

Abstract
The ability to regenerate damaged tissues is a common characteristic of multicellular organisms. We report a role for apoptotic cell death in promoting wound healing and tissue regeneration in mice. Apoptotic cells released growth signals that stimulated the proliferation of progenitor or stem cells. Key players in this process were caspases 3 and 7, proteases activated during the execution phase of apoptosis that contribute to cell death. Mice lacking either of these caspases were deficient in skin wound healing and in liver regeneration. Prostaglandin E2, a promoter of stem or progenitor cell proliferation and tissue regeneration, acted downstream of the caspases. We propose to call the pathway by which executioner caspases in apoptotic cells promote wound healing and tissue regeneration in multicellular organisms the “Phoenix Rising” pathway.
Introduction
The ability to repair damaged tissues is essential for metazoan organisms (1,2). Some organisms (for example, salamanders) possess the remarkable ability to completely regenerate entire amputated limbs (3,4,5). In contrast, other organisms, such as humans, can only partially replace damaged organs (for example, liver regeneration) (6,7,8). Wound healing and tissue regeneration are complicated processes involving the coordinated efforts by many different cell types. The initial response in mammals suffering tissue injuries is generally considered an inflammatory one. Subsequently, new tissue formation and tissue remodeling complete the wound healing process. Because inflammatory cells were thought to be the “first responders” at the site of tissue injury, a long-held notion was that cells of the immune system, especially macrophages and neutrophils, were crucial in initiating and coordinating the wound healing/tissue regeneration process (9,10). However, observation of intact wound healing in PU.1 knockout mice, which lack macrophages and neutrophils, showed that neither cell type is necessary for skin excision wound healing (11). Therefore, the initiating cellular and molecular events in wound healing and tissue regeneration remain unclear.
The stem cells in and around damaged tissues play a critical role in wound healing and tissue regeneration (1). It was generally assumed factors released from damaged tissues mobilize and recruit these stem and progenitor cells to the damaged site, where they proliferate, differentiate, and eventually replace the damaged tissue. The released factors were thought to be arise during the inflammatory process ensuing from the initial tissue damage. Previous studies have focused on the roles of immunoeffector cells, such as macrophages, that are activated by tissue injury and that secrete cytokines and growth factors that promote wound healing and tissue regeneration (1,12). Growth factors, such ashepatocyte growth factor (HGF)(13), fibroblast growth factors FGF7, FGF10, FGF22(14,15), and transforming growth factor β (TGF-β) (16,17) play important roles wound healing and tissue regeneration, as do small molecule hormones, such as acetylcholine(18), catecholamine (19,20), and polyunsaturated fatty acids(21). However, it is unclear which cell types and molecular mechanisms initiate the signaling cascades responsible for wound healing.
In this study, we hypothesized that dying cells in the wounded tissues send signals to stimulate the proliferation of stem or progenitor cells that starts the process of tissue regeneration and wound healing.
Results
Stimulation of stem and progenitor cells by dying cells in vitro and in vivo
We used irradiated mouse embryonic fibroblasts (MEFs) to simulate dying cells in wounded tissues and determined whether these cells stimulated the proliferation of co-cultured firefly luciferase (Fluc)-labeled stem or progenitor cells (see fig. S1A for validation the intensity of luciferase corresponds to cell numbers and fig. S1B for verification that the change in cell numbers is due to proliferation, not inhibition of cell death, of the stem or progenitor cells). In the co-culture system, dying MEFs (4 × 104) significantly stimulated the proliferation of a small number (200) of Fluc-labeled murine epidermal keratinocyte progenitor (EKP) cells, neural stem cells (NSC), or mesenchymal stem cells (MSC) when compared with Fluc-labeled cells cultured alone or with nonirradiated, live MEF cells (p<0.05) (Fig. 1A). We observed similar growth-promoting properties from various lethally irradiated human and mouse cells (Fig. S1C), suggesting that growth-promoting activity towards stem or progenitor cells is a general property of dying mammalian cells.
Fig. 1. Stimulation of stem or progenitor cell proliferation by dying cells.

(A) Lethally irradiated MEF cells were mixed with Fluc-labeled mouse EKP, NSC, or MSC cells and Fluc-labeled cell proliferation was quantified with bioluminescence imaging (see fig. S1). (B) EKP-Fluc cells were injected subcutaneously into the hind legs of nude mice either alone (left leg) or with lethally irradiated MEF cells (right leg). The EKP-Fluc cells were then imaged. The relative luciferase activity normalized against day 0, right after injection, is graphed (P<0.01, t test, n=5 total, days 1, 3, 5). (C) About 2 × 105 EKP-Fluc cells were injected subcutaneously into left (unirradiated) or right (irradiated with 18 Gy) hind legs of nude mice and monitored for growth. Signals at later time points were normalized to the signals on day 0 (P<0.05 the entire course of experiment one-way ANOVA test, n=5 total,).
We also confirmed the ability of dying cells to support the proliferation of progenitor cells in vivo. When 2 × 105 lethally irradiated MEFs were co-injected with 1000 nonirradiated Fluc-labeled mouse EKP cells into nude mice (subcutaneously into the right hind legs), the intensity of the luciferase signal, representing cell proliferation, was significantly increased compared to the luciferase signal of EKP-Fluc cells injected alone (subcutaneously into the left hind legs) Fig. 1B). In fact, proliferation was only observed for EKP-Fluc cells that were co-injected with irradiated MEF cells; for EKP-Fluc cells injected alone, no proliferation was observed. In both groups, the luciferase signal weakened over time, presumably due to a generally nonpermissive host tissue environment.
