

Abstract
The heterogeneity and distribution of GABAA receptor subunits mediates differential roles in behavior. It is thought that particular behavioral responses to benzodiazepine (BZ) ligands might be associated with an action at a regionally defined receptor subtype. However, the role of specific GABAA receptor subtypes in particular brain regions is less clear. Such detailed knowledge of regional α1-GABAA receptor function will advance our understanding of the neural circuitry underlying the role of GABAA receptors and the effects of GABAA-modulating drugs on behavior. By combining inducible, site-specific α1 subunit deletion, using a lentivirus expressing Cre-recombinase in mice with the α1 subunit gene flanked by loxP sites, we examine baseline and pharmacological effects of deletion of amygdala α1-GABAA receptors. We find that amygdala-specific reduction of α1 receptor subunits does not affect mRNA or protein levels of amygdala α2 or α3 subunit receptors. Nor does this inducible reduction affect baseline locomotion or measures of anxiety. However, we also find that this inducible, site-specific deletion does disrupt the normal sedative-locomotor inhibition as well as the anticonvulsive effects, of two distinct BZ-site ligands, diazepam and zolpidem, which is relatively α1-subunit selective. These data, using inducible, region and subunit-specific deletion, combined with pharmacogenetic approaches, demonstrate that amygdala expression of the α1-GABAA receptor subunit is required for normal BZ effects on sedation, locomotion, and seizure inhibition, but not for anxiolysis.
Introduction
Fast synaptic inhibition in the mammalian forebrain is mediated by GABA interacting with postsynaptic GABAA receptors (GABAARs), which are pentameric complexes composed of multiple subunits (α1-6, β1-3, γ1-3, ρ1-3, ε, π, and δ) (Burt and Kamatchi, 1991; Barnard et al., 1998; Mehta and Ticku, 1999). The various combinations of subunits result in a heterogeneous population of GABAARs, although most contain at least one member of the α, β, and γ subunit classes in a proposed stoichiometry of two α-, two β-, and one γ-subunit (Backus et al., 1993; Sieghart et al., 1999; Möhler et al., 2002; Sieghart and Sperk, 2002). Defined subunit compositions display specific regional and cellular distribution (Fritschy and Mohler, 1995) which together confer distinct electrophysiological and pharmacological properties (Barnard et al., 1998; Sanger, 2004; Möhler, 2006; Nutt, 2006; Sieghart, 2006). Receptors containing the α1, α2, α3, or α5 subunits in combination with any of the β-subunits and the γ2-subunit are sensitive to benzodiazepine (BZ) modulation (Wisden et al., 1991; Hadingham et al., 1996; Wafford et al., 1996).
By far the largest population of benzodiazepine-sensitive (BZ)-GABAAR subtypes contain α1subunits (McKernan and Whiting, 1996). The behavioral and pharmacological functional roles of α1-GABAAR subtypes have been revealed using subtype-selective drugs (for review, see Atack, 2003). One such agent is the imidazopyridine zolpidem, among the most common clinically used sedative-hypnotics. Studies using recombinant GABAARs demonstrate zolpidem has a high affinity for the GABAARs containing α1 subunits, an ∼20-fold-lower affinity at α2- and α3-containing GABAARs, and no affinity at α5-containing GABAARs (Pritchett and Seeburg, 1990; Sanger et al., 1996). When compared with nonselective benzodiazepines, zolpidem and other agents with preferential affinity for α1-GABAAR subtypes (e.g., zaleplon, zopiclone) show great potency in inducing motor-sedative effects in rodents (Griebel et al., 1996; Sanger et al., 1996; Drover, 2004). α1-selective agonists also possess anticonvulsant properties (Sanger et al., 1996; Velísková et al., 1998; Drover, 2004). For example, zolpidem reduces pentylenetetrazole (PTZ) and electroconvulsive shock-induced convulsions (Depoortere et al., 1986; Sanger et al., 1996; Crestani et al., 2000).
The behavioral and pharmacological roles of α1-GABAARs have been further elucidated by pharmacological studies using mice with targeted point mutations rendering them insensitive to allosteric modulation by BZ-site ligands due to the replacement of a conserved histidine residue with arginine at position 101 of the α1-GABAA subunit gene (Benson et al., 1998; Rudolph et al., 1999; Crestani et al., 2000; Marowsky et al., 2004). These corresponding α1(H101R) mice fail to show a motor-sedative effect to diazepam or zolpidem (Rudolph et al., 1999; McKernan et al., 2000). The ability of diazepam and zolpidem to prevent PTZ-induced seizures was also reduced in α1(H101R) mice (Rudolph et al., 1999; Crestani et al., 2000). In contrast, α2 and α3 point mutated mice display dose-dependent increases in the seizure thresholds which are comparable to wild type controls (Löw et al., 2000).
Evidence from mice with deletion of the α1 subunit gene also supports the view that α1-containing receptors in part mediate the anticonvulsant effect of diazepam. Global knock-outs lack all α1-containing receptors as assessed by high-affinity [3H]zolpidem-binding sites (Kralic et al., 2002a), yet exhibit a viable phenotype despite a resulting loss of a majority of GABAARs in the brain (Sur et al., 2001; Vicini et al., 2001; Kralic et al., 2002a). Behavioral studies reveal these mutants exhibited a tremor when handled and were more susceptible to bicuculline-induced seizures (Sur et al., 2001; Kralic et al., 2002a). Furthermore, while diazepam dose-dependently increased the seizure threshold of wild-type mice, it failed to increase the seizure threshold in α1 knock-outs (Kralic et al., 2002a).
While the use of conventional knock-out and point-mutation strategies have provided vital insights into the various roles of α1-GABAARs, there are a number of limitations with traditional knock-out strategies. Relatively little is known about the role of this subtype in regionally defined areas of the brain (cf. Kralic et al., 2006; Zeller et al., 2008). In addition, there are often compensatory changes with other GABAAR subtypes (Sur et al., 2001; Rudolph and Mohler, 2004; Kralic et al., 2006). In certain instances, the compensatory changes are unexpected based on known pharmacology. For example, benzodiazepines and α1-selective compounds show great potency for suppressing locomotor activity (Crestani et al., 2000; McKernan et al., 2000); however, due to compensation or developmental effects, α1 KO mice are more sensitive to the sedative effects of diazepam (Kralic et al., 2002b; Zeller et al., 2008). Such complex compensatory changes make it difficult to assess whether phenotypic consequences result from the lack of normal gene expression in the adult or compensatory abnormalities caused by α1-GABAAR gene deletion during early prenatal or postnatal periods.
There is a need to examine the role of different GABAAR genes with an inducible method, allowing time-, regional-, and subunit-specific deletion of specific receptor subunits. The lateral and basolateral nuclei of the amygdala express high levels of α1-GABAARs, and are primary sites involved in many behavioral responses induced by BZs (Hevers and Luddens, 1998; Teuber et al., 1999; Pirker et al., 2000; Smith, 2001; Kaufmann et al., 2003; Kopp et al., 2004; Savić et al., 2005; Heldt and Ressler, 2006; Engin and Treit, 2008). To address these issues, the current study examined the behavioral, pharmacological and compensatory consequences of targeted α1-GABAARs deletion in the amygdala of adult mice.
Materials and Methods
Animals.
Adult Gabra1-tm1Geh male mice (9–15 weeks) weighing 23–28 g were used in this study. Mice were housed in standard group cages (<6/cage) and given ad libitum access to food and water on a 12 h light/dark cycle. All experiments were performed during the light cycle and were conducted on mice between 5 and 10 weeks of age. The experiments were approved by our Institutional Protocol Approval Committee and were in accordance with Yerkes Primate Research Center Regulations.
The Gabra1-tm1Geh targeted knock-in mice (The Jackson Laboratory) possess loxP sites on both side of the α1 exon encoding an essential transmembrane domain. Normal expression of the α1 gene is observed in mice that are homozygous for this “floxed” allele, and these mice do not display any deficiencies in α1-GABAA. In the presence of Cre recombinase (CRE), the transmembrane domain of the α1-GABAA gene is deleted, resulting in a nonfunctional α1 subunit (Vicini et al., 2001).
Lentivirus production.
Localized deletion of α1-GABAARs was accomplished by microinjections of a CRE recombinase-expressing lentivirus vector. This vector was derived from the HIV-based lentivirus backbone pLV-CMV-GFP-U3Nhe (Tiscornia et al., 2003) which allows for virally mediated expression of green fluorescent protein (GFP) driven by a cytomegalovirus (CMV) promoter. The Cre-recombinase-expressing viral vector (LV-Cre) was created by replacing the GFP coding sequence in pLV-CMV-GFPU3-Nhe with the coding sequence for Cre-recombinase. Viral particles were produced by cotransfecting these lentiviral packaging constructs with plasmids coding for delta 8.9 and VSV-G into HEK-293T cells following standard methods (Tiscornia et al., 2006; Heldt et al., 2007; Jasnow et al., 2009). The packaged, unconcentrated virus was collected over a period of 5 d posttransfection and then concentrated using ultracentrifugation and resuspended in sterile PBS/1% BSA. The resulting titer was assessed in HEK-293T cells. The observed titer of the GFP and Cre-expressing viruses typically ranged from 1 × 108 to 1 × 109 infectious particles per ml.
Cell culture experiments.
Primary cultures of hippocampal neurons from Gabra1-tm1Geh floxed mice (P10) were prepared using a modified protocol based on the method of Brewer (1997). Briefly, the hippocampus was dissected on ice and dissociated in Hibernate A medium (BrainBits). Neurons were plated onto poly-d-lysine-precoated 24-well plates at a density of 2.5 × 105 cells/cm2 in Neurobasal A media (Invitrogen). The cultures were kept in a humidified incubator at 37°C and 5% CO2. Two weeks after plating, wells received either 1 μl of LV-Cre, LV-GFP or were left untreated followed by 3 d of incubation at 37°C and 5% CO2. Neurons were then fixed with 4% Paraformaldehyde in PBS and blocked for nonspecific binding in PBS containing 1% BSA and 3% normal goat serum. α1-GABAAR subunits were stained with rabbit anti-α1-GABAAR subunits antibody (1:500; Fisher) and goat anti-rabbit Alexa Fluor 568 (1:1000; Invitrogen). After PBS rinse, cells were treated with Hoechst 33342 stain (10 μg/ml) for 10 min to identify nuclei of both neuronal and glial cells. Wells were viewed using a Leica DMRA microscope and random images from LV-GFP, LV-Cre (n = 8), and untreated wells (n = 8, area = 0.175 cm2/each) were captured using a Nikon Digital Sight DS-U1 camera system and NIS Elements BR 2.30 Software. Hoechst and α1-GABAAR-positive neurons from the same area were counted, and the level of α1-GABAAR subunit expression was calculated as a percentage of the total Hoechst-stained nuclei.
