

Abstract

A tandem dimerization/macrocyclization reaction utilizing the Prins cyclization has been developed. This reaction develops molecular complexity through the formation of highly substituted dimeric tetrahydropyran macrocycles. Mild conditions utilizing rhenium(VII) catalysts were explored for aromatic substrates while harsher Lewis acidic conditions were used for aliphatic substrates. Both aldehydes and acetals are shown to be viable substrates for this reaction.
Oxacyclic macrodimers are an important class of natural products that offer a wide array of structural complexity and bioactivity.1 These macrolides have become popular targets for synthetic chemists.2 The most direct way to construct these molecules is through the union of two monomeric species in a tandem dimerization and macrocyclization reaction. The most common version of this strategy utilizes two esterification reactions between an activated acid and an alcohol, the initial dimerization followed by a macrolactonization.3 While often successful, this approach does not greatly enhance the complexity of the intermediate through the formation of carbon-carbon bonds. Alternative bond-forming reactions, such as Suzuki coupling and olefin metathesis have been used with occasional success in dimerization and macrocyclization strategies.4,5 Herein we describe a new dimerization and macrocyclization strategy based on the Prins cyclization reaction.
The Prins cyclization is a powerful reaction that forms cis-2,6-disubstituted tetrahydropyrans (THPs) through the addition of an olefin to an oxocarbenium ion generated from the condensation of an aldehyde with a homoallylic alcohol.6 Recently, intramolecular Prins cyclizations have been utilized as key macrocyclization steps in the synthesis of several THP containing natural products (Figure 1 A).7 We report the extension of this methodology to the formation of oxacyclic macrodimers through sequential Prins dimerization and macrocyclization reactions (Figure 1 B). This synthetic strategy has been applied to a model for the marine macrodiolide clavosolide A.
Figure 1.

Comparision of Prins macrocyclization reactions.
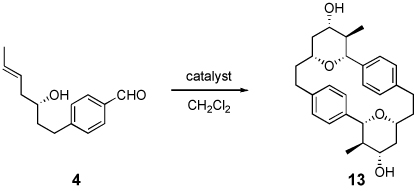
The [n,n]-cyclophanes were chosen as targets for the initial study of this reaction, since the aromatic ring of these substrates provides rigidity that precludes the undesired macrocyclization reaction, and the starting materials were accessible from simple precursors. The Prins cyclization substrates 4, 8, and 12 were synthesized from the corresponding commercially available para-, meta-, or ortho-substituted ethyl iodobenzoate, respectively (Scheme 1). Palladium-mediated coupling of allyl alcohol and ethyl 4-iodobenzoate (1) yielded known aldehyde 2 in 85% yield.8 Nokami’s menthone derived allyl-transfer reagent was utilized to yield optically pure E-homoallylic alcohol 3.9 The ethyl ester was reduced with LiAlH4 to provide the benzylic alcohol, which was selectively oxidized with MnO2 to afford aldehyde 4 in 61% overall yield. The meta- and ortho-substituted substrates (8 and 12, respectively) were synthesized in a similar manner.
Scheme 1.