The proliferation-promoting effects of dying cells was also evident from experiments in which EKP-Fluc cells were injected subcutaneously into lethally irradiated (18 Gy) or nonirradiated hind legs of nude mice (Fig. 1C). EKP-Fluc cells injected into irradiated hind legs showed a significant initial proliferation (increase in luciferase intensity) in the first week and, although the luciferase signals subsequently weakened, the cells were still detectable beyond day 103 after injection (Fig. 1C), indicating long-term engraftment. In comparison, EKP-Fluc cells injected into nonirradiated left hind legs showed minimal proliferation (Fig. 1C) and rapid signal attenuation to undetectable. The difference in the luciferase intensity between EKP-Fluc-injected irradiated (right hind legs) and the nonirradiated (left hind legs) were statistically significant throughout the entire period of observation (P<0.05, one-way ANOVA test, n=5).
These results underscored the importance of tissue damage and cell death in stimulating the proliferation of stem or progenitor cells. They also agree with a report showing proliferation of individual myogenic stem cells in radiation- or drug-treated mouse tissues (22), and are consistent with long-established bone marrow transplant procedures in which donor hematopoietic stem cells only engraft into hosts that have been ablated of their original bone marrow cells through irradiation or chemical treatment (23).
A key role for caspases 3 and 7 in cell death-induced proliferation of stem and progenitor cells
Because of the requirement for regenerative capability among different tissues, the factor(s) regulating cell death-stimulated stem and progenitor cell proliferation may be derived from a common process operating in dying cells. Therefore, we hypothesized that the molecular machinery involved in apoptosis, one of the major mechanisms of cell death in many different types of mammalian cells, might play a key role in initiating the paracrine cascades that lea to the stimulation of stem and progenitor cell proliferation.
We examined the possibility that apoptotic caspases contribute to cell death-stimulated progenitor and stem cell proliferation, because these enzymes are essential proteases that function as the nexus of the apoptotic process (24,25,26,27,28,29,30). We used caspase 3-deficient (Casp3−/−), caspase 7-deficient (Casp7−/−), or with the doubly deficient Casp3−/− Casp7−/− MEFs (31,32). Irradiation of these cells reduced clonogenicity of these cells to a similar extent as wild type cells, despite inducing significantly less apoptosis markers, indicating cells die through other mechanisms. We evaluated the proliferation of various Fluc-labeled stem or progenitor cells when cocultured with lethally irradiated caspase 3-deficient (Casp3−/−), caspase 7-deficient (Casp7−/−), or with the doubly deficient Casp3−/−Casp7−/− MEFs (31,32). Compared with wild-type fibroblasts, both lethally irradiated Casp3−/−(Fig. 2A) and Casp7−/− (fig. S2A) fibroblasts were less effective at stimulating stem or progenitor cell proliferation. The doubly deficient MEFs were even less effective at promoting stem or progenitor cell proliferation than were either of the singly caspase-deficient MEFs, consistent with the overlapping functions of caspase 3 and 7 (Fig. 2A).
Fig. 2. Critical role of caspase 3 in cell death stimulation of stem and progenitor cell proliferation.

(A) Proliferation of 200 Fluc-labeled stem or progenitor cells mixed with lethally irradiated MEF cells were monitored in 24-well plates serially with bioluminescence imaging. The error bars represents SEM. In all three groups, the differences between the wild-type and Casp3−/− groups are statistically significant (P<0.05, t-test, n=4 independent samples). The differences between the Casp3−/− groups and the Casp3−/−Casp7−/− groups are also significant (P<0.05, t-test, n=4). (B) EKP-Fluc cells injected alone or with lethally irradiated MEFs were monitored with bioluminencence imaging. Relative luciferase activities from co-injected EKP-Fluc (in the right hind legs) normalized against those from cells injected alone (in the left hind legs) are graphed. The difference between the two groups are significant (P<0.01, one-way ANOVA test, n=5,).
(C) Growth of EKP-Flu cells in wild-type and Casp3−/− mice. Relative luciferase activity in the irradiated legs normalized against those in the non-irradiated legs (P<0.05, n=5 each in wild type and knockout mice, one-way ANOVA test). (D) MEF cells induced tissue growth in a modified DIVAA assay. Host tissue growth into silicone cylinders was quantified by FITC-lectin staining of endothelial cells in the vasculature. Signals were normalized against phosphate buffered saline (PBS) control. Cylinders were prepared with PBS, wild-type MEFs, or Casp3−/− MEFs mixed with BME prior to implantation. The differences in tissue growth into the cylinders containing the wild-type MEFs versus the PBS control and versus the knockout MEFs were significant (*P<0.05,t test, n=6 cylinders in 6 different mice for each group).
We also examined the importance of caspase 3 and caspase 7 in vivo for promoting stem or progenitor cell proliferation with three paradigms. The first experiment was similar to that described for Figure 1B. 2 × 105 lethally irradiated wild-type or Casp3−/− MEFs were co-injected with 1000 EKP-Fluc cells into the right hindlimbs of mice, and compared to the left hindlimbs, which were injected with the same pair of cells only nonirradiated. The animals co-injected with the wild-type MEFs exhibited a significantly greater increase in luciferase intensity, indicating proliferation of the EKP-Fluc cells, compared to the animals co-injected with the Casp3−/− MEFs (p<0.05, n=5), which showed minimal proliferation (Fig. 2B). Similar results were also obtained when lethally irradiated wild-type and Casp7−/− MEF cells were co-injected with EKP-Fluc cells, although the difference in the ability to stimulate proliferation was smaller than that observed between Casp3−/− and wild type MEF cells (fig. S2B).
