with PV1(Mahoney) at a multiplicity of 10 either in the absence or presence of 5-methyluridine (1 µM to 1mM). When a cytopathic effect was observed, the viral titers were determined by plaque assay.[39]
Structure deposition
The structure and related spectra have been deposited in the BMRB, #20110.
Results
Uridylylation stabilizes PV1-VPgpU
In contrast to the unmodified VPg, which required the addition of the stabilizing co-solvent Trimethyl amine N-oxide (TMAO) to obtain sufficient NOE constraints(PDB file 2BBL)[55], the 2D-NOESY spectrum of VPgpU in low molarity phosphate buffer at neutral pH showed many medium and long range NOE cross-peaks (e.g., from the tyrosine ring δ and ε protons of TYU3 to many other side chains (Figure 2a)). These indicated stable interactions between TYU3 and residues such as Leu6, Pro7, Asn8, Lys9, and Arg17 which are essential for PV replication in vivo, and uridylylation in vitro. [59] Peptide chemical shifts, assignment of which was aided by natural abundance 15N-HSQC (Figure 2b) and 13C-HMQC (Figure 2c) were overall quite similar to those for VPg in buffer or in 1 M TMAO, with only a few showing major differences (>0.15 ppm). These included (where the first number in parentheses is the chemical shift in ppm for VPg in TMAO and the second number is for VPgpU in buffer): Tyr3 HE1 (6.72 to 7.06), Gly5-HN (7.4 to 8.03). Leu6 HN (8.09 to 8.22) and HB2(1.5 to 1.73), Lys 9 HN (8.18 to 8.33), Pro11 HA (4.309 to 4.125), Val13 HN (8.06 to 8.2), Thr15 (8.28 to 8.43), Ile16 HN (8.2 to 8.35), Arg17 QH (6.7 to 6.9), Lys20 HN (8.3 to 8.49) and Gln22 HE21 (7.05 to 7.63). All the chemical shifts for VPgpU in buffer were closer to the values measured for VPg in buffer than in 1M-TMAO. These differences in the chemical shifts correlate for the most part with residues whose positions in the VPgpU structure differ from those for VPg, or are found close to the UMP. Thanks to the natural abundance 3D spectra and the apparently increased stability of the uridylylated peptide, even the more labile side chain protons could be assigned for VPgpU. The only problems encountered were with the two alanines, which were difficult to assign unambiguously as both showed some cross peaks with the Tyr3 residue (see Figure 2a, top).
Figure 2. Images from spectra used for assignment.

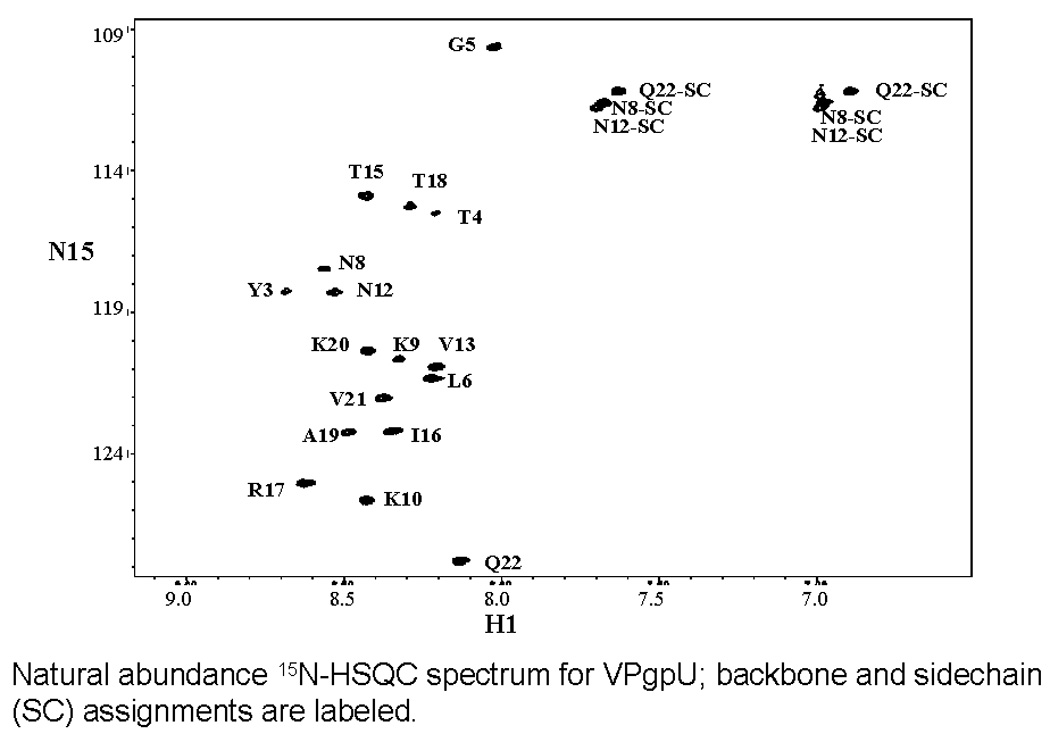
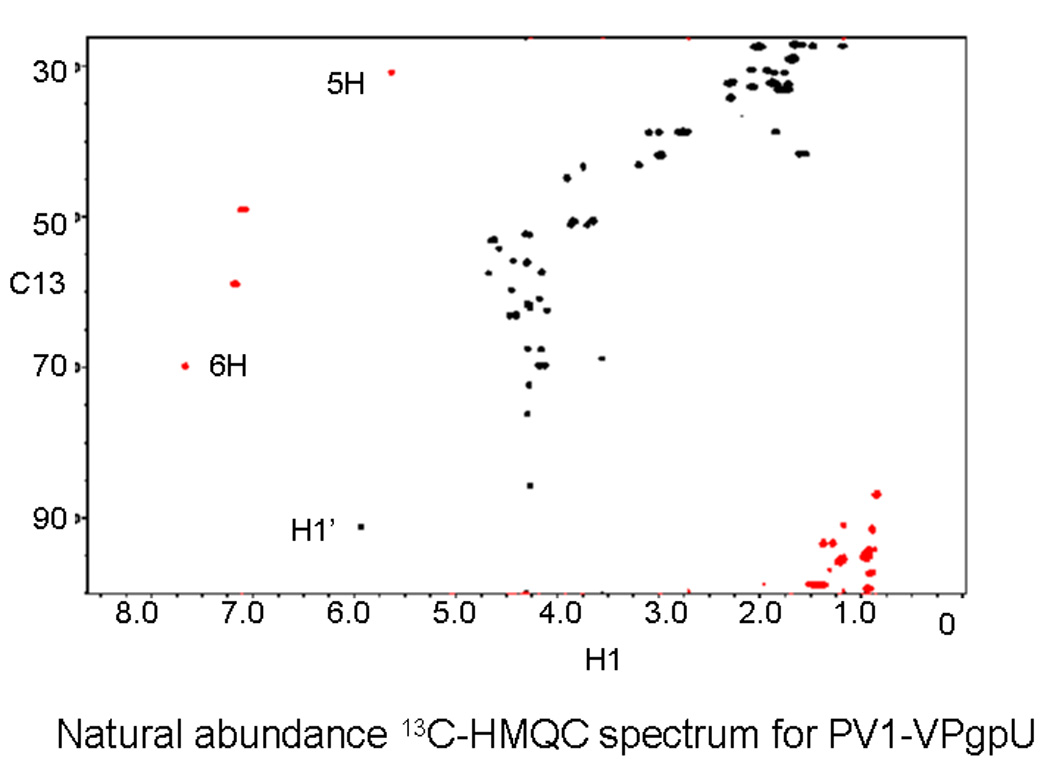
A. section of the NOESY spectrum, showing NOE cross peaks from the aromatic (δ and ε) protons of the uridylylated Tyr3 side chain (TYU3) to the rest of the peptide. A hand assignment is shown for the more obvious peaks. The position of crosspeaks to the side chain amines of ASN8 and ASN12 are also indicated. B. Natural abundance 15N-HSQC spectrum, showing the assignments for the sidechains (SC) of Asn8, Asn12, and Q22 and the backbone amide assignments (found for all except G1 and A2). C. The well resolved natural abundance 13C-HMQC spectra was particularly useful for refining the assignment of the sidechain and UMP atoms (labels).