Surgery and histology.
Gabra1-tm1Geh mice received bilateral amygdala microinjections of either the CRE lentivirus or the control pLV-CMV-GFPU3-Nhe lentivirus that does not produce CRE. Mice were anesthetized by injections of Ketamine-Metomidine (80 mg/kg: 1.0 mg/kg, i.p.), then mounted in a stereotaxic apparatus. Small holes were drilled in the skull above the injection site, and a 30-gauge Hamilton microsyringe was lowered to the following coordinates from bregma based on the mouse brain atlas of Paxinos and Franklin (2001): AP = −1.4, ML = ±3.3, DV = −5.0. The microsyringe was left in place 10 min before and after each injection, and a total volume of 1.0 μl of LV-CRE or LV-GFP was administered at a rate of 0.05 μl/min at each site. After surgery the incision was closed with cyanoacrylate glue and each mouse was placed on a heated pad after a postsurgical injection of Antisedan (4.0 mg/kg, i.p.). The narcotic analgesic, buprenorphine (0.05 mg/kg, s.c.), was administered to mice after recovery from anesthesia. Postsurgical monitoring was performed before testing.
One week after behavioral testing, mice were anesthetized with isoflurane and the brains were rapidly dissected and frozen on crushed dry ice. Coronal sections (20 μm) of brains were cut on a Leica cryostat at −20°C, mounted on gelatin-coated slides, and stored at −80°C until processed for histochemistry. For each brain, sections were placed on 10 consecutive sets of slides such that each set contained similar sequential sections of the brain. Four sets of slides from each brain were used for in situ hybridization analyses of mRNA (Cre, α1, α2, and α3), and 1 set of slides was stained with cresyl violet.
Control LV-Cre and LV-GFP mice behavioral experiments.
To control for any actions of Cre other than specific cre-loxP-mediated recombination (Schmidt-Supprian and Rajewsky, 2007), we examined the sedative-locomotor inhibition and the anticonvulsive properties of zolpidem after LV-GFP and LV-Cre microinjections in mice without the loxP-flanked target gene. In this experiment, C57BL/6J received bilateral LV-GFP (n = 10) or LV-Cre (n = 10) amygdala microinjections (ns = 8) as outlined Surgery and Histology section. 14 d after surgery, all mice were exposed for 30 min to a novel open-field apparatus acclimation session. One day later, mice received Session1 of a 2-session random crossover experimental designed to evaluate the effect of zolpidem treatment on open-field activity. On Session 1, approximately one-half of the LV-GFP and LV-Cre mice were administered a low-dose pretreatment 30 min before testing (0.25 mg/kg); the remaining mice were given a high-dose (0.5 mg/kg). Test Session 2 was conducted following a seven-d drug washout period to limit pretreatment crossover effects. On this session, drug pretreatment for each mouse was reversed. Analyses excluded one LV-Cre animal due to poor injection. Two weeks after open-field testing mice the anticonvulsant effects of zolpidem were tested by administration of 2.5 mg/kg, i.p. 30 min before PTZ administration (85 mg/kg). Eight mice from each surgery group were used for the anticonvulsant test.
Test drugs and doses.
Zolpidem, a preferential GABAARα1 subtype modulator, and diazepam, a nonselective BZ-site agonist (Sigma) were dissolved in a 15% DMSO/saline solution. The chemo-convulsant PTZ (Sigma) was dissolved in saline. All drugs were administered in a volume of 8 ml/kg after animal weights were recorded. Test drug doses levels for examining their motor-impairing effects were based on preliminary showing that the lower test drug dose (diazepam, 5.0 mg/kg; zolpidem, 0.25 mg/kg) reliably decreases activity from baseline levels, and the high dose (diazepam, 10.0 mg/kg; zolpidem, 0.5 mg/kg) causes a significant decrease in activity compared with the low dose (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Thus, for the motor-impairing effects, mice received either diazepam (5.0 or 10 mg/kg) or zolpidem (0.25 or 0.5 mg/kg). Examination of potential group differences in the convulsant action of PTZ was conducted with two different doses (55 and 85 mg/kg, i.p.). A PTZ dose of 85 mg/kg was used to examine the anticonvulsive effects based on preliminary data showing highly reliable decreases in both myoclonic and tonic seizure latencies during a 20 min observation period (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). For drug-induced seizure inhibition, mice were pretreated with either diazepam (0.25 or 1.0 mg/kg) or zolpidem (2.5 or 4.0 mg/kg). Dose levels were based on preliminary showing that the lower test drug dose (diazepam,0.25 mg/kg; zolpidem, 2.5 mg/kg) was effective at inhibiting tonic seizures, whereas the high dose (diazepam, 1.0 mg/kg; zolpidem, 4.0 mg/kg) was effective at inhibiting both myoclonic and tonic seizures (supplemental Figs. S3, S4, available at www.jneurosci.org as supplemental material).
In situ hybridization.
In situ hybridization was performed to examine the expression of mRNA using procedures and probes previously described (Ressler et al., 2002; Heldt and Ressler, 2007). Briefly, hybridization riboprobes are prepared from linearized clones using appropriate RNA polymerase and radioactive 35S-UTP in the polymerase reaction. Radiolabeled antisense RNA strands are base hydrolyzed and isolated using a Sephadex spin column (Roche). Probes are diluted to a concentration of 100,000 cpm/μl in hybridization buffer, and sections are incubated overnight in humid chambers at 52°C with 75 μl of probe-buffer solution covered with a Parafilm coverslip. Slides are then washed, air dried, and apposed to Biomax MR autoradiography film (Eastman Kodak) for 1–5 d.
The α1 subunit plasmid was constructed by TOPO subcloning of a cDNA PCR product using the following customized primers: a1 sense, GGAGTGACGACTGTTCTGACTATG; a1 antisense, TTCTGGAACCACGCTTTTG (Sigma). Plasmid DNA sequencing (Iowa State University, Ames, IA) confirmed subclone sequence and orientation. The resulting a1 PCR transcript corresponds to nucleotides 1307–1509 of the a1 cDNA which is flanked by loxP sites and deleted by CRE recombinase. These nucleotides encode amino acids included in the second transmembrane domain and intercellular loop between transmembranes 3 and 4. Previous analyses have demonstrated signal specificities and density distributions using both sense and antisense RNA riboprobes (Heldt and Ressler, 2007).
Image analysis.
Each film was scanned using a high-resolution Epson 3700 flat-bed scanner (3000 dpi) and saved in JPEG format at a size of 32,000 × 18,000 pixels. To estimate levels of receptor mRNA transcript, the total area of each target site subregion (in pixels) was first outlined by freehand tool of Adobe PhotoShop with reference to Nissl-stained section from the same mouse and the atlas of Paxinos and Franklin (2001). The hybridization signal intensities of brain regions were defined on the basis of gray values (GVs) between 0 (brightest) and 255 (darkest) obtained from the luminosity histogram feature of Adobe Photo. For each section, GVs were recorded for the regions of interest, as well as an adjacent background area with little or no hybridization signal. For the quantification, signal intensities were computed by subtracting background area GVs from region of interest GVs. Independent statistical comparisons were performed for LA and BL regions of the amygdala. Statistical analyses were performed with independent samples t tests (LSD). For ease of presentation, figures are presented as percentages of control group (LV-GFP).
Western blots.
Unilateral amygdala punches were obtained with a 1 mm brain punch tool, suspended in 25 μl of homogenized lysis buffer (5 mm HEPES, 1 μm EDTA) with a protease inhibitor cocktail kit (Roche), then homogenized with a sonicator on ice. The protein concentrations were measured using 2.5 μl samples of homogenates with BCA protein assay reagent and bovine serum albumin (BSA) as the reference standard (Pierce) (Bradford, 1976). Equal amounts of protein (20 μg) per animal were boiled for 5 min and loaded on 7.5% polyacrylamide gels (Bio-Rad). After electrophoresis, the samples were transferred to nitrocellulose membrane (0.45 μm, Bio-Rad) at 30 mA for 2 h at 4°C (25 mm Tris-HCl, 190 mm glycine, and 20% methanol). After transfer, the membranes were washed three times with blocking buffer (2% nonfat dry milk, 0.1% Tween 20, 50 mm NaCl, 10 mm HEPES, pH 7.4) with 5 min intervals. Membranes were then incubated with a primary antibody at 4°C overnight. Following three washes with blocking buffer with 5 min intervals, membranes were incubated with a corresponding HRP-coupled secondary antibody at room temperature for 60 min. The membranes were subsequently washed with water to remove the substrate solution and visualized by SuperSignal West chemiluminescence substrate system (Pierce). Reactive protein bands were visualized and analyzed using an Alpha Innotech Fluorchem imaging system. Rabbit polyclonal antibody to mouse α1-GABAAR (1:1000) and α3-GABAAR (1:1000) were obtained from Abcam. Rabbit polyclonal antibody to mouse α2-GABAAR (1:1000) was obtained from Alomone Labs. Monoclonal mouse anti-glyceraldehyde-3-phosphate dehydrogenase antibody (GAPDH, 1:3000) was obtained from Fitzgerald Industries. Total blotted protein levels were normalized to levels of GAPDH to control for variations in protein loading. As such, the relative values were quantified as the protein of interest divided by the loading control. Statistical analyses were performed with independent samples t tests (LSD). For ease of presentation, figures are presented as percentages of control group (LV-GFP).
Behavioral and histological group assignments.