A second in vivo model that we used to examine the importance of caspases in the promotion of stem or progenitor cell proliferation was a radiation-induced wound healing model with transgenic Casp3−/− and Casp7−/− knockout mice. These mice have decresed tissue apoptosis in reponse to various stimuli(31,32). Furthermore, Casp 3−/− mice have profound developmental defect in the brain due to reduced brain cell apoptosis(31) About 1000 EKP-Fluc cells were injected subcutaneously into irradiated (right) and nonirradiated (left) hind legs of mice. The irradiated tissues in wild-type mice (in C57BL/6 background) showed significantly more capacity to promote EKP-Fluc proliferation (Fig. 2C) compared with irradiated tissues in Casp3−/− mice (also in C57BL/6 background), which showed minimal growth-promoting ability towards the injected EKP-Luc cells during the observation period. Similar results were also obtained for Casp7−/− mice (fig. S2C), indicating that caspase 7 also participates in the same process.
We evaluated the importance of caspase 3 in an experimental model of tissue regeneration, the directed in vivo angiogenesis assay (DIVAA). We placed lethally irradiated wild-type or Casp3−/− MEFs (mixed with basement membrane extract) into silicone cylinders and implanted the cylinders subcutaneously into nude mice. After two weeks, we sacrificed the mice, removed the implanted cylinders, and quantified host tissue growth by measuring vascular growth into the cylinders. Whereas irradiated wild-type MEF cells induced significant vascular growth (P<0.05), irradiated Casp3−/− MEF cells induced minimal vascular growth (Fig. 2D). In fact, the vascular growth in cylinders containing irradiated Casp3−/− MEF cells was not significantly different from that of the negative control cylinders containing only PBS (phosphate-buffered saline).
Evidence for the involvement of caspases 3 and 7 in skin wound healing
To determine the roles of caspases under more physiologically relevant circumstances, we performed two additional experiments in vivo. In the first experiment (Fig. 3A and B), we examined if the effect of caspase 3 or caspase 7 deficiency on the healing excision wounds in mouse skin. Excision wounds were created in wild-type, Casp3−/−, or Casp7−/− mice in the C57BL/6 background through punch biopsy (Fig. 3 and fig. S4). The rates of wound healing in these mice were quantified by measuring wound diameters. Casp3−/− mice exhibited reduced rate of repair of dorsal skin wounds when compared with that of wild-type mice (Fig. 3A). All of the wounds in wild-type mice were healed by day 9. In contrast, the time for complete wound closure in Casp3−/− mice was 14 days. We conducted immunofluorescence analyses to visualize the rate of wound closure by staining the skin at the wound site with an antibody against cytokeratin 14, which is a marker for skin epithelial cells, a key cell responsible for basal layer formation during epidermal closure. It is clear from the immunofluorescence staining (Fig. 3B) that skin epithelium recovery around the wound was markedly attenuated in caspase 3-deficient mice. By day 6, when there was already complete epidermal closure in wild-type mice, as demonstrated by continuous cytokeratin 14 staining across the wound gap, there was little sign of closure in the wound gap of caspase 3-deficient mice. Staining for cytokeratin 6, which is a marker for keratinocytes and indicates epidermal cell migration, showed a similar reduction in the progression of wound closure in the Casp3−/− animals (fig. S3). The exact pattern of staining was different than that of cytokeratin 14.
Fig. 3. Key roles for caspase 3 in skin wound healing.

(A) Healing of 4-mm circular excision wounds in C57BL/6-derived wild-type and Casp3−/− mice. Wound healing was monitored through periodic measurement with a caliper. Data are plotted as the changes in the average areas +SEM) of wounds from the two groups of 8 mice with two wounds each. (P<0.01, one-way ANOVA test, n=16 wounds in each group). (B) Full-thickness skin samples containing entire wound sites were biopsied from wild-type and Casp3−/− mice at different time points after punch biopsy. Paraffin-embedded samples were sectioned and analyzed with antibodies against cytokeratin 14 (K14), a marker for skin epithelial cellsHematoxylin and eosin (H&E) staining shows basic tissue and cellular structures. The scale bars represent 500 µm. (C) BrdU staining in skin biopsies containing full-thickness wounds. The scale bars represent 200 µm. BrdU staining was quantified by counting and averaging 6 randomly chosen 100x fields on 6 mice. The error bars represent SEM. (P<0.01, t test, n=6).
Staining with BrdU, which was administered to mice shortly before sacrificing, confirmed that cellular proliferation was significantly attenuated in wounded skin tissue of Casp3−/− mice (Fig. 3C). Because BrdU incorporation is a direct indicator of DNA synthesis during cellular proliferation, these data are consistent with a defect in Casp3−/− mice of the cellular proliferation that should occur during normal skin wound healing. Quantification of representative fields indicated that there was at least a 10-fold reduction in BrdU incorporation in Casp3−/− mice (Fig. 3C) on the first day after skin biopsy. Experiments with Casp7−/− mice showed a very similar defect in skin wound healing and cellular proliferation (BrdU incorporation) (fig. S4).