We were also able to include some dihedral angle constraints that suggested that Y3-G5 had β character (J>8.5 Hz), and L6, K10, A19-Q22 were α (so J<5); N12, T15, I16, R17, T18 were Intermediate/RC. (See Supplementary data table). Although several calculations with differing tolerances for the program variables converged (see supplementary data), the ensemble of structures selected (“V29”, Figure 3A; summarized in Table 1) conformed to the most constraints, had a very tight bundle, and agreed with hand assignments of the spectrum. The V29 calculation used a chemical shift tolerance of 0.02 ppm, with a distance constraint tolerance of 1.0 * d-RMSD (decreasing to 0.5 * d-RMSD in the later cycles); constraints assigned to ambiguous peaks were rejected that were violated in more than 70% of the generated structures (decreasing to >40% in later iterations). The average root mean square deviation (RMSD) of the bundle is 0.38Å +− 0.11Å for backbone and 0.54Å +− 0.14Å for all atoms.
Figure 3. Structure of PV1-VPgpU.
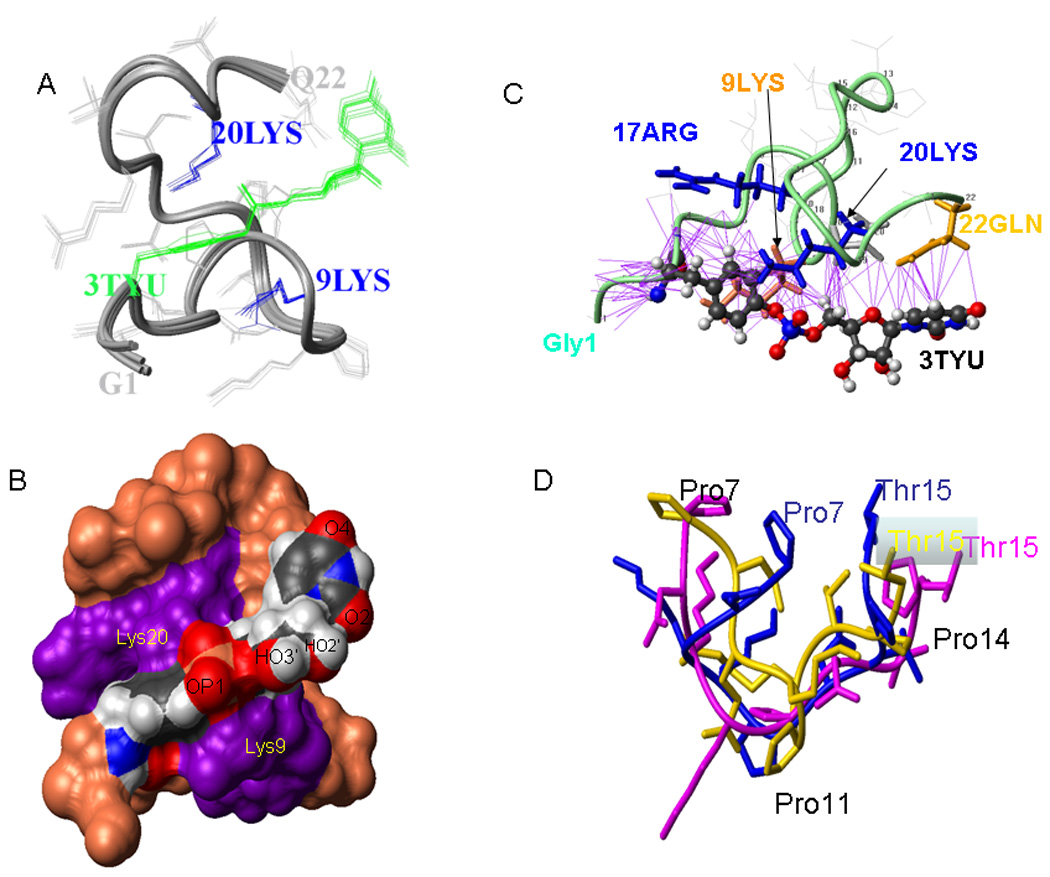
A) Bundle of the 10 top NMR structures for VPgpU; the TYU sidechain is green. B) Space filling depiction of the top structure, showing how two conserved positively charged residues residues Lys 9 and Lys 20 (violet)surround the TYU sidechain, which is colored according to atom type (Red:O, Blue: N, Black:C, Khaki: P, and gray:H), with atoms important for base-pairing and chain elongation labeled. C) Ribbon diagram showing distances < 4Å (purple lines) between sidechains from the rest of the VPg peptide to the TYU sidechain. The backbone is turquoise, the sidechains of Arg 17 and Lys 20 are blue, Lys 9 is orange and that of Gln22 is gold; the other sidechains are shown in light gray. The orientation of this figure is tilted slightly from that of PV1-VPgpU in Figure 3. D) overlay of the middle 9 residues of the solution structures of PV1-VPg (PDB file 2BBL; maroon), PV1-VPgpU (blue, current study), and the 9 residues visible in the structure of CVB3-VPg complexed with its polymerase[25] (gold).