Synthesis of the Aldehyde Monomers
With appropriate starting materials in hand, the dimerization/macrocyclization reaction of aldehyde 4 was explored (Table 1). Previously, O3ReOSiPh3 has been shown to effectively catalyze Prins reactions of aromatic aldehydes.10 When aldehyde 4 was exposed to O3ReOSiPh3 at room temperature for 22 h, 33% of cyclophane 13 was isolated (Entry 1). The remainder of the mass was attributed to rhenium(VII) mediated polymerization. To diminish this byproduct, the reaction was run at lower concentration, which increased the yield of the dimer (Entry 2). Gratifyingly, decreasing the catalyst loading to 25% had little effect on the yield (Entry 3), but decreasing the loading further resulted in diminished yields (Entry 4). Other rhenium(VII) catalysts proved less effective than O3ReOSiPh3 (Entries 5 and 6). Heating the reaction in CH2Cl2 decreased the reaction time but had minimal effect on the overall yield (Entries 7 and 8). Finally, alternative Prins promoters were investigated. Both TESOTf11 and trifluoroacetic acid12 produced acylated variations of cyclophane 13 (Entries 9 and 10), but ultimately proved inferior to the rhenium(VII) conditions.
Table 1.
Screening Prins Dimerization/Macrocyclization Conditions
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | conc | time | temp | yield |
| 1 | O3ReOSiPh3 (40 mol %) |
0.10 M | 22 h | 23 °C | 33% |
| 2 | O3ReOSiPh3 (50 mol %) |
0.050 M | 24 h | 23 °C | 53% |
| 3 |
O3ReOSiPh3 (25 mol %) |
0.050 M | 24 h | 23 °C | 51% |
| 4 | O3ReOSiPh3 (5 mol %) |
0.050 M | 24 h | 23 °C | 26% |
| 5 | aq O3ReOH (50 mol %) |
0.050 M | 24 h | 23 °C | 29% |
| 6 | Re2O7 (50 mol %) |
0.045 M | 25 h | 23 °C | 38% |
| 7 | O3ReOSiPh3 (25 mol %) |
0.050 M | 5 h | 40 °C | 51% |
| 8 | O3ReOSiPh3 (5 mol %) |
0.050 M | 5 h | 40 °C | 42% |
| 9 | TESOTfa (20 equiv) |
0.030 M | 3 h | 23 °C | 27%b |
| 10 | TFA (20 equiv) |
0.30 M | 3 h | 23 °C | 11%b |
Run in AcOH with 30 equiv TMSOAc.
Yield after hydrolysis of the crude acyl trapped product by MeOH/K2CO3.
Having developed optimized conditions for the dimerization/macrocyclization reaction of the para-substituted aldehyde, the ortho- and meta-substituted aldehydes were employed to further expand the scope of the reaction (Scheme 2). The ortho-substituted aldehyde 12 afforded cyclized monomer 14 via an intramolecular Prins reaction in 90% yield. Prins macrocyclizations can be very effective, when not precluded by the geometry of the substrate.7 In contrast, when aldehyde 8 was exposed to the Prins conditions, dimeric cyclophane 15 was obtained in 28% yield. Surprisingly, the trimeric macrocycle 16 was also isolated in 13% yield. The remainder of the mass was associated with a small amount of starting material and a complex mixture of other products. The Prins reaction led to significant yields of dimer in two of the three cases examined, and in each case a single enantiomer and diastereomer of the product was obtained.
Scheme 2.

Optimized Prins Macrocylization/Dimerization Reactions
The C2-symmetric macrodiolide (−)-clavosolide A, Figure 2, was isolated from the marine sponge Myriastra clavosa gathered off the coast of the Philippines.13 The unique architecture of this THP-containing macrodimer inspired several total syntheses14 and a structural revision.15 All reported approaches utilized Yamaguchi's macrolactonization conditions to form the dimer from a complex, THP containing monomer.16 The interesting architecture of clavosolide A appeared to be well suited to a Prins dimerization and macrocyclization strategy.
Figure 2.

Two alternative strategies for the dimerization and cyclization of (−)-clavosolide A
Due to concern over the lability of the cyclopropylcarbinyl moiety of clavosolide A,17 a model system was designed that replaced the problematic cyclopropyl side-chain with a cyclohexane ring. The cyclohexyl monomer was synthesized from two simple fragments 20 and 24 (Scheme 3 and 4). Enzymatic resolution of known β-lactone 17 with Lipase PS Amano in the presence of benzyl alcohol afforded the resolved (S)-lactone (18) and β-hydroxy ester 19.18 Both substrates were then reduced with LiAlH4, and the resultant enantiomeric diols were monoprotected as TBS-ethers 20 and 21.
Scheme 3.

Synthesis of the cyclohexyl alcohols 20 and 21
Scheme 4.

Synthesis of the ester monomer 25
The synthesis of the acid fragment 24 began with DDQ-mediated deprotection of the p-methoxybenzyl ether of known silyl ether 22 to provide an inseparable mixture of alcohol 23 and p-anisaldehyde.19 The p-anisaldehyde was then removed by reduction and chromatography. Surprisingly, oxidation to the carboxylic acid proved challenging. Oxidation of alcohol 23 to the aldehyde followed by Lindgren–Pinnick oxidation afforded very poor yields of acid 24. Additionally, TEMPO-mediated bleach oxidations20 resulted in chlorination of the substrate. Instead, a large excess of pyridinium dichromate was employed over three days to produce acid 24 in 66% yield.21 The acid was then coupled to both alcohol enantiomers 20 and 21, either through esterification to maintain the alcohol stereochemistry, or Mitsunobu reaction to invert the stereochemistry, to afford bis-TBS ether 25.
With the completion of the monomer framework, a few simple manipulations were needed to test the key Prins dimerization/macrocyclization reaction (Scheme 5). Removal of the two silyl groups proved to be surprisingly difficult. Under standard deprotection conditions, only the primary silyl group was cleaved. Unfortunately, unmasking the primary alcohol under more forcing basic or acidic conditions led to a deleterious acyl migration. Ultimately, a large excess of HF·pyridine in THF was needed to avoid this side reaction, and the diol 26 (not shown) was formed in 88% yield. Finally, TEMPO-catalyzed selective oxidation successfully produced the requisite aldehyde 27. Aldehyde 27 was labile and decomposed upon silica gel chromatography. Attempts to perform a Prins dimerization and macrocyclization reaction on the crude aldehyde under various conditions led to decomposition of the starting material, presumably through acid promoted elimination of the β-acyl aldehyde.
Scheme 5.