Evidence for the involvement of caspases 3 and 7 in liver regeneration
We examined the importance of caspases on liver regeneration in a well-established partial hepatectomy model in mice (33). Two of the four lobes of liver (the median and left lateral lobes, which account for about 2/3 of total liver weight) were surgically removed from wild-type, Casp3−/−, or Casp7−/− mice and the mice were allowed to recover (Fig. 4 and fig. S5). When measured in terms of liver weight, wild-type mice regenerated lost liver tissue effectively, reaching about 78% of pre-surgery weight by day 10 (Fig. 4A). In comparison, liver weight in Casp3−/− mice remained at only 60% of pre-surgery level (Fig. 4A) and in Casp7−/− remained at 57% (fig. S5A), indicating a substantial defect in liver regeneration. Quantification of BrdU staining indicated that, when compared with wild-type mice, there was at least a 50% reduction in cellular proliferation in Casp3−/− mice (Fig. 4B) as well as in Casp7−/− mice (fig. S5B).
Fig. 4. Key roles of caspase 3 in liver regeneration after partial hepatectomy.

(A) Assessment of liver regeneration after partial hepatectomy by measuring liver weight. The ratios of the average weight (+/− SEM) of regenerated livers (from mice that have undergone partial hepatectomy) against un-resected liver weight are graphed. (P<0.01 on and after day 2, t test, n=5). (B) Cell proliferation (BrdU-positive cells) at different time points after partial hepatectomy. Representative images are shown in the upper panels (scale bars, 100 µm). Nuclei were stained with DAPI. Lower panel shows quantitation of BrdU-positive cells by counting the average number of BrdU-positive cells in 4 randomly chosen 200x fields. Average and SEM are plotted. The difference between the two groups is statistically significant on days 2 and 5 (P<0.01, t test, n=4 mice in each group).
The role of calcium-independent phospholipase A2 in cell death-induced tissue regeneration
Thus far, our in vivo results provide evidence for the involvement of caspase 3 and 7 in wound healing and tissue regeneration. We wanted to determine the downstream factors by which caspase 3 and 7 stimulated cellular proliferation and the tissue regeneration process. It seemed likely that the dying cells released factors that promoted stem or progenitor cell proliferation, thus we focused on caspase-activated pathways that would release growth-promoting factors. One candidate is calcium-independent phospholipase A2 (iPLA2), whose activity is enhanced by caspase 3 or 7 cleavage and the activation of which increases the synthesis and release of arachidonic acid and lysophosphocholine from apoptotic cells (34,35,36). Arachidonic acid is of particular interest, because it is a precursor to prostaglandin E2 (PGE2) (37,38), a known stimulator of stem cell proliferation (39,40,41,42,43,44), tissue regeneration, and wound healing (45,46,47). Therefore, we determined the rate of arachidonic acid release from wild-type and Casp3−/− MEFs. Ionizing radiation, which induces increasing amount of apoptosis in wild type MEF cells from 24 hrs post-irradiation, induced a significant amount of arachidonic acid release by 48 hours from wild-type MEFs (P<0.01 n=3) (Fig. 5A, left panel). However, this release was significantly less for irradiated Casp3−/− MEFs, which confirms the previous studies (34,35,36). Western blot analysis confirmed a caspase 3-dependent cleavage of iPLA2 (fig. S6A), which is also consistent with previous observations (34,35,36).
Fig. 5. A role for caspase 3-mediated activation of iPLA2 in cell death stimulation of stem or progenitor cell proliferation.

(A) Left panel: Arachidonic acid (AA) release from wild-type and Casp3−/− MEF cells with or without irradiation. The difference in AA release between irradiated wild-type and casp3−/− MEF cells are statistically significant at 48 hrs (t-test, n=3, P<0.01). The graph was plotted as relative levels of [3H]-archidonic acid in the supernatant, which were derived by normalizing [3H]-AA levels at different time points against those in the supernatant taken right before irradiation. Right panel: Prostaglandin E2 (PGE2) concentration in the supernatants of nontreated and irradiated wild-type and Casp3−/− MEF cultures. Data are plotted as the average and SEM and statistically significant differences are noted (t test, n=3). (B) Role of iPLA2 in cell death-mediated stimulation of EKP cell proliferation in vitro. Data are plotted as the average and SEM (n=5, t-test). (C)The importance of iPLA2 in tissue growth in a modified DIVAA assay. Top panel: Quantification of host tissue vasculature from cylinders containing various MEF cells. All the data were normalized against the PBS (phosphate-buffered saline) control group, which serves as the negative control. Statistically significant differences are noted (XXX test, n=X). Lower panel: Photographs of representative cylinders after removal from the mice. Tissue growth (yellowish color) into the cylinder is apparent. The red smears are red blood cells that are part of the host tissue that has grown into the cylinder.
Because arachidonic acid is a precursor for prostaglandin E2, we examined PGE2 concentration in the supernatants of wild-type and Casp3−/− MEF cultures before and after irradiation. Our results showed a strong correlation of supernatant PGE2 concentration with cellular caspase 3 status (Fig. 5A, right panel). The supernatant from wild-type MEF cultures had a higher basal concentration of PGE2 than did the cultures of Casp3−/− MEFs and showed a strong increase after radiation. In addition, radiation-induced concentrations of PGE2 in the supernatant from Casp3−/− MEF cultures were significantly lower than those from irradiated wild-type cell cultures. A role for iPLA2 in mediating radiation-induced increase in PGE2 concentration was clearly demonstrated in two different ways. Short-hairpin RNA (shRNA)-mediated knockdown of iPLA2 significantly reduced induced concentrations of PGE2 in wild-type MEF cultures (Figure 5A, right panel). (The effectiveness of the shRNA in knocking down iPLA2 is shown in fig. S6B). Overexpression of a truncated iPLA2 gene, which encodes a truncated protein (Fig. S6B) that is identical to a putative, constitutively active caspase 3 or 7 cleavage product of iPLA2, significantly restored both the basal concentration and the irradiation-induced increase in the concentration of PGE2 in Casp3−/− MEF cultures (Fig. 5A, right panel). These results provide evidence that caspase 3, and not caspase 7, regulates PGE2 concentrations through the activation of iPLA2 in dying cells.