All mice (n = 144) were tested in the open-field apparatus 14 d after surgery to evaluate baseline motor behavior in a novel environment. Baseline anxiety-like behavior, as assessed by evaluating the percentage of time mice spent in the central zone of the open field, was obtained from 105 of these mice. Time spent in the central zone was omitted from the remaining 39 animals due to data recovery difficulties. Fifteen LV-GFP mice and 16 LV-Cre mice were used to examine the convulsant action of PTZ (55 and 85 mg/kg, i.p.). Western blot analyses were performed on homogenized amygdala punches collected from LV-GFP mice (n = 10) and LV-Cre mice (n = 10). The effect of test-drug treatment on open-field activity was evaluated using a 2-session random crossover experimental design using the remaining animals (n = 93). Separate animals were used to examine the behavioral effects of diazepam and zolpidem at each of two dose levels. On Session 1, approximately one-half of the LV-GFP and LV-Cre mice were administered a low-dose pretreatment 30 min before testing (diazepam, 5 mg/kg; zolpidem, 0.25 mg/kg); the remaining mice were given a high-dose (diazepam, 10 mg/kg; zolpidem, 0.5 mg/kg). Test Session 2 was conducted following a seven-d drug washout period to limit pretreatment crossover effects. On this session, drug pretreatment dose level for each mouse was reversed. Two weeks after open-field testing mice were assigned to one of eight groups to test the anticonvulsant effects of test drugs. Mice in the diazepam groups were pretreated with either 0.25 mg/kg, i.p. (LV-GFP, n = 10; LV-Cre, n = 12) or 1.0 mg/kg (LV-GFP, n = 10; LV-Cre, n = 12) 30 min before PTZ administration. Mice in the zolpidem groups were pretreated with either 2.5 mg/kg, i.p. (LV-GFP, n = 12; LV-Cre, n = 16) or 4.0 mg/kg (LV-GFP, n = 11; LV-Cre, n = 10) before PTZ administration. Histological examination and in situ hybridization analyses of mRNA levels were performed on coronal brain sections of the remaining animals 1 week after behavioral testing.
Separate animals were used to examine the behavioral effects of diazepam on elevated plus maze performance (LV-GFP, n = 9; LV-Cre n = 10). On Session 1, approximately one-half of the LV-GFP and LV-Cre mice were administered diazepam (1.5 mg/kg, i.p.) 30 min before testing; the remaining mice were given vehicle. Test Session 2 was conducted following a two-week drug washout period to limit crossover effects. On this session, drug pretreatment dose level for each mouse was reversed.
Motor activity and motor-impairing effects of test drugs.
Baseline motor activity and the motor-impairing effects of test drugs were measured by examining the total ambulatory distance (in cm) during the 30 min open field test session. In rodents, drug-induced changes in both ambulatory distance and counts represent a standard behavioral assay for testing the motor effects of drugs (Vogel and Vogel, 2002). The anxiety-like behaviors were evaluated by computing percentage of time mice spent in the central zone of the open field. An increase in the percentage of central zone time is indicative of an anxiolytic-like phenotype and best takes into account potential confounding changes in locomotive activity (Prut and Belzung, 2003). The central zone was defined as the central compartment of the floor centrally located 6 cm from the perimeter of the chamber walls. Activity for each mouse was measured 30 min after test drug injection using individual activity chambers constructed from clear polycarbonate and equipped with four 24-beam infrared arrays across the base of each chamber wall (MED Associates, model OFA-MS). Activity data are collected via computer and was analyzed with the MED Associates' Activity Monitor Data Analysis software. All testing was conducted under standard room lighting.
Assessing the anxiolytic-like activity of diazepam on elevated plus-maze.
The elevated plus maze consisted of two open arms (50 × 6.5 cm) and two closed arms with a wall (50 × 6.5 × 15 cm) attached to a common central platform (6.5 × 6.5 cm) to form a cross. The maze was elevated 65 cm above the floor. Test sessions were conducted under standard room lighting (100 lux) where behaviors were continuously videotaped by a video camera placed over the apparatus. Activity was collected, and analyzed via computer using automated Limelight software. Before each test, the plus maze was cleaned with Quatricide (Pharmacal).
Elevated plus-maze testing was conducted with a separate cohort of animals. Thirty minutes before elevated plus-maze testing, each animal was weighed then injected intraperitoneally with diazepam (1.5 mg/kg). At the start of each session, a mouse was placed in the central “hub” of the maze with its head facing an open arm and was allowed to freely explore for 5 min. The percentage of open arm entries [open arm/(open + closed arm) entries] × 100 and percentage time in open arms [time in open arms/(time in open + closed arms)] × 100 were computed. Both of these parameters are indicators of anxiolytic-like activity (Pellow and File, 1986; Hogg, 1996). The total number of arm entries (open + closed) was used as an indicator of locomotor activity (Rodgers and Dalvi, 1997). Arm entry was considered complete if all four paws entered a closed or open arm from the central platform.
PTZ-induced seizures and anticonvulsant effects of test drugs.
The systemic administration of PTZ for induction of generalized clonic seizures in rodents is widely used to identify potential anticonvulsants (Löscher and Schmidt, 1994; Meldrum, 2002). Immediately following PTZ injection, mice were placed individually in acrylic observation chambers cage (450 mm × 350 mm × 300 mm) for a 20 min (1200 s) observation period. Because the appearance of myoclonic and clonic seizures indicate the initiation and spreading of the seizures, respectively, we evaluated both parameters in the present study. The time between the injection of PTZ and the appearance of myoclonic jerks or “jumps” was defined as the myoclonic seizure-onset time (Kaputlu and Uzbay, 1997). Clonic seizures were defined as convulsions involving the whole body and loss of righting ability. Mice were given an “anticonvulsant rescue” dose of diazepam (5 mg/kg) 5 s after clonic seizure onset or immediately after the observation period, whichever came first. To examine the anticonvulsant effects of test drugs, mice were given test drugs 30 min before administration of PTZ (85 mg/kg PTZ). If no seizure occurred during the observation period, the mice were considered protected. Each mouse was used only once in an experiment. Duration of 1200 s was taken as a cutoff time in calculation of the onset time of PTZ-induced seizures. Data were analyzed with one-way ANOVA to examine relationships between experimental groups and seizure onset latency. Pearson correlations were used to examine relationships between α1-GABAAR mRNA level and response to diazepam (0.25 mg) in delaying myoclonic seizure onset latencies. Note that one animal was removed from the correlation analyses due to a latency that was >3 SD above the mean.
Statistical analyses of behavioral measures.
Group differences in baseline motor activity and percentage central zone time were examined using two-sample t tests. Two-way ANOVAs were used to examine group differences in seizures latencies, ambulatory distances, and elevated plus-maze-dependent measures. The motor-impairing effects of test drugs, as measured by ambulatory distance (in cm), were analyzed by means of a 2 × 2 ANOVA with group (LV-Cre, LV-GFP) as between-subjects factor and drug level as the within-subjects factors. For diazepam drug levels were 5.0 and 10 mg/kg; for zolpidem, drug levels were 0.25 and 0.5 mg/kg. Group differences in PTZ-induced myoclonic and clonic latency to seizures were each assessed using a 2 × 2 ANOVA with group (LV-Cre, LV-GFP) and PTZ drug level (55 or 85 mg/kg) as between-subjects factors. To examine the anticonvulsant effects of test drugs, myoclonic and clonic seizures latencies (in sec) were analyzed separately by means of 2 × 2 ANOVAs with group (LV-Cre, LV-GFP) and drug level as between-subjects factors. Drug levels for the zolpidem were 2.5 and 4.0 mg/kg; levels for diazepam were 0.25 and 1.0 mg/kg. Elevated plus-maze-dependent measures, including total arm entries and percentages of open arm entries and open arms time were analyzed separately by means of a 2 × 2 ANOVA with group (LV-Cre, LV-GFP) as the between-subjects factor and treatment (Vehicle, Diazepam) as the within-subjects factor. Follow-up comparisons between dose levels or group were completed with pairwise or two-sample t test as needed. The level of significance was set at p = 0.05.
Results
Robust deletion of α1-GABAAR subunits in vitro
Figure 1 images represent the same field visualized separately illustrating bright-field with Hoechst-stained nuclei (top panels) and α1-GABAAR subunits antibody (bottom) in primary cultures of hippocampal neurons. As seen in Figure 1, a and b, the number of Hoechst-stained neurons in both LV-Cre and untreated cells were similar (t(14) = 0.36, p > 0.05). The mean (SEM) number of total Hoechst-stained neurons was 52.5 (4.0) in untreated wells and 55.1 (5.9) in LV-Cre-treated wells. In addition, the average number of Hoechst-stained neurons with primary dendrites was similar between wells of untreated and LV-Cre neurons (untreated: 19.9 (3.9); LV-Cre: 20.6 (2.0); t(14) = 0.17, p > 0.05). Because Hoechst stains the condensed chromatin in apoptotic cells more than normal chromatin, the similarity in number, intensity and density of staining suggests an absence of apoptosis in LV-Cre-infected cells. As seen in Figure 1, c and d, the number of α1-GABAAR-positive neurons was significantly higher in vehicle-treated cells. In untreated wells, the level of α1-GABAAR subunit expression as a percentage of the total Hoechst-stained nuclei was 35% (3%). In contrast, no α1-GABAAR-positive neurons were identified in LV-Cre-treated wells, p < 0.01.
Figure 1.

α1-GABAAR deletion in cultured mouse neurons with Cre and in vivo lentivirus infectivity of mouse amygala. Panels a-f demonstrate the level of α1-GABAAR subunit expression visualized with immunohistochemistry in LV-GFP-infected, untreated (no LV), and LV-Cre-infected primary mouse neurons from the floxed mouse line used for the remaining in vivo studies. a, Overlay of green fluorescent and Hoechst stain (blue) image depicting LV-GFP infection and chromatin-positive nuclei of neuronal cells. b, Immunocytochemistry for α1-GABAAR (red) in same field as in a. c, Overlay of bright field Hoechst stain (blue) image of primary neuronal culture with no infection. d, Immunocytochemistry for α1-GABAAR (red) in same field as in c. e, Overlay of bright field Hoechst stain (blue) image of primary neuronal culture with LV-Cre infection. f, Immunocytochemistry for α1-GABAAR (red) in same field as in e. These data demonstrate the complete removal of α1-GABAAR signal from cells infected with LV-Cre from this targeted, inducible, knock-out mouse strain. g–i, Low-power micrograph of cresyl violet staining (g), GFP fluorescence (h), and GFP fluorescence overlayed with Hoechst stain (i) in mouse with LV-GFP infection, demonstrating the dense infectivity of lentivirus in the mouse basolateral amygdala (BLA). Large arrow demonstrates target of infection in BLA. Also shown are central nucleus of amygdala (CeA) and lateral amygdala (LA).
Amygdala-specific reduction of α1-GABAAR mRNA, with no effect on α2 or α3 levels
Qualitatively, the selective decrease in α1-GABAAR subunit expression within the amygdala was observed following LV-Cre infection. Shown in Figure 1a–f are representative photomicrographs of parallel sections following LV injection into mouse amygdala. Figure 2a–c shows the decreased expression of α1 subunit mRNA and Cre recombinase expression pattern in LV-Cre mice. In comparison, Figure 2d shows the α1-GABAAR mRNA expression pattern in LV-GFP mice. Photomicrographs revealed no change in the expression of α2 or α3 subunits in LV-Cre mice with decreased α1 subunit mRNA (Fig. 2e,f). Nissl-stained sections also revealed no discernable histological abnormalities in the amygdala from animals infected with LV-Cre (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
Figure 2.