Prins dimerization and macrocyclization
To circumvent this decomposition, aldehyde 27 was protected as dimethyl acetal 28. This substrate proved much more manageable and was easily purified by column chromatography. It has been shown that Lewis acid-promoted Prins cyclizations of acetals are particularly effective with TESOTf in acetic acid.11,22 Gratifyingly, when acetal 28 was subjected to these conditions, dimeric macrolide 29 was produced in 43% yield. Using acetal 28 avoids the decomposition of the sensitive aldehyde 27.
The first example of a Prins dimerization and macrocyclization reaction has been developed. This powerful methodology affords macrocyclic dimers while simultaneously enhancing molecular complexity. Several complex [n.n]-paraphanes were prepared in only five steps from commercially available material. A model for the synthesis of clavosolide A was successfully demonstrated, and studies will continue to apply this strategy to natural product synthesis.
Supplementary Material
Acknowledgment
This work was supported by the National Institute of General Medicine (GM-43854) and by a generous gift from the Schering-Plough Research Institute. K.T. was supported by a fellowship from the Royal Thai Government and M.R.G. was supported by a Lilly Graduate Research Fellowship.
Footnotes
Supporting Information Available Characterization data and experimental procedures for all compounds described are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Norcross RD, Paterson I. Chem. Rev. 1995;95:2041–2114. [Google Scholar]
- 2.Kang EJ, Lee E. Chem. Rev. 2005;105:4348–4378. doi: 10.1021/cr040629a. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wakamatsu T, Yamada S, Ban Y. Heterocycles. 1986;24:309–312. [Google Scholar]; (b) Furstner A, Mlynarski J, Albert M. J. Am. Chem. Soc. 2002;124:10274–10275. doi: 10.1021/ja027346p. [DOI] [PubMed] [Google Scholar]; (c) Panek JS, Porco JA, Jr., Lobkovsky E, Beeler AB. Org. Lett. 2003;12:2149–2152. doi: 10.1021/ol034608z. [DOI] [PubMed] [Google Scholar]; (d) Barth R, Mulzer J. Angew. Chem., Int. Ed. 2007;46:5791–5794. doi: 10.1002/anie.200701065. [DOI] [PubMed] [Google Scholar]
- 4.(a) Cheung LL, Marumoto S, Anderson CD, Rychnovsky SD. Org. Lett. 2008;10:3101–3104. doi: 10.1021/ol8011474. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Nold AL, Milburn RR, Schindler CS. Angew. Chem., Int. Ed. 2006;45:6527–6532. doi: 10.1002/anie.200601867. [DOI] [PubMed] [Google Scholar]
- 5.(a) HWE olefinations: Hoye TR, Humpal PE, Moon B. J. Am. Chem. Soc. 2000;122:4982–4983. (b) Olefin metathesis: Smith AB, III, Adams CM, Kozmin SA, Paone DV. J. Am. Chem. Soc. 2001;123:5925–5937. doi: 10.1021/ja0106164. (c) Condensations: Hoye TR, Ye Z, Yao LJ, North JT. J. Am. Chem. Soc. 1996;118:12074–12081.
- 6.Recent reviews on the Prins cyclization: Pastor IM, Yus M. Curr. Org. Chem. 2007;11:925–957. Snider BB. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Heathcock CH, editors. Vol. 2. New York: Pergamon Press; 1991. pp. 527–561. Adams DR, Bhatnagar SP. Synthesis. 1977:661–672. Overman LE, Pennington LD. J. Org. Chem. 2003;68:7143–7157. doi: 10.1021/jo034982c.