Exposure of EKP-FLuc cells to either PGE2 or lysophophostidylcholine (LPC), which are both products of activated iPLA2, showed that PGE2 stimulated cell proliferation, but that LPC did not (Fig. S6C). Because PGE2 has been implicated in stem cell mobilization and tissue regeneration, we examined whether caspase 3-mediated iPLA2 activation was important for stimulating stem and progenitor cell proliferation. We evaluated wild-type MEF cells transduced with a shRNA-encoding gene against iPLA2 and Casp3−/− MEF cells transduced with a truncated, constitutively active version of iPLA2 (that was identical to a previously identified caspase-generated fragment, see fig.e S6B) for the ability to support proliferation of EKP-Fluc cells. Reducing iPLA2 abundance in lethally irradiated wild-type MEFs significantly diminished their ability to support EKP-Fluc proliferation in vitro, whereas transduction of the constitutively active iPLA2 into Casp3−/− MEFs significantly increased their capacity to support EKP-Fluc proliferation when lethally irradiated (Fig. 5B).
A role for iPLA2 in tissue regeneration was also obtained with a modified DIVAA tissue regeneration assay in nude mice (Fig. 5C). In this assay, silicone cylinders containing cells or growth factors are placed into the host and the growth of the host tissue the implanted silicone cylinders is quantified. Knocking down iPLA2 significantly attenuated the ability of irradiated wild-type MEF to induce host vascular growth into MEF cell-embedded silicone cylinders, whereas exogenous expression of a constitutively active, truncated iPLA2 significantly enhanced the ability of Casp3−/− MEF cells to stimulate host tissue growth into the cylinders. Therefore, these experiments provide evidence for a pathway involving caspase 3-activated iPLA2 in stem or progenitor cell proliferation and tissue regeneration.
Discussion
Our study reveals a key role for programmed cell death in tissue regeneration in mice. Furthermore, we found that caspases 3 and 7, the “executioner” proteases that are instruments of cellular death and that function at the terminal stages of apoptosis, hold the key for mobilizing tissue stem and progenitor cells and promoting tissue regeneration. Caspases 3 and 7 cleaved and activated iPLA2 to ultimately trigger the production of the growth signal PGE2, thus showing that these executioner caspases not only contribute to the death process, but also participate in the production of paracrine signals.
Examples of the importance of apoptosis in tissue regeneration have been reported in earlier studies. For example, tail regeneration in Xenopus laevis requires apoptosis (48), as does tissue regeneration in planaria (flat worms) (49). However, the molecular mechanisms by which apoptosis contributed to regeneration were not determined. A study demonstrating the importance of apoptosis in Hydra head regeneration (50) showed that positive, growth signals (in this case, Wnt3) were generated from apoptotic cells. However, the molecular mechanism for Wnt3 generation is not clear. It is, therefore, interesting to note a relationship between PGE2 and Wnt signaling pathways in regulating developmental specification and stem cell growth in mammalian cells (51). From these studies and our own results, it appears that the involvement of apoptosis-caspase-PGE2 (+ Wnt?) pathways in wound healing and tissue regeneration is evolutionarily conserved.
Because our study suggests a key role for cell death in promoting tissue regeneration in mammalian cells, we propose to name this mechanism the “Phoenix Rising” pathway of tissue regeneration (Fig. 6). We believe that it is a fundamental biological process that metazoan organisms employ to sense tissue damage and initiate repair, regeneration, or both. The existence of such a pathway is a manifestation of the inextricable link between the Yin and Yang of cellular life and death in metazoan organisms.
Fig. 6. A schematic representation of the “Phoenix Rising” pathway of cell death-induced tissue regeneration pathway.

In damaged tissues, apoptotic cells activate caspase 3 and 7 through either the intrinsic pathways, involving APAF and caspase 9, or extrinsic pathways, involving caspase 8. Activated caspase 3 and 7 subsequently cleave and activate iPLA2, which generates arachidonic acid. Arachidonic acid is then converted into PGH2 by cyclooxygenases 1 and 2 (Cox1&2). PGE2 synthase converts PGH2 into PGE2, which stimulates stem cell proliferation and tissue regeneration.
Materials and Methods
Cell lines and culture conditions
EKP cells derived from C57BL/6 mice were purchased from Millipore (Billerica, MA). NSCs, derived from Balb/C mice, were purchased from American Type Culture Collection (Manassas, VA). MSCs were obtained from freshly isolated bone marrow following established protocols (MSCs, Technical Manual, Stem Cell Technologies, Vancouver, BC, Canada). Wild-type, Casp3−/, −Casp7−/−, and Casp3−/−Casp7−/− double knockout MEFs were obtained from Dr. Richard Flavell of Yale University (New Haven, CT). EPK cells were maintained in Epidermal Keratinocyte medium from Millipore (catalog#CNT-02). NSC cells were grown in neural stem cell medium (Millipore, catalog#SCM003). Wild-type and caspase-knockout MEF cells were cultured in DMEM medium with 5% FBS.