Table 1.
Summary of structure calculation for VPgpU
| Total NOE (before diagonalization) | 578 |
| Total number of distance constraints | 284 |
| Intra-residue | 139 |
| Inter-residue | 145 |
| Sequential (|i - j| = 1) | 78 |
| Medium-range (|i - j| < 5) | 32 |
| Long-range (|i - j| > = 5) | 35 |
| Total dihedral angle restraints (phi) | 11 |
| Violations (mean and S.D.) of distance constraints | (0.18 ± 0.23) Å |
| Violations (mean and S.D.) of dihedral angle constraints | (3.97 ± 5.65)° |
| Maximum deviations from idealized bond lengths | 0.027 Å |
| Maximum deviations from idealized bond angles | 5.0° |
| Pairwise RMSDa for heavy atoms (mean and S.D.) | (0.54 ± 0.14) Å |
| Pairwise RMSDa for backbone atoms (mean and S.D.) | (0.38 ± 0.11) Å |
Pairwise RMSD was calculated for the 10 best structures.
Conserved residues position the bound UMP
The UMP lies across the front face, with residues that are conserved in the enterovirus VPgs holding it in position for base pairing. For example, the conserved, positively charged sidechains of Lys9, Arg17, and Lys 20 are all close to the phosphoester (Fig. 3B). The atoms of the uridine involved in Watson-Crick pairing (O2, N3 and O4 atoms) are solvent exposed, as is the 3’-OH of the ribose that will form the nucleotide-phosphate bond during RNA elongation. The other side of the ring is close to the side chain of the terminal Gln22 residue (Figure 3C), which also showed a large increase in its chemical shift when compared to the free VPg. This interaction suggested that the peptide could position the uridine for base pairing.
Comparison of the VPgpU structure to the NMR and partial crystal structures of picornaviral VPgs
The structures determined for picornavirus VPgs (Figure 4) show considerable variability. The 3D-structure of PV1-VPgpU shown here differs considerably from that of PV1-VPg (PDB file 2BBL). Upon uridylylation, the N- and C-termini of PV1-VPg move inward to surround the TYU sidechain. The side chain of Tyr3 is on a different face. The most similar areas of the two structure bundles are residues P7-R17; the mean structure of PV1-VPgpU in this area can be overlaid with the bundle of structures of PV1-VPg with bb-RMSDs of < 3.22Å (Figure 2D).
Figure 4.
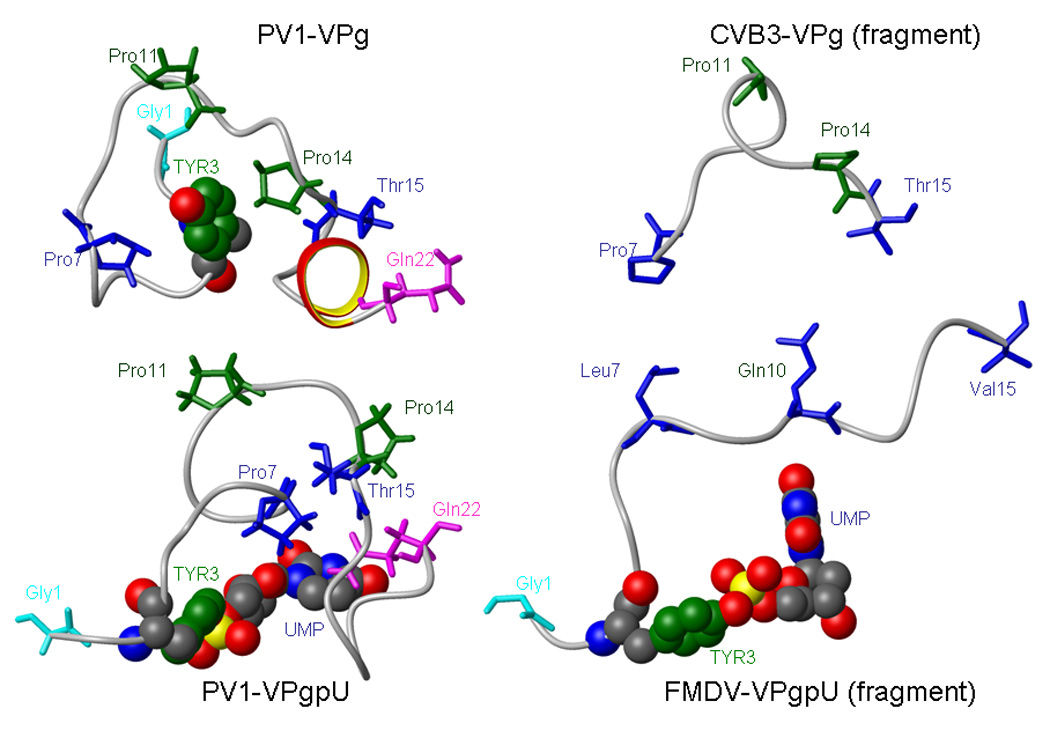
Side by side comparison of PV1-VPg and PV1-VPgpU solution NMR structures with the partial crystal structures of CVB3 VPg (residues 7–15) and FMDV- VPgpU (residues 1–15).
The middle 9 residues (P7-Thr15) of PV1 VPg and the partial structure of CVB3-VPg can be overlaid with a backbone RMSD of 2.8Å (a similar overlay with PV1-VPgpU has an RMSD of 3.8Å). The three proline residues are in very similar locations. The enterovirus VPg structures cannot be superimposed in any meaningful way with the partial crystal structures of FMDV-VPg (PDB file 2D7S) or VPgpU (PDB file 2F8E). As the partial structures of FMDV-VPg and VPgpU are so similar (backbone RMSD of only 0.6Å), only the FMDV-VPgpU structure is shown in Figure 4.