Region- and subunit-specific reduction of α1-GABAAR subunit mRNA in the adult brain. a, b, In situ hybridization analyses of mRNA expression indicated that local LV-Cre injections decreased the expression of α1 subunit relative to LV-GFP-infected mice (a) and produced reliable expression of Cre recombinase (b). c, Overlay of Cre recombinase with α1-GABAAR mRNA expression. d–f, Local LV-GFP injections produced no discernable decreases in α1-GABAAR subunit mRNA expression (d). LV-Cre injections produced no evident effect on the expression of the α2 subunit (e) or the expression of the α3 subunit (f). Arrows denote basolateral amygdala in all sections.
Quantitative analyses of mRNA expression levels reveal that the LV-Cre approach to site-specific GABA subunit deletion resulted in statistically significant reduction of functional α1-GABAAR subunits that was largely limited to the amygdala. Comparisons of α1-subunit mRNA transcript levels identified significantly less α1-GABAAR subunit expression in LV-Cre mice in comparison with LV-GFP mice. In particular, reliable mRNA changes were noted for α1 within both the LA, t(38) = 4.17, p < 0.01, and BL, t(38) = 6.13, p < 0.01. With reference to GFP animals, the relative level of α1-GABAAR mRNA expression within the LA and BL were 52.2% and 44.7%, respectively, in LV-Cre-infected animals (Fig. 3a). No significant group differences were identified for α2 or α3 subunits in either the LA or BL (p values >0.05).
Figure 3.

mRNA and protein analyses of GABAAR subunit expression. a, mRNA levels are expressed as percentages with reference to LV-GFP control animals. Lateral (LA) and basolateral (BLA) nuclei of the amygdala. b, Western blot analysis of α1-, α2-, and α3-GABAAR protein levels from amygdala tissue homogenates. c, Digital image example of the Western blot membrane showing protein bands of α1-, α2-, and α3-GABAARs as well as GAPDH protein in LV-Cre (+) and LV-GFP (−) mice. Protein amounts determined from these Western blots were normalized to levels of a GAPDH to loading control. Protein levels are expressed as percentages with reference to LV-GFP control animals. Error bars denote 1 SEM. Stars indicated that the difference was statistically significant, p values <0.05.
Amygdala-specific reduction in α1-GABAAR protein, with no effect on α2 or α3 levels
Previous studies have revealed that both global and targeted deletion of α1 subunits during development leads to compensatory increases in the expression of α2- and α3-GABAAR subtypes that may function to protect against the α1-GABAAR loss (Sur et al., 2001; Kralic et al., 2002b, 2006; Zeller et al., 2008). Thus, we performed Western blot analyses of tissue homogenates to examine α1-, α2-, and α3-GABAAR subunit protein expression in mice given either LV-Cre or LV-GFP microinjections (Fig. 3b,c). A comparison of α1-subunit protein levels in the amygdala revealed significantly less α1-GABAAR protein expression in LV-Cre mice in comparison with LV-GFP mice, t(12) = 7.62, p < 0.01. No group differences were detected in α2 or α3 subunit protein levels t(12) = 0.22, p > 0.05 and t(12) = 0.98, p > 0.05, respectively. With reference to GFP control animals, the level of α1-GABAAR amygdala protein expression in LV-Cre-infected animals was 62.4%. The relative levels of α2- and α3-GABAAR protein were 99.3% and 93.1%, respectively. Together with the mRNA in situ findings, these results indicate that Cre-mediated inducible gene reduction of the α1-GABAA subunit in adult mice leads to no significant compensatory increases in the expression of α2- and α3-GABAAR subtypes within the timeframe of these experiments.
No difference in baseline and central zone locomotion in LV-Cre and LV-GFP mice
Two weeks after microinjection surgery, the sedative effects of diazepam and zolpidem were examined in both α1-GABAA deleted mice (LV-Cre) and controls (LV-GFP). As seen in Figure 4, when LV-Cre and LV-GFP mice were tested in open-field, both groups showed similar levels of locomotor activity as measured by ambulatory distance, t(142) = 0.81, p > 0.05. Thus, in the open field, a reduction of α1-GABAARs located within the amygdala does not critically affect baseline motor behavior as assessed by percentage central zone time.
Figure 4.

Baseline activity and motor effects of test drugs in LV-Cre and LV-GFP mice. a, Diazepam effects on locomotor activity. Baseline motor activity (control) was measured by examining the total ambulatory distance (in cm) during the 30 min open field test session. To examine the motor-impairing effects of diazepam, mice were given test drug injection (intraperitoneally) 30 min before open field testing. The motor-impairing effects of diazepam are significantly blunted in mice with decreased α1-GABAARs in the amygdala at the 10 mg/kg dose. b, With zolpidem, motor-impairing effects are significantly blunted at both the 0.25 mg/kg and 0.5 mg/kg dose. Total ambulatory distances are expressed as mean + SEM. Star indicates that the difference between virus groups was statistically significant (p < 0.05).
LV-Cre-infected mice have decreased sensitivity to diazepam on motor-sedative behavior
Following baseline locomotion testing in the open-field, LV-Cre and LV-GFP mice were treated with diazepam and subsequently tested in the open-field. At doses of 5 and 10 mg/kg, diazepam produced a decrease in locomotor activity in both groups when compared with previous drug-free open-field testing, p values >0.05. However, as seen in Figure 4a, LV-GFP mice showed significantly less motor activity than LV-Cre mice as reflected by a significant main effect of group, F(1,42) = 4.49, p < 0.05. Direct comparisons between groups at each dose level revealed that the motor activity of LV-GFP mice was significantly lower than LV-Cre mice at 10 mg/kg but not 5 mg/kg dose level, t(42) = 2.26, p < 0.05, t(42) = 1.39, p > 0.05, respectively. These finding indicate that the reduction of amygdala α1-GABAARs impairs the motor-impairing effects of diazepam at 10 mg/kg.
LV-Cre-infected mice have decreased sensitivity to α1-selective zolpidem on motor-sedative behavior
To examine whether deficits in diazepam-induced motor deficits seen in LV-Cre mice were due to a reduction in α1-GABAARs, we examined the motor-impairing effects of zolpidem in LV-Cre and LV-GFP mice. Overall, the administration of zolpidem at doses of either 0.25 or 0.5 mg/kg induced a reduction in motor activity in both LV-Cre and LV-GFP mice when compared with drug-free novel open-field testing, p values <0.05. As seen with diazepam, zolpidem induced a greater motor activity reduction in LV-GFP mice than in LV-Cre mice as reflected by a significant main effect of group, F(1,47) = 13.76, p < 0.01 (Fig. 4b). The zolpidem-induced motor impairment was significantly greater in LV-GFP mice compared with LV-Cre mice at both 0.25 mg/kg, t(47) = 2.25, p < 0.05, and 0.5 mg/kg t(47) = 3.72, p < 0.01, dose levels as measured by ambulatory distance. Together these findings suggest that the motor-impairing actions of zolpidem are in part mediated by α1-GABAARs located within the amygdala.
No difference in the proconvulsant effect of pentylenetetrazole in LV-Cre and LV-GFP mice
To assess in vivo the consequences of reduced amygdala α1-GABAARs on the convulsant actions of PTZ, the latency to myoclonic and clonic seizures was examined in LV-Cre and LV-GFP mice following intraperitoneal injections (55 or 85 mg/kg). As shown in Figure 5, a and b, no group differences in latency to myoclonic and clonic seizures were detected at either dose of PTZ (p values >0.05). These findings indicate that a localized decrease of amygdala α1-GABAARs does not affect PTZ-induced latency to seizures.
Figure 5.

The convulsant actions of PTZ in LV-Cre and LV-GFP mice. Seizure onset latencies were defined as the time (in sec) between the injection of PTZ and the first appearance of myoclonic and clonic seizure activity. Behaviors were assessed during 20 min (1200 s) observation period. a, Local amygdala deletion of α1-GABAARs does not affect PTZ-induced myoclonic seizure onset time. b, Local amygdala deletion of α1-GABAARs does not affect PTZ-induced clonic seizure onset time. c, Despite the lack of effect on PTZ-induced seizure latency, there was a significant correlation between lateral amygdala α1-GABAAR mRNA level and response to diazepam (0.25 mg) in delaying seizure onset (n = 12, r = 0.61, p < 0.05). d, Basolateral amygdala α1-GABAAR mRNA level also correlates with response to diazepam in delaying seizure onset (n = 12, r = 0.81, p < 0.005). Error bars denote 1 SEM.
LV-Cre-infected mice have decreased sensitivity to diazepam with pentylenetetrazole seizures
One week after open-field testing, the anticonvulsant effects of diazepam were tested in LV-Cre and LV-GFP mice. Mice were first given systemic injections of diazepam (0.25 or 1.0 mg/kg) followed 30 min later by administration of PTZ (85 mg/kg). With the combined LV-Cre and LV-GFP groups, there was a significant relationship between lateral amygdala (Fig. 5c) (r = 0.61, p < 0.05) and basolateral amygdala (Fig. 5d) (r = 0.81, p < 0.005) α1-GABAAR mRNA level and response to 0.25 mg of diazepam in delaying myoclonic seizure onset. This suggests that animals with reduced α1-GABAAR mRNA following LV-Cre infection would have shorter seizure onset latencies.
Compared with 0.25 mg/kg, the pretreatment of 1 mg/kg diazepam increased the latency to the first myoclonic episode in both LV-Cre and LV-GFP mice as reflected by a significant main effect of dose, F(1,40) = 12.42, p < 0.01 (Fig. 6a,b). Overall, diazepam increased myoclonic latency in LV-GFP mice significantly more than in LV-Cre mice as reflected by a significant main effect of group, F(1,40) = 23.35, p < 0.01. Comparisons between groups at each dose level revealed the anticonvulsant effect of 0.25 mg/kg was greater in LV-GFP mice than LV-Cre as measure by latency to myoclonic onset, t(20) = 2.44, p < 0.05. LV-GFP mice also showed significantly greater myoclonic latency at the 1.0 mg/kg dose level compared with LV-Cre mice, t(20) = 4.71, p < 0.01.
Figure 6.