Mouse strains
Nude mice were obtained from National Cancer Institute (Bethesda, MD). Wild-type C57BL/6, −Casp3−/−, and −Casp7−/− mice, and enhanced green fluorescent protein (EGFP)-expressing transgenic mouse (all in C57/BL6 background) were obtained from the Jackson Laboratory (Bar Harbor, Maine, US).The transgenic mice were deposited by Dr. Richard Flavell. All animal procedures were approved by the University of Colorado Denver Institutional Animal Use and Care Committee. Mice were kept in the vivarium located on the Anschutz Medical Campus of UC Denver, Aurora, CO.
Genetic manipulation of the cell strains
For many experiments, we used the pLEX lentiviral vector system (52) from Open Biosystems (Huntsville, AL) for transduction of various genes into MEF cells and various stem or progenitor cells. The genes transduced with pLEX include (i) firefly luciferase gene, which was transferred from the plasmid pGL4.31-luc2 from Promega (Madison, Wisoconsin); (ii) a truncated version of the mouse iPLA2, which was amplified through RT-PCR from murine mRNA with the following primers:
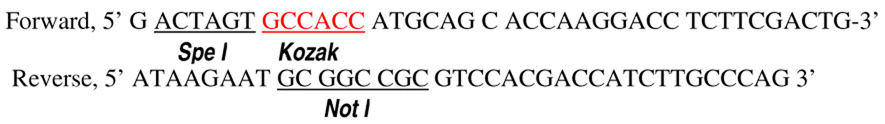
We used the Pfx polymerase (Invitrogen, Carlsbad, CA) for the PCR amplification. The amplified fragment encodes aa453–679, which is equivalent to aa514–733 of human iPLA2(35,36), of murine iPLA2 (accession# NM-016915). This fragment is a constitutively active, caspase-cleavage product (34,35,36). The fragment was cloned into the Spe I and Not sites of a modified pLEX plasmid. The modified pLEX plasmid has an influenza hemagglutinin (HA) tag inserted between Not I and Mlu I (two of the unique restriction sites in pLEX) so that genes inserted into the NotI site can be fused with the HA tag.
We obtained lentiviral vectors encoding shRNA-encoding minigenes targeted against the murine iPLA2 gene from Open Biosystems (Huntsville, AL). These minigenes were carried in the pLKO.1 lentiviral vectors system. The most effective one had the following targeting sequence: 5’GCGTATGAAGGACGAGGTGTTTCTCGAGAAACACCTCGTCCTTCATACGTTTTTTG-3’ (catalog#Rmmu534-NM-016915 from Open Biosystems), in bold red are the sense and antisense targeting sequences and in underlined blue is loop sequence). We followed the manufacturer’s instructions to produce live recombinant lentiviral vectors in 293T cells.
Bioluminescence imaging of cultured cells
Proliferation of Fluc-labeled stem or progenitor cells on lethally irradiated feeder cells was tracked though bioluminescence imaging. Imaging of the cells was performed with an IVIS200 instrument from Caliper Life Sciences (Hopkinton, MA). Luciferin (obtained from Caliper Life Sciences) was added to the mediaat a concentration of 150 µg/ml. After 5–10 minutes of incubation, cells cultured in Petri dishes or multiwell plates were imaged in the instrument. Quantification of signal strength was performed with manufacturer supplied software.
Bioluminescence imaging of cells implanted in mice
Growth of Fluc-labeled stem or progenitor cells in vivo was followed through noninvasive bioluminescence imaging with the IVIS200 instrument. Mice were injected with 150 mg/kg of D-luciferin in 200 µl of PBS and imaged 10 minutes later. The time between injection and imaging was kept constant among different batches of mice.
Irradiation of cells and mice
X-ray irradiation of cells and mice were carried out in a RS-2000 Biological Irradiator [Rad Source Corporation (Atlanta, GA)] located in the vivarium of University of Colorado Denver Anschutz Medical Campus.. The dose rate for the machine is about 1 Gy/minute.
Measurement of arachidonic acid release
To measure arachidonic release, we plated cells in 6-well plates at a density of 2.0 × 105 cells/well. About 1.0 µCi of [3H]-archidonic acid (GE Healthcare Life Sciences) was added to the cells cultured in about 1 ml of serum-free DMEM with 0.5 mg/ml lipid-free bovine serum albumin (Sigma Chemical Co., St Louis, USA). After 16 hrs, the cells were washed with fresh media 3 times and incubated with 3 ml of DMEM supplemented with 5% serum. After 5 hrs, when archidonic acid in the supernatant reaches a steady state level, the cells were exposed to 8 Gy of x-rays. Supernatants were then removed at 4, 24, and 48 hrs from the cells and counted with a scintillation counter for quantification of [3H]-arachidonic acid.
ELISA measurement of PGE2
To evaluate the PGE2 secretion from cells, we plated about 4 × 104 cells/well in 24-well plates. Cells were cultured in 1 ml of DMEM supplemented with 2% fetal bovine serum. Cells were then irradiated with x-rays (10 Gy). Supernatant from the cells was removed before and 48 hrs after cellular irradiation. PGE2 in the supernatants was measured with an ELISA kit from R&D Systems (Minneapolis, MN, USA).
Skin excision wound healing assay
To characterize the rate of wound healing in different strains of mice, punch biopsies were created in the dorsal skin of mice. The biopsy procedure was carried out using VisiPunch devices from Huot Instruments (Menomonee Falls, WI, USA). Usually, two 4-mm excision wounds were created in the dorsal skin of each mouse. Monitoring of the rate of healing was performed by measuring the diameter of the wounds with a caliper every other day. For histological analysis, skin biopsies containing the full-thickness wounds were taken from mice sacrificed at different time points after generating the excision wounds. About 60 minutes before the sacrifice, mice were injected with BrdU at 50 mg/Kg. The tissue samples were paraffin-embedded at the Histology Core in the Dept of Dermatology at University of Colorado Denver.