Glutamine22 interactions with the UMP
A crystal structure for a different enzyme, N-acetylglucosamine-1-phosphate uridyltransferase (GlmU) in complex with UDP-N-acetylglucosamine, shows that the side chain of a conserved Gln residue (that is essential for activity) forms hydrogen bonds to the uridine base[38]. Similarly, our VPgpU structure shows the side chain of Gln22 positioned on the “back side” of the uridine base, i.e. forming hydrogen bonds at positions that are not involved in Watson-Crick basepairing. This suggested that the Gln22 side chain might be important for stabilizing UTP during uridylylation, or the bound UMP of VPgpU during priming. While the 5-position of uracil, which is methylated in thymidine, is not involved in base pairing, an interaction with the side chain of Gln22 could aid in determining the specificity of base addition. Sequence analysis shows that in addition to the C-terminal Gln that characterizes all the enteroviral VPgs, many picornaviral VPg’s also have Gln residues in the middle of the peptide. In the partial structure of the FMDV-VPgpU, the glutamine residue at position 10 of FMDV-VPg is near the bound UMP. However, unlike the C-terminal Gln22 in PV1-VPgpU, the Gln10 sidechain in FMDV-VPgpU is turned away from the base (Figure 4 details). Our structure would suggest that this sidechain stabilizes the reverse side of the uridine ring during base pairing, and then moves out of position to facilitate other polymerase contacts during elongation. We previously showed that deletions in the C-terminus diminished, but did not eliminate, uridylylation of PV1-VPg in vitro[47]. While Gln22 is an essential amino acid for growth of PV1, the importance of its interactions directly in priming by VPgpU is difficult to determine, as this C-terminal residue is also essential for correct processing by the PV-proteinase 3CDpro. We needed to demonstrate that the PV-polymerase indeed distinguished between bases with minor changes in the base at the 5 or 6 positions, which are not involved in base pairing
Inhibition studies with 5-MeUTP
To determine whether bonds at the 5-position could affect base incorporation, we turned to 5meUTP, which could also be called ribo-TTP. Thymidine differs from uridine only in that it is methylated at the 5 position of the base. Although this base could exist in nature, more typically the 5-position of UMP is posttranscriptionally methylated, as for example in many bacterial tRNAs[9, 50]. There were however no reports of 5-meUMP (or ribosyl-TMP) in viral RNA or whether 5-meUMP could inhibit PV growth. To test the specificity of the uridylyation reaction, we determined whether 5meUTP would compete with labeled UTP for addition to VPg in an in vitro assay. Using polyA RNA with a Mn2+ cofactor, conditions which “relax” base incorporation specificity[3], 5meUTP competed quite efficiently and greatly reduced labeled UMP incorporation into VPgpU (Figure 5, right). However, using the more stringent assay, which better correlates with in vivo conditions, using the real viral template, cre(2C) RNA and Mg2+,[42, 48, 66] a large (5–20x) excess of 5meUTP over UTP was required to achieve 50% inhibition (Figure 5, left). This suggests that the C5-atom of the uridine base is recognized during uridylylation in an in vivo-like situation, and thus the interaction we observed with the glutamine side chain might indeed be important for this reaction. Had we seen that the polymerase did not discriminate under either condition, we could have assumed that the interaction was not important.
Figure 5. 5-MeUTP does not inhibit VPg uridylylation in vitro when normal viral growth conditions are mimicked.

Increasing amounts of cold 5-MeUTP, at the concentrations shown, were added to VPg uridylylation reactions with a constant amount (10 µM) of labeled UTP. The reaction was either specific, with the cre(2C) RNA (the hairpin loop that is the template in the viral RNA) with Mg2+ (left) or non-viral specific with a poly(A) template and Mn2+ (right). Activity is calculated relative to the amount of UTP incorporated in the absence of 5-MeUTP.
We also examined the effect of 5-methyluridine, which can penetrate cells, on the growth of PV1 in tissue culture. HeLa R19 cells were infected with PV1 in the presence of increasing amount of 5-methyluridine (1 µM to 1 mM). Cytopathic effect was observed in all the treated cells. The final virus titer, 1–2×109 plaque forming units/ml even at the highest level of 5-methyluridine, showed no significant inhibition of virus yield.
Modeling the priming reaction: VPgpU docks near the active site residues of the polymerase
To model the interactions during priming, we docked VPgpU to the PV-polymerase crystal structure[61]. All of the 10 best structures had ZDock scores greater than 50, and all placed VPgpU into the active site of the polymerase. This was in marked contrast to the dockings with the more elongated PV1-VPg structure, discussed previously[56]. The position of VPgpU in the highest ranked complexes were at a significantly different position from that seen for non-uridylylated VPg in the CVB3 complex (dotted ovals in Figure 6 mark this area in PV1- and FMDV polymerases). As discussed previously[56], a similar docking with the non-uridylylated structure of PV1-VPg did not enter the active site, but did have a stable binding site on the PV1-3Dpol surface.
Figure 6. Docking places PV1-VPgpU into the active site of PV1-polymerase.
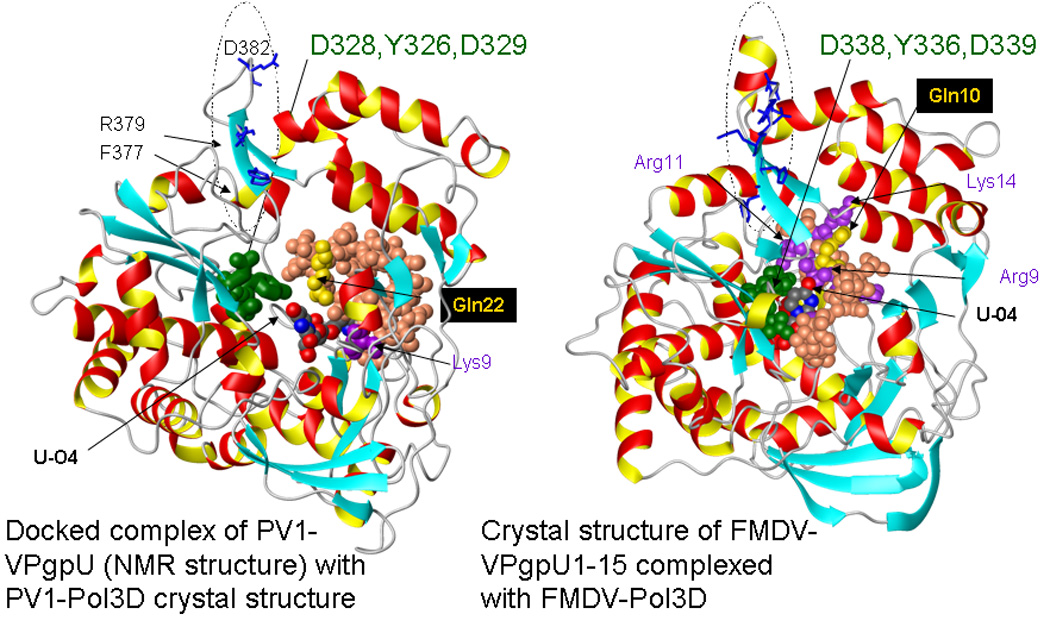
but further from the nucleotide/ metal binding site (green residues in the polymerases) than FMDV-VPgpU is in a crystal structure with its polymerase. Left: A docked complex (Zdock score >50) of PV1-VPgpU (space filling, colored as in Figure 3B) with a crystal structure of PV1-Pol3D (PDB file 1RA6). Right: For comparison, a snapshot is shown of the position of the N-terminal 15 amino acids of FMDV-VPgpU (see Figure 3) in a crystal structure of its complex with the FMDV polymerase (PDB file 2F8E) [18]. Note the O4 of the Uridine base is in a different position relative to the active site residues of the polymerase in the docked complex, The polymerases are shown in ribbon format in a similar orientation, with the side chains of the three residues conserved in the active sites of all polymerases, that mediate metal ion binding (PV1: D328,D329 or for FMDV, D338,D339) and incoming base stabilization (Y326, for FMDV Y336)) colored green. Sidechains on the polymerase are labeled in the one letter code, those of VPgpU are three letter code.