The anticonvulsant effects of benzodiazepines on PTZ-induced seizures in LV-Cre and LV-GFP mice. Seizure onset latencies were defined as the time (in seconds) between the injection of PTZ and the first appearance of myoclonic (a, c) and clonic (b, d) seizure activity assessed during a 20 min (1200 s) observation period. Diazepam or zolpidem was given 30 min before administration of PTZ. The anticonvulsant effects of diazepam (a, b) and zolpidem (c, d) are blunted in mice with decreased α1-GABAARs in the amygdala. In bar graphs, latencies to seizure onset are expressed as means. Error bars denote 1 SEM. Stars indicated that the difference was statistically significant, p values <0.05.
For clonic seizure episodes, the LV-GFP group displayed a higher mean latency than the LV-Cre group after pretreatment of diazepam, as reflected by a significant group effect, F(1,40) = 9.67, p < 0.01. Direct comparisons revealed that the clonic latencies of LV-GFP mice were significantly higher at both 0.25 mg/kg and 1.0 mg/kg dose level, t(20) = 2.21, p < 0.05 and t(20) = 2.26, p < 0.05, respectively. Together, these results suggest that the anticonvulsant effects of diazepam are blunted in mice with decreased α1-GABAARs in the amygdala.
LV-Cre-infected mice have decreased sensitivity to zolpidem with pentylenetetrazole seizures
To test the anticonvulsant effect of zolpidem, mice were first given systemic injections of the zolpidem (2.0 or 4.0 mg/kg) followed 30 min later by the administration of PTZ (85 mg/kg). A significant main effect of dose, F(1,45) = 52.83, p < 0.01, revealed that the anticonvulsant effect of 4.0 mg/kg was significant >2.5 mg/kg as measured by the latency to myoclonic episode. The ability of zolpidem to prevent PTZ-induced myoclonic seizures was reduced in LV-Cre mice as reflected by a significant group effect, F(1,45) = 11.70, p < 0.01. As seen in Figure 6c, this group main effect was largely influenced by the greater anticonvulsant effect on myoclonic seizure at the zolpidem dose level of 2.5 mg/kg in LV-GFP mice, t(26) = 3.26, p < 0.01. LV-Cre and LV-GFP mice pretreated with 4 mg/kg showed a similar latency to the first myoclonic jerk episode following the PTZ injection, t(19) = 1.79, p > 0.05.
An examination of clonic seizure latencies indicated that overall, the anticonvulsant effects of zolpidem were greater in LV-GFP mice, as exhibited by a reliable group effect, F(1,45) = 7.88, p < 0.01. As seen in Figure 6d, when compared with LV-GFP mice, the latency to clonic seizure onset was significantly lower in LV-Cre at both 2.5 mg/kg and 4.0 mg/kg dose levels, t(26) = 2.30, p < 0.05 and t(19) = 2.55, p < 0.05, respectively. Together, these findings are consistent with the view that α1-GABAARs in the amygdala play an important role in mediating the anticonvulsant effect of zolpidem.
No difference in anxiolytic effect of diazepam in LV-Cre and LV-GFP mice
In addition to their motor-sedative and anticonvulsant effects, benzodiazepines are renowned for their capacity to reduce anxiety. There is considerable evidence indicating that that the amygdala is a major site of action. In rodents, the anxiolytic effects of benzodiazepines in the amygdala have been demonstrated on a number of animal models of anxiety, including the elevated-plus maze (Green and Vale, 1992; Pesold and Treit, 1995; Nunes-de-Souza et al., 2000). On the other hand, microinjections of benzodiazepine antagonists and inverse agonists into the amygdala induce anxiogenic-like behaviors (Hodges et al., 1987; Da Cunha et al., 1992) and infusions of antagonists can also prevent the anxiolytic effects of peripheral injections of benzodiazepine agonists (Petersen et al., 1985; Sanders and Shekhar, 1995). Because past pharmacological and behavioral studies suggest that α2-, but not α1-containing receptors, mediate the anxiolytic effect of diazepam (Rudolph et al., 1999; Löw et al., 2000; McKernan et al., 2000; Atack, 2003), we examined whether the amygdala-specific lack of α1-GABAAR expression leads to differential responsiveness of diazepam's anxiolytic effects.
As seen in Figure 7, a and b, diazepam produced an anxiolytic-like effect in both LV-GFP and LV-Cre mice. An evaluation of the percentage time in open arms revealed a significant treatment effect F(1,17) = 15.83, p < 0.01. Neither the group effect nor group × treatment interaction were significant, F values (1,17) <1.21, p values >0.28. Paired t test indicated that both LV-GFP mice (p = 0.03) and LV-Cre mice (p = 0.02) displayed an increased percentage time in open arms after diazepam administration. Likewise the evaluation of the percentage open arm entries also revealed a significant treatment effect F(1,17) = 12.78, p < 0.01 but no reliable group effect nor group × treatment interaction, F(1,17) <1.24, p values >0.28. Diazepam reliably increased the percentage open arm entries in both LV-GFP (p = 0.04) and LV-Cre (p = 0.03) groups. As seen in Figure 7c, assessment of total arm entries (open + closed) indicated no significant group differences in number of entries during drug or vehicle testing, t values (17) <2.1, p values >0.06.
Figure 7.
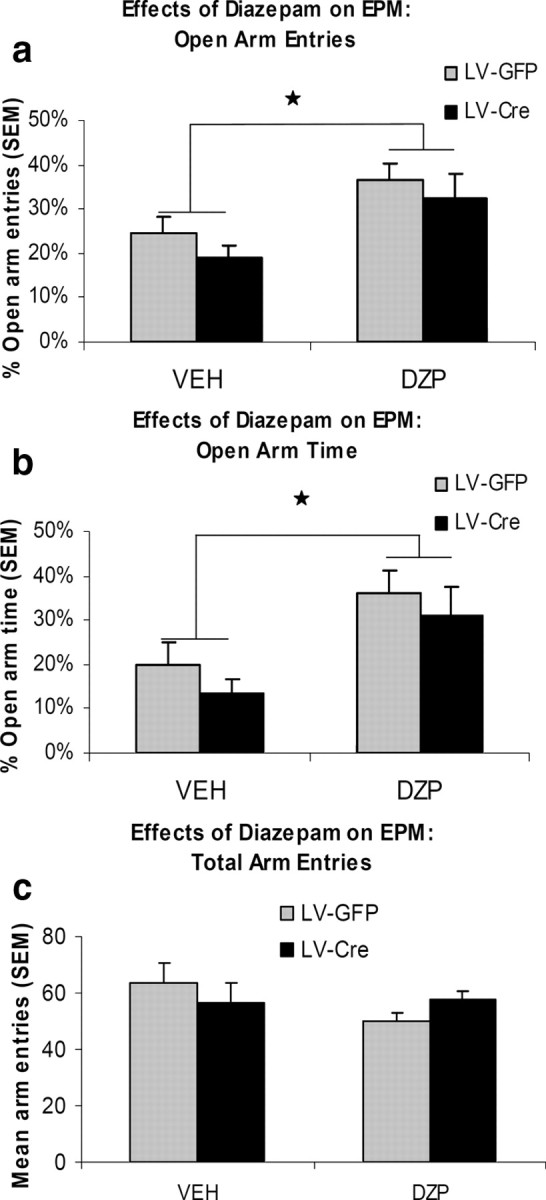
The effects of diazepam on elevated plus-maze (EPM) performance. Mice were given 1.5 mg/kg diazepam (DZP) or vehicle (VEH) 30 min before testing. Bar graphs show the percentage of open arm entries (a), percentage of open arm time (b), and total (open + closed) number of arm entries (c) on the elevated plus-maze. Error bars denote 1 SEM. Stars indicate significant difference from vehicle treatment, p values <0.05.
Open field exploratory behavior (as examined for locomotion in Fig. 4), was also examined for anxiety-like behavior with regards to percentage time in center versus surround of the open field. Assessment of the percentage central zone time revealed no differences between groups (LV-GFP, 59.5%; LV-Cre, 59.3%, t(103) = 0.94, p > 0.05).
Motor and anticonvulsant effects of zolpidem in control LV-Cre and LV-GFP mice
We next examined the sedative-locomotor inhibition and the anticonvulsive properties of zolpidem after LV-GFP and LV-Cre microinjections in wild-type mice without the loxP-flanked target gene. Overall, the administration of zolpidem induced a reduction in motor activity similarly in both LV-Cre and LV-GFP mice when compared with drug-free novel open-field testing (p values <0.05; LV-Cre: Mean = 4284, SEM = ±847; LV-GFP: mean = 4477, SEM = ±601). As seen in supplemental Figure S5 (available at www.jneurosci.org as supplemental material), no differences in zolpidem-induced motor impairment were identified in control LV-GFP and LV-Cre mice at either 0.25 or 0.5 mg/kg dose levels, t values (17) <0.14, p values >0.05, 0.01, as measured by ambulatory distance. For the anticonvulsant effect of zolpidem, an examination of seizure latencies revealed no differences control LV-GFP and LV-Cre mice in myoclonic onset t(14) = 0.468, p > 0.05, or clonic seizure onset t(14) = 0.139, p > 0.05 (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Together with the results from Gabra1-tm1Geh floxed mice, these findings suggest the deficits in the zolpidem-induced motor and anticonvulsive effects were not due to actions of Cre other than specific cre-loxP-mediated recombination.
Discussion
Baseline activity and the motor sedation effects of test drugs
The present studies were undertaken in order examine the role of amygdala α1-GABAARs in the motor-sedative and anticonvulsant actions of diazepam. The reduction of amygdala α1-GABAARs in LV-Cre mice produced no differences in baseline activity in the open field. Likewise, global deletion of the α1 subunit reportedly does not alter baseline locomotor activity despite the reported existence of tremors (Sur et al., 2001; Kralic et al., 2002b). However the motor-sedative effects of diazepam were blunted at a dose level of 10 mg/kg in LV-Cre mice compared with LV-GFP mice. The motor-impairing effects of zolpidem were reduced at both 0.25 mg/kg and 0.5 mg/kg in LV-Cre mice. This finding is consistent with past reports indicating that a point mutation of the α1 subunit resulted in the complete loss of diazepam- and zolpidem-induced motor impairment (Rudolph et al., 1999; Crestani et al., 2000; McKernan et al., 2000). In contrast, the motor-impairing/sedative effects of diazepam were not impaired in either α2- or α3-point mutated mice (Löw et al., 2000). In fact, when tested in a novel environment, McKernan et al. (2000) found that diazepam (3 mg/kg) increased locomotor activity considerably in α1(H101R) mice. In contrast, global deletion of the α1 subunit increased the sensitivity to the motor-impairing effects of diazepam in the open field (Kralic et al., 2002a,b). This latter effect was possibly caused by the compensatory increase in levels of α2 and/or α3 subunit peptide expression in forebrain glutamatergic cells (Zeller et al., 2008), which was notably not seen in our current in vivo study. Together, these findings indicate that the motor-sedative effects of diazepam and zolpidem are partially due to their actions on amygdala α1-GABAARs. The sedative effects which remained after test drug injections are likely mediated by α1-GABAARs located in other neuronal regions or possibly by any remaining α1-GABAARs within the amygdala. Notably, since no effects were seen when the same viruses were injected into wild-type, non-floxed mice, as discussed previously by Schmidt-Supprian and Rajewsky (2007), we can be reasonably assured that the behavioral phenotypes observed were in fact specifically due to the targeted deletion of the α1-GABAAR within the amygdala.