For BrdU immunofluorescence staining, an Alexa Fluor 594 conjugated anti-BrdU, mouse monoclonal antibody from Invitrogen (Carlsbad, CA) was used at a dilution of 1:20. To quantify the number of cells positive for BrdU staining, four randomly chosen 100X fields were counted.
To analyze skin epithelial cells and keratinocytes through immunofluorescence staining, antibodies against cytokeratin 14 and cytokeratin 6, respectively, were used. These were rabbit polyclonal antibodies produced in-house and described previously (53).
Partial hepatectomy-liver regeneration assay
To quantify liver regeneration, we adopted a published protocol (33). Briefly, mice (8–10 weeks old) were anesthetized with isofluorane. After restraining the mouse, a 3-cm incision was created in the midline of abdominal skin and muscle to expose the xiphoid process. The median and left lateral lobes were tied around the top of the lobes using 4-0 silk surgical thread. The lobes were then removed surgically. This removes about 2/3 of the total liver weight. After suturing the abdomen closed, the mice were placed on a warm pad to recover. At different times after hepatectomy, mice were sacrificed for evaluation. About 60 minutes before the sacrifice, BrdU (50mg/kg) were injected i.p. into the mice to label proliferating cells. Regeneration rate of the liver was quantified with two methods.The weight of the two lobes of liver left intact in mice were determined and normalized to the weight of the same two lobes right after the initial surgery to derive a relative liver weight. In theory, the range should be from 1.0 (right after surgery) to 3.0 (100% recovery). Alternatively, immunofluorescence staining of BrdU-positive cells was performed in sections of paraffin-embedded liver tissues. Liver regeneration was then quantified through enumeration BrdU-positive cells in 4 randomly chosen 200x fields.
Modified DIVAA assay for tissue regeneration
To quantify the ability of different cells to induce tissue regeneration, we adapted the DIVAA™ from Trevigen (Gaithersburg, MD). The original purpose of this kit was to measure the ability of various growth factors to induced angiogenesis in vivo. The basic procedure involves mixing growth factors at different concentrations with basement membrane extract (BME) and loading the mixture (in a total volume of 20 µl) into silicone cylinders called the “angioreactor”. In our experiments, instead of mixing growth factors with the BME, we mixed various MEFs with BME. For each angioreactor, we used about 2 × 105 cells in a final volume of 20 µl. We surgically implanted up to four the angioreactors subcutaneously into each nude mouse. Because the angioreactor has only one sealed end, the host tissue and vasculature can grow into it. Two weeks after implantation, host mice were sacrificed and the angioreactors were removed. The contents of the angioreactors were then transferred to microtubes and their endothelial cell content was quantified by FITC-lectin staining following manufacturer (Trevigen)’s instructions.
Supplementary Material
Acknowledgements
We thank Dr. Richard Flavell of Yale University for providing the MEFs with caspase 3 and 7 deficiencies. We also want to thank the anonymous reviewers for the excellent suggestions/criticisms.
Funding: This study was supported in part by grant EB001882 (to C–Y Li) from US National Institute of Bioimaging and Bioengineering, grant CA131408 and CA CA136748 from the US National Cancer Institute (to C–Y Li), and grant NNX09AH19G (to C–Y Li) from NASA Space Radiation Biology Research Program. Qian Huang was supported by Grant 2010CB529900 from the National Basic Research Project of China and Outstanding Young Scientist grants 30325043 & 30428015 from China National Natural Science Foundation.
Footnotes
Competing interests: The authors declare no financial interest in the published study.
References and Notes
- 1.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 2.Birnbaum KD, Sanchez Alvarado A. Slicing across kingdoms: regeneration in plants and animals. Cell. 2008;132:697–710. doi: 10.1016/j.cell.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slack JM, Savage S. Regeneration of mirror symmetrical limbs in the axolotl. Cell. 1978;14:1–8. doi: 10.1016/0092-8674(78)90295-7. [DOI] [PubMed] [Google Scholar]
- 4.Brockes JP, Kumar A. Plasticity and reprogramming of differentiated cells in amphibian regeneration. Nat Rev Mol Cell Biol. 2002;3:566–574. doi: 10.1038/nrm881. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez Alvarado A. Planarian regeneration: its end is its beginning. Cell. 2006;124:241–245. doi: 10.1016/j.cell.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Sandgren EP, et al. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell. 1991;66:245–256. doi: 10.1016/0092-8674(91)90615-6. [DOI] [PubMed] [Google Scholar]
- 7.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 8.Zaret KS. Genetic programming of liver and pancreas progenitors: lessons for stem-cell differentiation. Nat Rev Genet. 2008;9:329–340. doi: 10.1038/nrg2318. [DOI] [PubMed] [Google Scholar]
- 9.Grose R, Werner S. Wound-healing studies in transgenic and knockout mice. Mol Biotechnol. 2004;28:147–166. doi: 10.1385/MB:28:2:147. [DOI] [PubMed] [Google Scholar]
- 10.Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Martin P, et al. Wound healing in the PU.1 null mouse--tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;13:1122–1128. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- 12.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chmielowiec J, et al. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177:151–162. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner S, et al. The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science. 1994;266:819–822. doi: 10.1126/science.7973639. [DOI] [PubMed] [Google Scholar]