A dotted oval on each polymerase structure marks the area of the possible binding site for VPg on PV1-pol3D[56] (equivalent to that seen in the crystal structure of CVB3 complex[25]). Note that the corresponding area, also circled by an oval, on the FMDV polymerase (right) differs in sequence and secondary structure.
Comparing one of the dockings for PV1-VPgpU (Fig. 6, left) with the position of FMDV-VPgpU in the co-crystal structure with its polymerase[18] (Fig. 6, right), indicates both approach the active site from the front. However, the UMP in the FMDV structure is much closer to the active site residues that mediate metal and incoming nucleotide binding.
Discussion
The structure we determined for the PV1-VPgpU (Figure 2–Figure 4, Table 1) gives tantalizing evidence of how structural flexibility can aid in initiating RNA synthesis by the RNA-polymerase. Comparing PV1-VPgpU with partial structures of other VPgs (Figure 4) indicates the enterovirus VPg’s are more compact, and may have a different mode of interaction with their polymerases. We determined that PV1-3DPol can distinguish between UTP and 5meUTP, which would suggest a role for residues of VPg, including Gln22 in selectivity (Figure 3D and Figure 5). Finally, we found that the full structure of PV1-VPgpU has a stable docking site within the active site cleft of PV1-3DPol, consistent with a “front-loading” VPgpU priming position[2, 18] (Figure 6).
The docked structure may represent an intermediate stage in the priming reaction. In later stages, VPgpU may reconfigure and the UMP could rotate into the position seen for the FMDV-VPgpU. The change in configuration could be driven by binding of the metal ions around the reactive phosphate, to allow addition of another nucleotide to the bound UMP. As to the differences seen in the structures, it is significant that FMDV polymerase accepts more diversity in the sequences of VPg (Fig. 1) than that of PV1 does and uridylylates PV1-VPg nearly as well as it does all three of its own genomic VPgs. PV1-polymerase cannot uridylylate any of the FMDV-VPgs.[43] Consistent with the variety of sequences it can accept, the structural comparison suggests that the FMDV polymerase requires a less structured VPg than that does PV1 RNA polymerase.
Differences between FMDV and enteroviruses structures
Although PV is considered the prototype picornavirus, there are profound differences between PV and other enteroviruses compared to the aphthoviruses represented by FMDV. These differences extend to VPg sequence and structure and perhaps to the mechanism of uridylylation or even priming. The enterovirus structures (CVB3 partial and both NMR structures for PV1) are more structured and compact than those for FMDV (Figure 4). We saw changes in the solution structures of PV1-VPg before and after uridylylation, whereas the partial structures of FMDV-VPg and FMDV-VPgpU were very similar (bbRMSD ∼ 0.6A), and unfolded, with no discernable secondary structure elements. FMDV-VPg, even before uridylyation, was located in the active site of the of the FMDV polymerase. In contrast, the CVB3-VPg binding site seen in the crystal structure of the complex is identical to that previously predicted for PV1-VPg based on mutagenesis data[35] and docking of the NMR structure on the PV1-polymerase structure[56]. This binding site for VPg in PV1-polymerase (dotted ovals in Figure 5) differs greatly in sequence (FMDV:389HFHMDYG395 vs. PV1:377FFRADEK383; residues directly implicated in VPg binding are bold) and structure from that in FMDV polymerase [25].
Sequence heterogeneity in this area is not surprising: the two RNA polymerases share many motifs and a similar 3D-structure, but are only 29% identical[19]. Divergent VPg sequences (Fig. 1) and structures (Fig. 4) also correspond to differences in the viral RNA template used for uridylylation. While PV uses a stem loop [(cre(2C)] located in the 2C coding region[48], the FMDV cre with the same function is located in the 5'NTR[37] and has a different consensus sequence than that of enteroviruses[44]. PV-polymerase can use poly-A RNA as a template for uridylylation in the presence of Mn2+, while FMDV-polymerase cannot[7] (however, as Figure 5 shows, PV1 polymerase using this type of reaction is error prone). FMDV replicates its RNA at a different site within the cytoplasm than PV, as suggested by its insensitivity to Brefeldin A, a drug that modifies cellular membranes and inhibits PV replication[14, 45]. Finally, the cis-acting elements required for RNA replication are very different at the 5'-terminii of the PV and FMDV genomes. The 5'-end of PV RNA forms a cloverleaf like structure while that of FMDV RNA forms a large hairpin, called the S structure [1, 10].
One should thus not assume that the overall similarity of the polymerase structures indicate identical VPgpU dependent mechanisms for both genera, although the docking in Figure 6 does suggest that they are similar. The calicivirus polymerases also have many conserved sequence elements in common with those of picornaviruses[21] (see also Fig. 2 in [19]), but they use completely different primers, also called VPgs, that are between120–135 amino acids long [5, 36]. Our task is to discern what the picornavirus VPg-dependent mechanisms may have in common, that may help to shed light on these more distantly related reactions.
Changes in the structure of VPg upon uridylylation permit it to enter the active site and prime RNA synthesis
An inherently flexible (“disordered”[52, 53]) structure may be required for the activity of all VPgs. One might also speculate that the VPg-polymerase interactions are semi-stable and transient, with VPg changing its conformation according to the demands of uridylylation and during extension of the RNA. Too stable an interaction of VPg with the polymerase could slow RNA replication. We previously suggested a mechanism in which the presumed precursor of VPg, the 3AB protein, would bind at the position shown by the oval in Fig. 6, right[56] that had been previously identified by mutagenesis studies[34] and now confirmed by the CVB3 structure of the complex[25]. VPg would be cleaved off and uridylylated. The change in conformation would allow VPgpU to release from the polymerase, and leave the replication complex or enter the active site and prime RNA synthesis[56]. We initially hypothesized that uridylylation would cause the molecule to be less structured, more like that of FMDV-VPg before and after uridylylation [18], especially as VPgs show characteristics of disordered proteins[28, 52, 53]. However, after uridylylation, PV1-VPgpU becomes more compact. This compact structure could then dock within the active site of the polymerase (Fig. 6, left). The comparison in Figure 6 suggests that PV1 and FMDV could have a similar priming mechanism, even if they differ in the binding site for initial uridylylation. Atomic details of the exact priming reaction will require a crystal structure of a complex consisting of at least 4 different molecules: VPgpU, the viral polymerase(s), the correct RNA (which is different in PV1 and FMDV, as noted above), and incoming nucleotide (s). Efforts are underway in several labs to obtain such complexes in sufficiently high resolution.