Seizure susceptibility to PTZ
The susceptibility to PTZ-induced convulsions was unaltered in LV-Cre mice in this study. Both LV-GFP and LV-Cre groups displayed similar latencies to myoclonic and clonic PTZ-induced seizures at 55 and 85 mg/kg dose levels. The lack of group differences in our study suggests that amygdala α1-GABAARs are not critically involved in the proconvulsant actions of PTZ. Past evidence, however, suggests that α1-GABAARs likely mediate the proconvulsant effects of BZ-site convulsants and GABAAR antagonists. In wild-type mice, BZ-site inverse agonists such DMCM and Ro 15-4513 display proconvulsant properties (Crestani et al., 2002) whereas they produce no proconvulsant properties in the α1(H101R) mice. Global deletion of the α1-GABAARs results in an increased seizure susceptibility to the GABAAR antagonist bicuculline which competes with GABA for its binding site (Kralic et al., 2002a). The lack of group differences may likely be due to the mechanisms involved in PTZ seizure initiation. In addition to the disinhibition of the GABA system (Macdonald and Barker, 1977), PTZ-induced activation of the glutamatergic system is involved in seizure initiation (Nevins and Arnolde, 1989). Thus, in the absence of α1GABAAR subtypes, glutamatergic-induced overexcitation is likely contained by the synaptic inhibition provided by remaining GABAARs. Consistent with this interpretation, the sensitivity to kainic acid-induced seizure activity is unaffected in global α1 KOs (Schneider Gasser et al., 2007). Furthermore the sensitivity to PTZ-induced seizure activity is unaltered in α1(H101R) mice (Crestani et al., 2002). Thus it appears that the role of the α1-GABAARs in mediating the proconvulsant actions is restricted to benzodiazepine site ligands. To the degree that the proconvulsant actions of bicuculline and inverse agonists are due to their action on amygdala α1-GABAARs, we would predict that the LV-Cre mice may also show an increased seizure susceptibility to these compounds. Additional studies are underway to compare the proconvulsant effects of a variety of GABAergic ligands.
Anticonvulsant effects of test drugs
In contrast to the similar susceptibility to PTZ-induced convulsions, significant group differences were detected in the anticonvulsant effects of test drugs. Compared with LV-GFP mice, LV-Cre mice showed shorter myoclonic and clonic latencies when either 0.25 or 1.0 mg/kg diazepam was administered 30 min before PTZ (85 mg/kg). The ability of zolpidem to inhibit convulsant actions of PTZ was also blunted in LV-Cre mice. At the dose of 2.5 mg/kg, LV-Cre mice showed shorter myoclonic latencies, and at both 2.5 and 4.0 mg/kg zolpidem, clonic seizure latencies were longer in LV-GFP mice compared with LV-Cre mice. Our results are consistent with reports from KO and point mutated studies. For example, in contrast to wild type mice, α1-GABAA knock-outs also show attenuated diazepam-dependent increases in seizure threshold levels (Kralic et al., 2002a). In point-mutated α1(H101R) mice, the ability of zolpidem to prevent PTZ-induced seizures is eliminated (Crestani et al., 2000), and the anticonvulsant activity of diazepam is partially lost in α1(H101R) mice (Rudolph et al., 1999). This later finding indicates that diazepam's anticonvulsant effects are also mediated by other GABAAR subtypes (α2-, α3-, α4-, α5-), and indeed can be antagonized by the benzodiazepine antagonist, flumazenil (Rudolph et al., 1999). However, Löw et al. (2000) have reported no changes in the anticonvulsant activity of diazepam in point-mutated mice with α2- or α3-GABAARs that have been rendered diazepam-insensitive. Likewise, the anticonvulsant effects of diazepam are unaffected in point-mutated α5(H101R) mice (Crestani et al. 2002) and diazepam's anticonvulsant effects are unaltered by pretreatment with RO4938581, a novel α5-GABAAR inverse agonist (Ballard et al., 2009).
Our amygdala-specific deletion studies are also consistent with previous studies. Animals treated chronically with benzodiazepines exhibited compensatory decreases in cortical α1 subunit expression, and were found to be tolerant to the anticonvulsant effects of diazepam (Tietz et al., 1999). Together, these findings suggest that α1-GABAARs in the amygdala play a role in the anticonvulsant effects of both diazepam and zolpidem and that a reduction of amygdala α1-GABAARs reduces the effectiveness of diazepam and zolpidem to inhibit PTZ-induced convulsions. The anticonvulsant effects of zolpidem that remain are likely mediated by remaining amygdala α1-GABAARs or receptors located in other brain areas involved in the development of generalized seizures. One potential site is the hippocampus. For example, it has been shown that enhancing α1-GABAAR subunit levels in the dentate gyrus of the hippocampus inhibits the development of status epilepticus in rats (Raol et al., 2006).
Specificity and efficiency of α1-GABAAR subunit deletions within the amygdala
Genetic ablation of α1-GABAAR subunits using the Cre/loxP system in combination with a Cre recombinase-expressing lentivirus allowed us to address the question of whether compensatory changes result from diminished gene expression in adult animals or from the lack of normal expression during development. In the current study, quantitative analyses of mRNA expression levels in LV-Cre mice revealed significantly less α1-GABAAR subunit expression in LV-Cre mice in comparison with LV-GFP mice, whereas no differences were identified for α2 or α3 subunits. Thus, the reduction of α1-mRNA levels does not result in increased transcription levels of α2 or α3 subunits. At the translational level, the expression of α1-GABAAR amygdala protein expression in LV-Cre-infected animals was also decreased. Thus, the partial α1-mRNA deficit in LV-Cre mice was sufficient to disrupt protein production. The α1-GABAAR mRNA expression levels in the amygdala represented 45–52% of control values whereas Western blot analysis indicated that α1-GABAAR subunit expression was reduced by only 40% relative to LV-GFP mice. It is worth noting that Western blot results were conducted on tissue homogenates tissue punches centered at the level of the amygdala and likely included noninfected tissue located outside the lateral and basolateral amygdala. The relative levels of α2- and α3-GABAAR protein were unchanged. Thus, our results indicate that Cre-mediated gene deletion of the α1-GABAA subunit in adult mice leads to no significant compensatory increases in the expression of α2- and α3-GABAAR subtypes. It seems unlikely that alterations in the motor and anticonvulsant actions of test drugs seen in the present study can be attributed to an upregulation of α2- and/or α3-GABAAR subtypes. However, it cannot be ruled out that regional changes in subtypes other than α2 and α3 may have influenced our behavioral results.
Previous studies have revealed that both global and targeted deletion of α1 subunits can lead to compensatory changes that may function to protect against the α1-GABAAR loss. For example, in vivo studies conducted with global KO strains displayed increased expression of α2 and α3 subunit peptides in brain regions where the α1 subunit is absent (Sur et al., 2001; Kralic et al., 2002a; Schneider Gasser et al., 2007), which in some cases are gradually lost in successive generations of mice (Sur et al., 2001). Likewise forebrain-specific α1 knock-out and heterozygote mutants also show increased levels of α3 subunit peptide expression (Zeller et al., 2008), indicating that the reduction of mRNA caused by the loss of a single α1 subunit allele can also cause compensatory changes. The upregulation of GABAA R subunits is not the result of increased mRNA expression and likely takes place at the level of translation or posttranslationally, at the level of receptor assembly or receptor trafficking (Bosman et al., 2005; Ogris et al., 2006). In contrast, Kralic et al. (2002a) revealed that α1 subunit gene-deficient heterozygous mice show an approximate 40% decrease in α1 subunit peptide expression yet no significant changes in α2 or α3 peptide despite significant decrease in β2/3 peptide. Hence, the vital factors which dictate when and whether such compensatory changes occur are unclear. The conditional KO approach used in this study may be particularly useful for investigating the fundamental temporal and spatial processes that govern normal brain development.
Baseline anxiety-like behavior and anxiolytic-like effects of diazepam
In the elevated plus maze, diazepam produced an anxiolytic effect as indexed by an increase in the percentage time in the open arms and percentage of open arm entries in both LV-GFP and LV-Cre mice. Vehicle-treated animals from both groups displayed similar plus maze behavior, suggesting no differences in baseline anxiety-like behavior. In support, we observed similar percentage of central time between groups in the open field test. Together the findings indicate that a reduction in amygdala α1-GABAARs does not affect baseline anxiety-like behavior or the anxiolytic-like activity of diazepam. These findings are consistent with experiments showing that diazepam effectively reduces anxiety in both α1 knock-out and α1 point mutated mice as measured in the elevated plus maze (Rudolph et al., 1999; Kralic et al., 2002a). Diazepam also reliably reduces anxiety-related behaviors in these global α1 point mutated mice as measured in the light-dark choice test (Rudolph et al., 1999). In contrast, the anxiolytic effect of diazepam was undetected in α2 point mutated mice, suggesting that α2-, but not α1-containing receptors, mediate the anxiolytic effect of diazepam (Löw et al., 2000). Indeed, subtype selective agonists that have relatively strong effects at receptors containing the α2, α3, and α5 subunits when compared with receptors containing the α1 subunits show anxiolytic-like activity. For example, SL651498, a novel pyridoindole derivative, behaves as a full agonist at GABAARs containing α2 and α3 subunits and as a partial agonist at GABAARs containing α1subunits (Griebel et al., 2001). Similar to diazepam, SL651498 induces anxiolytic-like activity in a variety of behavioral tests including the elevated plus-maze, light/dark test, and punished lever pressing (Griebel et al., 2001, 2003; Atack, 2003; Griebel et al., 2003). Likewise, the benzodiazepine-site ligand L-838,417, which preferentially activates GABAARs containing the α2, α3 and α5 subunits, also shows potent anxiolytic action in animal models of anxiety (McKernan et al., 2000). However, unlike α1-selective agonists, both L-838,417 and SL651498 produce little or no sedation at doses which produce anxiolytic-like activity (McKernan et al., 2000; Griebel et al., 2003).