- 7.(a) Custar DW, Zabawa TP, Scheidt KA. J. Am. Chem. Soc. 2008;130:804–805. doi: 10.1021/ja710080q. [DOI] [PubMed] [Google Scholar]; (b) Woo SK, Kwon MS, Lee E. Angew. Chem., Int. Ed. 2008:3242–3244. doi: 10.1002/anie.200800386. [DOI] [PubMed] [Google Scholar]; (c) Wender PA, DeChristopher BA, Schrier AJ. J. Am. Chem. Soc. 2008;130:6658–6659. doi: 10.1021/ja8015632. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bahnck KB, Rychnovsky SD. J. Am. Chem. Soc. 2008;130:13177–13181. doi: 10.1021/ja805187p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Taylor EC, Gillespie P, Patel M. J. Org. Chem. 1992;57:3218–3225. [Google Scholar]; (b) Watson SE, Taylor EC, Patel H. Synth. Commun. 1998;28:1897–1905. [Google Scholar]
- 9.(a) Nokami J, Nomiyama K, Matsuda S, Imai N, Kataoka K. Angew. Chem., Int. Ed. 2003;42:1273–1276. doi: 10.1002/anie.200390327. [DOI] [PubMed] [Google Scholar]; (b) Nokami J, Ohga M, Nakamoto H, Matsubara T, Hussain H, Kataoka K. J. Am. Chem. Soc. 2001;123:9168–9169. doi: 10.1021/ja011257f. [DOI] [PubMed] [Google Scholar]
- 10.Tadpetch K, Rychnovsky SD. Org. Lett. 2008;10:4839–4842. doi: 10.1021/ol8019204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ko HM, Lee DG, Kim MA, Kim HJ, Park J, Lah MS, Lee E. Tetrahedron. 2007;63:5797–5805. [Google Scholar]
- 12.Barry CSJ, Crosby SR, Harding JR, Hughes RA, King CD, Parker GD, Willis CL. Org. Lett. 2003;5:2429–2432. doi: 10.1021/ol0346180. [DOI] [PubMed] [Google Scholar]
- 13.(a) Erickson KL, Gustafson KR, Pannell LK, Beutler JA, Boyd MR. J. Nat. Prod. 2002;65:1303–1306. doi: 10.1021/np020193z. [DOI] [PubMed] [Google Scholar]; (b) Rao MR, Faulkner DJ. J. Nat. Prod. 2002;65:386–388. doi: 10.1021/np010495l. [DOI] [PubMed] [Google Scholar]
- 14.(a) Carrick JD, Jennings MP. Org. Lett. 2009;11:769–772. doi: 10.1021/ol8028302. [DOI] [PubMed] [Google Scholar]; (b) Chakraborty TK, Reddy VR, Chattopadhyay AK. Tetrahedron Lett. 2006;47:7435–7438. [Google Scholar]; (c) Barry CS, Elsworth JD, Seden PT, Bushby N, Harding JR, Alder RW, Willis CL. Org. Lett. 2006;8:3319–3322. doi: 10.1021/ol0611705. [DOI] [PubMed] [Google Scholar]; (d) Smith AB, III, Simov V. Org. Lett. 2006;8:3315–3318. doi: 10.1021/ol0611752. [DOI] [PubMed] [Google Scholar]; (e) Son JB, Kim SN, Kim NY, Lee DH. Org. Lett. 2006;8:661–664. doi: 10.1021/ol052851n. [DOI] [PubMed] [Google Scholar]
- 15.(a) Chakraborty TK, Reddy VR. Tetrahedron Lett. 2006;47:2099–2102. [Google Scholar]; (b) Barry CS, Bushby N, Charmant JPH, Elsworth JD, Harding JR, Willis CL. Chem. Commun. 2005:5097–5099. doi: 10.1039/b509757f. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi MA, Katsuki T, Sacki H, Hirata K, Inanaga J. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- 17.Poulter DC, Winstein SJ. J. Am. Chem. Soc. 1969;13:3650–3652. [Google Scholar]
- 18.(a) Nelson SG, Spencer KL. J. Org. Chem. 2000;65:1227–1230. doi: 10.1021/jo9913412. [DOI] [PubMed] [Google Scholar]; (b) Nelson SG, Wan Z, Peelen TJ, Spencer KL. Tetrahedron Lett. 1999:6535–6539. [Google Scholar]
- 19.Eggen M, Mossman CJ, Buck SB, Nair SK, Bhat L, Ali SM, Reiff EA, Boge TC, Georg GI. J. Org. Chem. 2000;65:7792–7799. doi: 10.1021/jo000767+. [DOI] [PubMed] [Google Scholar]
- 20.Zhao MM, Li J, Mano E, Song ZJ, Tschaen DM. Org. Synth. 2005;81:195–203. [Google Scholar]
- 21.Corey EJ, Schmidt G. Tetrahedron Lett. 1979;20:399–402. [Google Scholar]
- 22.(a) Fujioka H, Okitsu T, Sawama Y, Murata N, Li R, Kita Y. J. Am. Chem. Soc. 2006;128:5930–5938. doi: 10.1021/ja060328d. [DOI] [PubMed] [Google Scholar]; (b) Tsunoda T, Suzuki M, Noyori R. Tetrahedron Lett. 1980;21:71–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.