In the meantime, this structure could be a starting point for designing mutations of VPg or mimetics of VPgpU that could interfere either with the uridylylation or priming steps, perhaps using a consensus approach[15, 16] to account for variability in the sequences of related enteroviruses. At least one mutant of PV1-VPg mutant can inhibit uridylylation in vitro (our unpublished data), and others have shown that VPg mutations can slow or eliminate PV replication in trans[13]. In conclusion, our structure for PV1-VPgpU suggests that its major role is to stabilize the bound UMP moiety to make initialization of RNA synthesis more efficient (and selective). Our findings also show that the PV1-polymerase distinguishes UTP from 5meUTP during uridylylation. The PV1-VPgpU can be a basis to design unique inhibitors that target the priming mechanism essential for replication, which could be novel therapies for human enteroviral infections. Perhaps of equal significance, VPgpU’s structural change upon nucleotidylation may help elucidate the consequences of other biologically important uridyl-transferase reactions, including cell wall synthesis in both G- and G+ bacteria [4], in RNA regulation[54], in “RNA editing” by terminal uridyltransferases (Tutases) and, in a process known primarily in trympanosomes, inserting U within existing mRNA sequences[27, 58].
Supplementary Material
Acknowledgements
We thank Drs. Junji Iwahara, David Volk and Tianzhi Wang for help with the NMR experiments. This work was supported in part by NIH grants R01 AI064913 to Werner Braun (PI) and Catherine Schein and R37AI015122 (Eckard Wimmer, PI). The facilities of the Sealy Center for Structural Biology and Molecular Biophysics at the UTMB were used in this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andino R, Rieckhof GE, Baltimore D. A Functional Ribonucleoprotein Complex Forms around the 5' End of Poliovirus Rna. Cell. 1990;63:369–380. doi: 10.1016/0092-8674(90)90170-j. [DOI] [PubMed] [Google Scholar]
- 2.Appleby T, Luecke H, Shim JH, Wu JZ, Cheney IW, Zhong W, Vogeley L, Hong Z, Yao N. Crystal structure of complete rhinovirus RNA polymerase suggests front loading of protein primer. J Virol. 2005;79:277–288. doi: 10.1128/JVI.79.1.277-288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold JJ, Gosh KB, Cameron C. Poliovirus RNA-dependent RNA polymerase 3Dpol. Divalent cation modulation of primer, template and nucleotide selection. J. Biol. Chem. 1999;274:37060–37069. doi: 10.1074/jbc.274.52.37060. [DOI] [PubMed] [Google Scholar]
- 4.Barreteau H, Kova A, Boniface DA, Sova M, Gobec S, Blanot D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiology Reviews. 2008;32:168–207. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 5.Belliot G, Sosnovtsev SV, Chang KO, McPhie P, Green KY. Nucleotidylylation of the VPg protein of a human norovirus by its proteinase-polymerase precursor protein. Virology. 2008;374:33–49. doi: 10.1016/j.virol.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belsham GJ. Distinctive Features of Foot-and-Mouth-Disease Virus, a Member of the Picornavirus Family - Aspects of Virus Protein-Synthesis, Protein Processing and Structure. Progress in Biophysics & Molecular Biology. 1993;60:241–260. doi: 10.1016/0079-6107(93)90016-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belsham GJ. Translation and replication of FMDV RNA. Foot and Mouth Disease Virus. 2005:43–70. doi: 10.1007/3-540-27109-0_3. [DOI] [PubMed] [Google Scholar]
- 8.Chen R, Li L, Weng Z. ZDOCK: An Initial-stage Protein Docking Algorithm. Proteins. Structure, Function and Genetics. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 9.Cicmil N. Crystallization and preliminary X-ray crystallographic characterization of TrmFO, a folate-dependent tRNA methyltransferase from Thermotoga maritima. Acta Crystallographica Section F-Structural Biology and Crystallization Communications. 2008;64:193–195. doi: 10.1107/S1744309108003825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke BE, Brown AL, Currey KM, Newton SE, Rowlands DJ, Carroll AR. Potential Secondary and Tertiary Structure in the Genomic Rna of Foot-and-Mouth-Disease Virus. Nucleic Acids Research. 1987;15:7067–7079. doi: 10.1093/nar/15.17.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cottam EM, Haydon DT, Paton DJ, Gloster J, Wilesmith JW, Ferris NP, Hutchings GH, King DP. Molecular epidemiology of the foot-and-mouth disease virus outbreak in the United Kingdom in 2001. Journal of Virology. 2006;80:11274–11282. doi: 10.1128/JVI.01236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford NM, Baltimore D. Genome-Linked Protein Vpg of Poliovirus Is Present as Free Vpg and Vpg-Pupu in Poliovirus-Infected Cells. Proceedings of the National Academy of Sciences of the United States of America-Biological Sciences. 1983;80:7452–7455. doi: 10.1073/pnas.80.24.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crowder S, Kirkegaard K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat Genet. 2005;37:701–709. doi: 10.1038/ng1583. [DOI] [PubMed] [Google Scholar]
- 14.Cuconati A, Molla A, Wimmer E. Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. Journal of Virology. 1998;72:6456–6464. doi: 10.1128/jvi.72.8.6456-6464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danecek P, Lu W, Schein CH. PCP consensus sequences of flaviviruses: correlating variance with vector competence and disease phenotype. J Mol Biol. 2010;396:550–563. doi: 10.1016/j.jmb.2009.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danecek P, Schein CH. Flavitrack analysis of the structure and function of West Nile non-structural proteins. Int J Bioinform Res Appl. 2010;6:134–146. doi: 10.1504/IJBRA.2010.032117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falk MM, Sobrino F, Beck E. Vpg-Gene Amplification Correlates with Infective Particle Formation in Foot-and-Mouth-Disease Virus. Journal of Virology. 1992;66:2251–2260. doi: 10.1128/jvi.66.4.2251-2260.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrer-Orta C, Arias A, Agudo R, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. The structure of a protein primer-polymerase complex in the initiation of genome replication. Embo Journal. 2006;25:880–888. doi: 10.1038/sj.emboj.7600971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. Journal of Biological Chemistry. 2004;279:47212–47221. doi: 10.1074/jbc.M405465200. [DOI] [PubMed] [Google Scholar]