It has been suggested that the compensatory increases in the α2 and/or α3 subunits of α1 KO mice may mask the lack of anxiety phenotype and altered sensitivity to BZ-induced anxiolytic-like effect (Sur et al., 2001; Kralic et al., 2002a). For example, homozygous α1 KO mice appear more sensitive to the anxiolytic effects of BZs when compared with heterozygous mice, possibly due the differential developmental upregulation of α2 and/or α3 subunits. Our results suggest that compensatory changes in the amygdala are not responsible for the normal BZ-induced anxiolytic-like effect seen in α1 KO mice.
In conclusion, these data demonstrate that the α1-GABAAR within the amygdala is required for sedative-locomotor inhibition and anticonvulsive effects, but not anxiolytic actions of benzodiazepines. We have shown that temporally and spatially restricted inducible reduction of the α1 subunit results in reduced agonist-induced motor-sedative and anticonvulsant effects without affecting baseline motor and anxiety levels or diazepam-induced anxiolytic behavior. Further research using region-, and cell type-selective manipulations of GABA receptors should further elucidate GABAergic mechanisms underlying complex behavior.
Footnotes
Support was provided by the National Institutes of Health (NIH) (DA019624 and F32 MH073389-01), the Center for Behavioral Neuroscience (National Science Foundation agreement IBN-987675), the Burroughs Wellcome Fund, and an NIH/National Center for Research Resources base grant (P51RR000165) to Yerkes National Primate Research Center. We thank Dr. Liping Mou for her excellent technical assistance with primary hippocampal neuronal cultures.
References
- Atack JR. Anxioselective compounds acting at the GABA(A) receptor benzodiazepine binding site. Curr Drug Targets CNS Neurol Disord. 2003;2:213–232. doi: 10.2174/1568007033482841. [DOI] [PubMed] [Google Scholar]
- Backus KH, Arigoni M, Drescher U, Scheurer L, Malherbe P, Möhler H, Benson JA. Stoichiometry of a recombinant GABAA receptor deduced from mutation-induced rectification. Neuroreport. 1993;5:285–288. doi: 10.1097/00001756-199312000-00026. [DOI] [PubMed] [Google Scholar]
- Ballard TM, Knoflach F, Prinssen E, Borroni E, Vivian JA, Basile J, Gasser R, Moreau JL, Wettstein JG, Buettelmann B, Knust H, Thomas AW, Trube G, Hernandez MC. RO4938581, a novel cognitive enhancer acting at GABA(A) alpha5 subunit-containing receptors. Psychopharmacology. 2009;202:207–223. doi: 10.1007/s00213-008-1357-7. [DOI] [PubMed] [Google Scholar]
- Barnard EA, Skolnick P, Olsen RW, Mohler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, Langer SZ. International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acid A receptors: classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- Benson JA, Löw K, Keist R, Mohler H, Rudolph U. Pharmacology of recombinant gamma-aminobutyric acidA receptors rendered diazepam-insensitive by point-mutated alpha-subunits. FEBS Lett. 1998;431:400–404. doi: 10.1016/s0014-5793(98)00803-5. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Heinen K, Spijker S, Brussaard AB. Mice lacking the major adult GABAA receptor subtype have normal number of synapses, but retain juvenile IPSC kinetics until adulthood. J Neurophysiol. 2005;94:338–346. doi: 10.1152/jn.00084.2005. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997 Feb;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Burt DR, Kamatchi GL. GABAA receptor subtypes: from pharmacology to molecular biology. FASEB J. 1991;5:2916–2923. doi: 10.1096/fasebj.5.14.1661244. [DOI] [PubMed] [Google Scholar]
- Crestani F, Martin JR, Möhler H, Rudolph U. Resolving differences in GABAA receptor mutant mouse studies. Nat Neurosci. 2000a;3:1059. doi: 10.1038/80553. [DOI] [PubMed] [Google Scholar]
- Crestani F, Martin JR, Möhler H, Rudolph U. Mechanism of action of the hypnotic zolpidem in vivo. Br J Pharmacol. 2000b;131:1251–1254. doi: 10.1038/sj.bjp.0703717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Assandri R, Täuber M, Martin JR, Rudolph U. Contribution of the alpha1-GABA(A) receptor subtype to the pharmacological actions of benzodiazepine site inverse agonists. Neuropharmacology. 2002;43:679–684. doi: 10.1016/s0028-3908(02)00159-4. [DOI] [PubMed] [Google Scholar]
- Da Cunha C, Wolfman C, Levi de Stein M, Ruschel AC, Izquierdo I, Medina JH. Anxiogenic effects of the intraamygdala injection of flumazenil, a benzodiazepine receptor antagonist. Funct Neurol. 1992;7:401–405. [PubMed] [Google Scholar]
- Depoortere H, Zivkovic B, Lloyd KG, Sanger DJ, Perrault G, Langer SZ, Bartholini G. Zolpidem, a novel nonbenzodiazepine hypnotic. I. Neuropharmacological and behavioral effects. J Pharmacol Exp Ther. 1986;237:649–658. [PubMed] [Google Scholar]
- Drover DR. Comparative pharmacokinetics and pharmacodynamics of short-acting hypnosedatives: zaleplon, zolpidem and zopiclone. Clin Pharmacokinet. 2004;43:227–238. doi: 10.2165/00003088-200443040-00002. [DOI] [PubMed] [Google Scholar]
- Engin E, Treit D. The effects of intra-cerebral drug infusions on animals' unconditioned fear reactions: a systematic review. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1399–1419. doi: 10.1016/j.pnpbp.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Green S, Vale AL. Role of amygdaloid nuclei in the anxiolytic effects of benzodiazepines in rats. Behav Pharmacol. 1992;3:261–264. [PubMed] [Google Scholar]
- Griebel G, Sanger DJ, Perrault G. The use of the rat elevated plus-maze to discriminate between non-selective and BZ-1 (omega 1) selective, benzodiazepine receptor ligands. Psychopharmacology (Berl) 1996;124:245–254. doi: 10.1007/BF02246664. [DOI] [PubMed] [Google Scholar]
- Griebel G, Perrault G, Simiand J, Cohen C, Granger P, Decobert M, Françon D, Avenet P, Depoortere H, Tan S, Oblin A, Schoemaker H, Evanno Y, Sevrin M, George P, Scatton B. SL651498: an anxioselective compound with functional selectivity for alpha2- and alpha3-containing gamma-aminobutyric acid(A) (GABA(A)) receptors. J Pharmacol Exp Ther. 2001;298:753–768. [PubMed] [Google Scholar]
- Griebel G, Perrault G, Simiand J, Cohen C, Granger P, Depoortere H, Françon D, Avenet P, Schoemaker H, Evanno Y, Sevrin M, George P, Scatton B. SL651498, a GABAA receptor agonist with subtype-selective efficacy, as a potential treatment for generalized anxiety disorder and muscle spasms. CNS Drug Rev. 2003;9:3–20. doi: 10.1111/j.1527-3458.2003.tb00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadingham KL, Garrett EM, Wafford KA, Bain C, Heavens RP, Sirinathsinghji DJ, Whiting PJ. Cloning of cDNAs encoding the human gamma-aminobutyric acid type A receptor alpha 6 subunit and characterization of the pharmacology of alpha 6-containing receptors. Mol Pharmacol. 1996;49:253–259. [PubMed] [Google Scholar]
- Heldt SA, Ressler KJ. Localized injections of midazolam into the amygdala and hippocampus induce differential changes in anxiolytic-like motor activity in mice. Behav Pharmacol. 2006;17:349–356. doi: 10.1097/01.fbp.0000224386.86615.e0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, Ressler KJ. Training-induced changes in the expression of GABA(A)-associated genes in the amygdala after the acquisition and extinction of Pavlovian fear. Eur J Neurosci. 2007;26:3631–3644. doi: 10.1111/j.1460-9568.2007.05970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevers W, Lüddens H. The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol Neurobiol. 1998;18:35–86. doi: 10.1007/BF02741459. [DOI] [PubMed] [Google Scholar]
- Hodges H, Green S, Glenn B. Evidence that the amygdala is involved in benzodiazepine and serotonergic effects on punished responding but not on discrimination. Psychopharmacology (Berl) 1987;92:491–504. doi: 10.1007/BF00176484. [DOI] [PubMed] [Google Scholar]
- Hogg S. A review of the validity and variability of the elevated plus-maze as an animal model of anxiety. Pharmacol Biochem Behav. 1996;54:21–30. doi: 10.1016/0091-3057(95)02126-4. [DOI] [PubMed] [Google Scholar]
- Jasnow AM, Rainnie DG, Maguschak KA, Chhatwal JP, Ressler KJ. Construction of cell-type specific promoter lentiviruses for optically guiding electrophysiological recordings and for targeted gene delivery. Methods Mol Biol. 2009;515:199–213. doi: 10.1007/978-1-59745-559-6_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaputlu I, Uzbay T. L-NAME inhibits pentylenetetrazole and strychnine-induced seizures in mice. Brain Res. 1997;753:98–101. doi: 10.1016/s0006-8993(96)01496-5. [DOI] [PubMed] [Google Scholar]
- Kaufmann WA, Humpel C, Alheid GF, Marksteiner J. Compartmentation of alpha 1 and alpha 2 GABA(A) receptor subunits within rat extended amygdala: implications for benzodiazepine action. Brain Res. 2003;964:91–99. doi: 10.1016/s0006-8993(02)04082-9. [DOI] [PubMed] [Google Scholar]
- Kopp C, Rudolph U, Löw K, Tobler I. Modulation of rhythmic brain activity by diazepam: GABA(A) receptor subtype and state specificity. Proc Natl Acad Sci U S A. 2004;101:3674–3679. doi: 10.1073/pnas.0306975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralic JE, Korpi ER, O'Buckley TK, Homanics GE, Morrow AL. Molecular and pharmacological characterization of GABAA receptor alpha 1 subunit knockout mice. J Pharmacol Exp Ther. 2002a;302:1037–1045. doi: 10.1124/jpet.102.036665. [DOI] [PubMed] [Google Scholar]