- 15.Braun S, auf dem Keller U, Steiling H, Werner S. Fibroblast growth factors in epithelial repair and cytoprotection. Philos Trans R Soc Lond B Biol Sci. 2004;359:753–757. doi: 10.1098/rstb.2004.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashcroft GS, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- 17.Amendt C, Mann A, Schirmacher P, Blessing M. Resistance of keratinocytes to TGFbeta-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J Cell Sci. 2002;115:2189–2198. doi: 10.1242/jcs.115.10.2189. [DOI] [PubMed] [Google Scholar]
- 18.Martin JW, Mousa SS, Shaker O, Mousa SA. The multiple faces of nicotine and its implications in tissue and wound repair. Exp Dermatol. 2009;18:497–505. doi: 10.1111/j.1600-0625.2009.00854.x. [DOI] [PubMed] [Google Scholar]
- 19.Gosain A, Jones SB, Shankar R, Gamelli RL, DiPietro LA. Norepinephrine modulates the inflammatory and proliferative phases of wound healing. J Trauma. 2006;60:736–744. doi: 10.1097/01.ta.0000196802.91829.cc. [DOI] [PubMed] [Google Scholar]
- 20.Pullar CE, Isseroff RR. The beta 2-adrenergic receptor activates pro-migratory and pro-proliferative pathways in dermal fibroblasts via divergent mechanisms. J Cell Sci. 2006;119:592–602. doi: 10.1242/jcs.02772. [DOI] [PubMed] [Google Scholar]
- 21.Michalik L, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Starzl TE, Zinkernagel RM. Transplantation tolerance from a historical perspective. Nat Rev Immunol. 2001;1:233–239. doi: 10.1038/35105088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horvitz HR, Worms life. and death (Nobel lecture) Chembiochem. 2003;4:697–711. doi: 10.1002/cbic.200300614. [DOI] [PubMed] [Google Scholar]
- 25.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 26.Yuan J, Horvitz HR. A first insight into the molecular mechanisms of apoptosis. Cell. 2004;116:S53–S56. doi: 10.1016/s0092-8674(04)00028-5. 51 p following S59. [DOI] [PubMed] [Google Scholar]
- 27.Horvitz HR, Shaham S, Hengartner MO. The genetics of programmed cell death in the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol. 1994;59:377–385. doi: 10.1101/sqb.1994.059.01.042. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 29.Li P, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 30.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 31.Kuida K, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 32.Lakhani SA, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 34.Atsumi G, et al. Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem. 1998;273:13870–13877. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 35.Lauber K, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, et al. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281:29357–29368. doi: 10.1074/jbc.M513105200. [DOI] [PubMed] [Google Scholar]
- 37.Langenbach R, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 38.Morham SG, et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 39.Feher I, Gidali J. Prostaglandin E2 as stimulator of haemopoietic stem cell proliferation. Nature. 1974;247:550–551. doi: 10.1038/247550a0. [DOI] [PubMed] [Google Scholar]
- 40.Fisher JW, Radtke HW, Jubiz W, Nelson PK, Burdowski A. Prostaglandins activation of erythropoietin production and erythroid progenitor cells. Exp Hematol. 1980;8 Suppl 8:65–89. [PubMed] [Google Scholar]
- 41.Hanson WR, Thomas C. 16, 16-dimethyl prostaglandin E2 increases survival of murine intestinal stem cells when given before photon radiation. Radiat Res. 1983;96:393–398. [PubMed] [Google Scholar]
- 42.Kriegler AB, Bradley TR, Hodgson GS. The effect of prostaglandins E1 and E2 on macrophage progenitor cells with high proliferative potential in mouse bone marrow in vitro. Blood. 1984;63:1348–1352. [PubMed] [Google Scholar]
- 43.Liou JY, et al. Cyclooxygenase-2-derived prostaglandin e2 protects mouse embryonic stem cells from apoptosis. Stem Cells. 2007;25:1096–1103. doi: 10.1634/stemcells.2006-0505. [DOI] [PubMed] [Google Scholar]
- 44.North TE, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–1011. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhrer I, Kuzmits R, Linkesch W, Ludwig H. Topical PGE2 enhances healing of chemotherapy-associated mucosal lesions. Lancet. 1986;1:623. doi: 10.1016/s0140-6736(86)92850-3. [DOI] [PubMed] [Google Scholar]
- 46.Jumblatt MM, Willer SS. Corneal endothelial repair. Regulation of prostaglandin E2 synthesis. Invest Ophthalmol Vis Sci. 1996;37:1294–1301. [PubMed] [Google Scholar]
- 47.Paralkar VM, et al. An EP2 receptor-selective prostaglandin E2 agonist induces bone healing. Proc Natl Acad Sci U S A. 2003;100:6736–6740. doi: 10.1073/pnas.1037343100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tseng AS, Adams DS, Qiu D, Koustubhan P, Levin M. Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev Biol. 2007;301:62–69. doi: 10.1016/j.ydbio.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hwang JS, Kobayashi C, Agata K, Ikeo K, Gojobori T. Detection of apoptosis during planarian regeneration by the expression of apoptosis-related genes and TUNEL assay. Gene. 2004;333:15–25. doi: 10.1016/j.gene.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 50.Chera S, et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell. 2009;17:279–289. doi: 10.1016/j.devcel.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 51.Goessling W, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kappes JC, Wu X. Safety considerations in vector development. Somat Cell Mol Genet. 2001;26:147–158. doi: 10.1023/a:1021082815013. [DOI] [PubMed] [Google Scholar]
- 53.Roop DR, et al. Synthetic peptides corresponding to keratin subunits elicit highly specific antibodies. J Biol Chem. 1984;259:8037–8040. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.