- 20.Garimella R, Xu Y, Schein CH, Rajarathnam K, Nagle GT, Painter SD, Braun W. NMR solution structure of attractin, a water-borne protein pheromone from the mollusk Aplysia californica. Biochemistry. 2003;42:9970–9979. doi: 10.1021/bi0274322. [DOI] [PubMed] [Google Scholar]
- 21.Garriga D, Navarro A, Querol-Audi J, Abaitua F, Rodriguez JF, Verdaguer N. Activation mechanism of a noncanonical RNA-dependent RNA polymerase. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20540–20545. doi: 10.1073/pnas.0704447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geen H, Freeman R. Band-Selective Radiofrequency Pulses. Journal of Magnetic Resonance. 1991;93:93–141. [Google Scholar]
- 23.Gomez RM, Berria MI, Levander OA. Host selenium status selectively influences susceptibility to experimental viral myocarditis. Biological Trace Element Research. 2001;80:23–31. doi: 10.1385/BTER:80:1:23. [DOI] [PubMed] [Google Scholar]
- 24.Gopalkrishna V, Patil PR, Kolhapure RM, Bilaiya H, Fulmali PV, Deolankar RP. Outbreak of acute hemorrhagic conjunctivitis in Maharashtra and Gujarat states of India, caused by Coxsackie virus A-24 variant. Journal of Medical Virology. 2007;79:748–753. doi: 10.1002/jmv.20886. [DOI] [PubMed] [Google Scholar]
- 25.Gruez A, Selisko B, Roberts M, Bricogne G, Bussetta C, Jabafi I, Coutard B, De Palma AM, Neyts J, Canard B. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. Journal of Virology. 2008;82:9577–9590. doi: 10.1128/JVI.00631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grzesiek S, Bax A. The Importance of Not Saturating H2o in Protein Nmr -Application to Sensitivity Enhancement and Noe Measurements. Journal of the American Chemical Society. 1993;115:12593–12594. [Google Scholar]
- 27.Guschina E, Benecke B-J. Specific and non-specific mammalian RNA terminal uridylyl transferases. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2008;1779:281–285. doi: 10.1016/j.bbagrm.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 28.Hebrard E, Bessin Y, Michon T, Longhi S, Uversky VN, Delalande F, Van Dorsselaer A, Romero P, Walter J, Declerk N, Fargette D. Intrinsic disorder in Viral Proteins Genome-Linked: experimental and predictive analyses. Virology Journal. 2009:6. doi: 10.1186/1743-422X-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King AMQ, Sangar DV, Harris TJR, Brown F. Heterogeneity of the Genome-Linked Protein of Foot-and-Mouth-Disease Virus. Journal of Virology. 1980;34:627–634. doi: 10.1128/jvi.34.3.627-634.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kramer M, Schulte BM, Toonen LWJ, de Bruijni MAM, Galama JMD, Adema GJ, van Kuppeveld FJM. Echovirus infection causes rapid loss-of-function and cell death in human dendritic cells. Cellular Microbiology. 2007;9:1507–1518. doi: 10.1111/j.1462-5822.2007.00888.x. [DOI] [PubMed] [Google Scholar]
- 31.Kriek N, Filippov DV, van den Elst H, Meeuwenoord NJ, Tesser GI, van Boom JH, van der Marel GA. Stepwise solid phase synthesis of uridylylated viral genome-linked peptides using uridylylated amino acid building blocks. Tetrahedron. 2003;59:1589–1597. [Google Scholar]
- 32.Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc. Natl. Acad. USA. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Chen R, Weng Z. RDOCK: Refinement of Rigid-body Protein Docking Predictions. PROTEINS: Structure, Function, and Genetics. 2003;53:693–707. doi: 10.1002/prot.10460. [DOI] [PubMed] [Google Scholar]
- 34.Lyle J, Clewell A, Richmond K, Richards O, Hope D, Schultz S, Kirkegaard K. Similar structural basis for membrane localization and protein priming by an RNA-dependent RNA polymerase. J Biol Chem. 2002;277:16324–16331. doi: 10.1074/jbc.M112429200. [DOI] [PubMed] [Google Scholar]
- 35.Lyle JM, Clewell A, Richmond K, Richards OC, Hope DA, Schultz SC, Kirkegaard K. Similar structural basis for membrane localization and protein priming by an RNA-dependent RNA polymerase. Journal of Biological Chemistry. 2002;277:16324–16331. doi: 10.1074/jbc.M112429200. [DOI] [PubMed] [Google Scholar]
- 36.Machin A, Alonso JMM, Dalton KP, Parra F. Functional differences between precursor and mature forms of the RNA-dependent RNA polymerase from rabbit hemorrhagic disease virus. Journal of General Virology. 2009;90:2114–2118. doi: 10.1099/vir.0.011296-0. [DOI] [PubMed] [Google Scholar]
- 37.Mason PW, Bezborodova SV, Henry TM. Identification and characterization of a cis-acting replication element (cre) adjacent to the internal ribosome entry site of foot-and-mouth disease virus. Journal of Virology. 2002;76:9686–9694. doi: 10.1128/JVI.76.19.9686-9694.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mochalkin I, Lightle S, Zhu Y, Ohren JF, Spessard C, Chirgadze NY, Banotai C, Melnick M, McDowell L. Characterization of substrate binding and catalysis in the potential antibacterial target N-acetylglucosamine-1-phosphate uridyltransferase (GlmU) Protein Science. 2007;16:2657–2666. doi: 10.1110/ps.073135107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molla A, Paul AV, Wimmer E. CELL-FREE, DENOVO SYNTHESIS OF POLIOVIRUS. Science. 1991;254:1647–1651. doi: 10.1126/science.1661029. [DOI] [PubMed] [Google Scholar]
- 40.Moura FEA, Ribeiro DCS, Gurgel N, Mendes ACD, Tavares FN, Timoteo CNG, da Silva EE. Acute haemorrhagic conjunctivitis outbreak in the city of Fortaleza, northeast Brazil. British Journal of Ophthalmology. 2006;90:1091–1093. doi: 10.1136/bjo.2006.098822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mumenthaler C, Güntert P, Braun W, Wüthrich K. Automated combined assignment of NOESY spectra and three-dimensional protein structure determination. J. Biomol. NMR. 1997;10:351–362. doi: 10.1023/a:1018383106236. [DOI] [PubMed] [Google Scholar]