- Kralic JE, O'Buckley TK, Khisti RT, Hodge CW, Homanics GE, Morrow AL. GABAA receptor alpha-1 subunit deletion alters receptor subtype assembly, pharmacological and behavioral responses to benzodiazepines and zolpidem. Neuropharmacology. 2002b;43:685–694. doi: 10.1016/s0028-3908(02)00174-0. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Sidler C, Parpan F, Homanics GE, Morrow AL, Fritschy JM. Compensatory alteration of inhibitory synaptic circuits in cerebellum and thalamus of gamma-aminobutyric acid type A receptor alpha1 subunit knockout mice. J Comp Neurol. 2006;495:408–421. doi: 10.1002/cne.20866. [DOI] [PubMed] [Google Scholar]
- Löscher W, Schmidt D. Strategies in antiepileptic drug development: is rational drug design superior to random screening and structural variation? Epilepsy Res. 1994;17:95–134. doi: 10.1016/0920-1211(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Löw K, Crestani F, Keist R, Benke D, Brünig I, Benson JA, Fritschy JM, Rülicke T, Bluethmann H, Möhler H, Rudolph U. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Barker JL. Pentylenetetrazol and penicillin are selective antagonists of GABA-mediated post-synaptic inhibition in cultured mammalian neurones. Nature. 1977;267:720–721. doi: 10.1038/267720a0. [DOI] [PubMed] [Google Scholar]
- Marowsky A, Fritschy JM, Vogt KE. Functional mapping of GABA A receptor subtypes in the amygdala. Eur J Neurosci. 2004;20:1281–1289. doi: 10.1111/j.1460-9568.2004.03574.x. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ, Castro JL, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- Mehta AK, Ticku MK. An update on GABAA receptors. Brain Res Brain Res Rev. 1999;29:196–217. doi: 10.1016/s0165-0173(98)00052-6. [DOI] [PubMed] [Google Scholar]
- Meldrum B. Do preclinical seizure models preselect certain adverse effects of antiepileptic drugs. Epilepsy Res. 2002;50:33–40. doi: 10.1016/s0920-1211(02)00066-9. [DOI] [PubMed] [Google Scholar]
- Möhler H. GABA(A) receptor diversity and pharmacology. Cell Tissue Res. 2006;326:505–516. doi: 10.1007/s00441-006-0284-3. [DOI] [PubMed] [Google Scholar]
- Möhler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- Nevins ME, Arnolde SM. A comparison of the anticonvulsant effects of competitive and non-competitive antagonists of the N-methyl-d-aspartate receptor. Brain Res. 1989;503:1–4. doi: 10.1016/0006-8993(89)91695-8. [DOI] [PubMed] [Google Scholar]
- Nunes-de-Souza RL, Canto-de-Souza A, da-Costa M, Fornari RV, Graeff FG, Pelá IR. Anxiety-induced antinociception in mice: effects of systemic and intra-amygdala administration of 8-OH-DPAT and midazolam. Psychopharmacology (Berl) 2000;150:300–310. doi: 10.1007/s002130000428. [DOI] [PubMed] [Google Scholar]
- Nutt D. GABAA receptors: subtypes, regional distribution, and function. J Clin Sleep Med. 2006;2:S7–11. [PubMed] [Google Scholar]
- Ogris W, Lehner R, Fuchs K, Furtmüller B, Höger H, Homanics GE, Sieghart W. Investigation of the abundance and subunit composition of GABAA receptor subtypes in the cerebellum of alpha1-subunit-deficient mice. J Neurochem. 2006;96:136–147. doi: 10.1111/j.1471-4159.2005.03509.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. New York: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Pellow S, File SE. Anxiolytic and anxiogenic drug effects on exploratory activity in an elevated plus-maze: a novel test of anxiety in the rat. Pharmacol Biochem Behav. 1986;24:525–529. doi: 10.1016/0091-3057(86)90552-6. [DOI] [PubMed] [Google Scholar]
- Pesold C, Treit D. The central and basolateral amygdala differentially mediate the anxiolytic effects of benzodiazepines. Brain Res. 1995;671:213–221. doi: 10.1016/0006-8993(94)01318-c. [DOI] [PubMed] [Google Scholar]
- Petersen EN, Braestrup C, Scheel-Krüger J. Evidence that the anticonflict effect of midazolam in amygdala is mediated by the specific benzodiazepine receptors. Neurosci Lett. 1985;53:285–288. doi: 10.1016/0304-3940(85)90552-x. [DOI] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Pritchett DB, Seeburg PH. Gamma-aminobutyric acidA receptor alpha 5-subunit creates novel type II benzodiazepine receptor pharmacology. J Neurochem. 1990;54:1802–1804. doi: 10.1111/j.1471-4159.1990.tb01237.x. [DOI] [PubMed] [Google Scholar]
- Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463:3–33. doi: 10.1016/s0014-2999(03)01272-x. [DOI] [PubMed] [Google Scholar]
- Raol YH, Lund IV, Bandyopadhyay S, Zhang G, Roberts DS, Wolfe JH, Russek SJ, Brooks-Kayal AR. Enhancing GABAA receptor {alpha}1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci. 2006;26:11342–11346. doi: 10.1523/JNEUROSCI.3329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, Davis M. Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci. 2002;22:7892–7902. doi: 10.1523/JNEUROSCI.22-18-07892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers RJ, Dalvi A. Anxiety, defence and the elevated plus-maze. Neurosci Biobehav Rev. 1997;21:801–810. doi: 10.1016/s0149-7634(96)00058-9. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Möhler H. Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol. 2004;44:475–498. doi: 10.1146/annurev.pharmtox.44.101802.121429. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Möhler H. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999a;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A. Anxiolytic effects of chlordiazepoxide blocked by injection of GABAA and benzodiazepine receptor antagonists in the region of the anterior basolateral amygdala of rats. Biol Psychiatry. 1995;37:473–476. doi: 10.1016/0006-3223(94)00183-4. [DOI] [PubMed] [Google Scholar]
- Sanger DJ. The pharmacology and mechanisms of action of new generation, non-benzodiazepine hypnotic agents. CNS Drugs. 2004;18(Suppl 1):S9–S15. doi: 10.2165/00023210-200418001-00004. [DOI] [PubMed] [Google Scholar]
- Sanger DJ, Morel E, Perrault G. Comparison of the pharmacological profiles of the hypnotic drugs, zaleplon and zolpidem. Eur J Pharmacol. 1996;313:35–42. doi: 10.1016/0014-2999(96)00510-9. [DOI] [PubMed] [Google Scholar]
- Saviæ MM, Obradoviæ DI, Ugresiæ ND, Bokonjiæ DR. Memory effects of benzodiazepines: memory stages and types versus binding-site subtypes. Neural Plast. 2005;12:289–298. doi: 10.1155/NP.2005.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007 Jul;8:665–668. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- Schneider Gasser EM, Duveau V, Prenosil GA, Fritschy JM. Reorganization of GABAergic circuits maintains GABAA receptor-mediated transmission onto CA1 interneurons in alpha1-subunit-null mice. Eur J Neurosci. 2007;25:3287–3304. doi: 10.1111/j.1460-9568.2007.05558.x. [DOI] [PubMed] [Google Scholar]
- Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol. 2006;54:231–263. doi: 10.1016/s1054-3589(06)54010-4. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Sperk G. Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr Top Med Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Höger H, Adamiker D. Structure and subunit composition of GABAA receptors. Neurochem Int. 1999;34:379–385. doi: 10.1016/s0197-0186(99)00045-5. [DOI] [PubMed] [Google Scholar]
- Smith TA. Type A gamma-aminobutyric acid (GABAA) receptor subunits and benzodiazepine binding: significance to clinical syndromes and their treatment. Br J Biomed Sci. 2001;58:111–121. [PubMed] [Google Scholar]
- Sur C, Wafford KA, Reynolds DS, Hadingham KL, Bromidge F, Macaulay A, Collinson N, O'Meara G, Howell O, Newman R, Myers J, Atack JR, Dawson GR, McKernan RM, Whiting PJ, Rosahl TW. Loss of the major GABAA receptor subtype in the brain is not lethal in mice. J Neurosci. 2001;21:3409–3418. doi: 10.1523/JNEUROSCI.21-10-03409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuber L, Wätjens F, Jensen LH. Ligands for the benzodiazepine binding site—a survey. Curr Pharm Des. 1999;5:317–343. [PubMed] [Google Scholar]
- Tietz EI, Huang X, Chen S, Ferencak WF., 3rd Temporal and regional regulation of α1, β2 and β3, but not α2, α4, α5, α6, β1 or γ2 GABAA receptor subunit messenger RNAs following one-week oral flurazepam administration. Neuroscience. 1999;91:327–341. doi: 10.1016/s0306-4522(98)00516-8. [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Ikawa M, Verma IM. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci U S A. 2003;100:1844–1848. doi: 10.1073/pnas.0437912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Velísková J, Löscher W, Moshé SL. Regional and age specific effects of zolpidem microinfusions in the substantia nigra on seizures. Epilepsy Res. 1998;30:107–114. doi: 10.1016/s0920-1211(97)00096-x. [DOI] [PubMed] [Google Scholar]
- Vicini S, Ferguson C, Prybylowski K, Kralic J, Morrow AL, Homanics GE. GABA(A) receptor alpha1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci. 2001;21:3009–3016. doi: 10.1523/JNEUROSCI.21-09-03009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel HG, Vogel WH. New York: Springer; 2002. Drug discovery and evaluation: pharmacological assays. [Google Scholar]
- Wafford KA, Thompson SA, Thomas D, Sikela J, Wilcox AS, Whiting PJ. Functional characterization of human gamma-aminobutyric acidA receptors containing the alpha 4 subunit. Mol Pharmacol. 1996;50:670–678. [PubMed] [Google Scholar]
- Wisden W, Herb A, Wieland H, Keinänen K, Lüddens H, Seeburg PH. Cloning, pharmacological characteristics and expression pattern of the rat GABAA receptor alpha 4 subunit. FEBS Lett. 1991;289:227–230. doi: 10.1016/0014-5793(91)81076-k. [DOI] [PubMed] [Google Scholar]
- Zeller A, Crestani F, Camenisch I, Iwasato T, Itohara S, Fritschy JM, Rudolph U. Cortical glutamatergic neurons mediate the motor sedative action of diazepam. Mol Pharmacol. 2008;73:282–291. doi: 10.1124/mol.107.038828. [DOI] [PubMed] [Google Scholar]