- 42.Murray K, Barton D. Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J. Virology. 2003;77:4739–4750. doi: 10.1128/JVI.77.8.4739-4750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nayak A, Goodfellow IG, Belsham GJ. Factors required for the uridylylation of the foot-and-mouth disease virus 3B1, 3B2, and 3B3 peptides by the RNA-dependent RNA polymerase (3D(pol)) in vitro. Journal of Virology. 2005;79:7698–7706. doi: 10.1128/JVI.79.12.7698-7706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nayak A, Goodfellow IG, Woolaway KE, Birtley J, Curry S, Belsham GJ. Role of RNA structure and RNA binding activity of foot-and-mouth disease virus 3C protein in VPg uridylylation and virus replication. Journal of Virology. 2006;80:9865–9875. doi: 10.1128/JVI.00561-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Donnell VK, Pacheco JM, Henry TM, Mason PW. Subcellular distribution of the foot-and-mouth disease virus 3A protein in cells infected with viruses encoding wild-type and bovine-attenuated forms of 3A. Virology. 2001;287:151–162. doi: 10.1006/viro.2001.1035. [DOI] [PubMed] [Google Scholar]
- 46.Oezguen N, Adamian L, Xu Y, Rajarathnam K, Braun W. Automated assignment and 3D structure calculations using combinations of 2D homonuclear and 3D heteronuclear NOESY spectra. Journal of Biomolecular NMR. 2002;22:249–263. doi: 10.1023/a:1014925824100. [DOI] [PubMed] [Google Scholar]
- 47.Paul AV, Peters J, Mugavero J, Yin J, van Boom JH, Wimmer E. Biochemical and genetic studies of the VPg-uridylylation reaction catalyzed by the RNA polymerase of poliovirus. J. Virol. 2003;77:891–904. doi: 10.1128/JVI.77.2.891-904.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 2000;74:10359–10370. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paul AV, van Boom JH, Filippov D, Wimmer E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature. 1998;393:280–284. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- 50.Persson BC, Gustafsson C, Berg DE, Bjork GR. THE GENE FOR A TRANSFER-RNA MODIFYING ENZYME, M5U54-METHYLTRANSFERASE, IS ESSENTIAL FOR VIABILITY IN ESCHERICHIA-COLI. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:3995–3998. doi: 10.1073/pnas.89.9.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ponnuraj EM, Mukundan P. Coxsackie-Virus-a-24 Variant as the Etiological Agent of the Acute Hemorrhagic Conjunctivitis Epidemic at Vellore, in 1986. Indian Journal of Medical Research Section a-Infectious Diseases. 1989;89:283–286. [PubMed] [Google Scholar]
- 52.Prischi F, Giannini C, Adinolfi S, Pastore A. The N-terminus of mature human frataxin is intrinsically unfolded. Febs Journal. 2009;276:6669–6676. doi: 10.1111/j.1742-4658.2009.07381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rantalainen KI, Uversky VN, Permi P, Kalkkinen N, Dunker AK, Makinen K. Potato virus A genome-linked protein VPg is an intrinsically disordered molten globule-like protein with a hydrophobic core. Virology. 2008;377:280–288. doi: 10.1016/j.virol.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rissland OS, Norbury CJ. The Cid1 poly(U) polymerase. Biochimica Et Biophysica Acta-Gene Regulatory Mechanisms. 2008;1779:286–294. doi: 10.1016/j.bbagrm.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 55.Schein CH, Oezguen N, Volk DE, Garimella R, Paul A, Braun W. NMR structure of the viral peptide linked to the genome (VPg) of poliovirus. Peptides. 2006;27:1676–1684. doi: 10.1016/j.peptides.2006.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schein CH, Volk DE, Oezguen N, Paul A. Novel, structure-based mechanism for uridylylation of the genome-linked peptide (VPg) of picornaviruses. Proteins-Structure Function and Bioinformatics. 2006;63:719–726. doi: 10.1002/prot.20891. [DOI] [PubMed] [Google Scholar]
- 57.Sklenar V, Piotto M, Leppik R, Saudek V. Gradient-Tailored Water Suppression for H-1-N-15 Hsqc Experiments Optimized to Retain Full Sensitivity. Journal of Magnetic Resonance Series A. 1993;102:241–245. [Google Scholar]
- 58.Stagno J, Aphasizheva I, Rosengarth A, Luecke H, Aphasizhev R. UTP-bound and Apo Structures of a Minimal RNA Uridylyltransferase. Journal of Molecular Biology. 2007;366:882–899. doi: 10.1016/j.jmb.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strauss DM, Wuttke DS. Characterization of protein-protein interactions critical for poliovirus replication: Analysis of 3AB and VPg binding to the RNA-dependent RNA polymerase. Journal of Virology. 2007;81:6369–6378. doi: 10.1128/JVI.02252-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Talluri S, Wagner G. An optimized 3D NOESY-HSQC. Journal of Magnetic Resonance Series B. 1996;112:200–205. doi: 10.1006/jmrb.1996.0132. [DOI] [PubMed] [Google Scholar]
- 61.Thompson AA, Peersen OB. Structural Basis for Proteolysis-Dependent Activation of the Poliovirus RNA-Dependent RNA Polymerase. Embo J. 2004;23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Nip AM, Wishart DS. A simple method to quantitatively measure polypeptide JHNHα coupling constants from TOCSY or NOESY spectra. Journal of Biomolecular NMR. 1997;10:373–382. doi: 10.1023/a:1018315729609. [DOI] [PubMed] [Google Scholar]
- 64.Xu Y, Jablonsky MJ, Jackson PL, Braun W, Krishna NR. Automatic assignment of NOESY cross peaks and determination of the protein structure of a new world scorpion neurotoxin using NOAH/DIAMOD. J. Magn. Reson. 2001;148:35–46. doi: 10.1006/jmre.2000.2220. [DOI] [PubMed] [Google Scholar]
- 65.Xu Y, Schein CH, Braun W. Combined automated assignment of NMR spectra and calculation of three-dimensional protein structures. In: Rama-Krishna N, Berliner LJ, editors. Biological Magnetic Resonance. New York, Boston, Dordrecht, London, Moscow: Kluwer Academic / Plenum Publishers; 1999. pp. 37–79. [Google Scholar]
- 66.Yang Y, Rijnbrand R, McKnight KL, Wimmer E, Paul A, Martin A, Lemon SM. Sequence requirements for viral RNA replication and VPg uridylylation directed by the internal cis-acting replication element (cre) of human rhinovirus type 14. J. Virol. 2002;76:7485–7494. doi: 10.1128/JVI.76.15.7485-7494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.