

Abstract
Membrane lipids are active contributors to cell function as key mediators in signaling pathways of inflammation, apoptosis, migration, and proliferation. Recent work on multimolecular lipid structures suggests a critical role for lipid organization in regulating the function of both lipids and proteins. Of particular interest in this context are the polyphosphoinositides (PPI’s), specifically phosphatidylinositol (4,5) bisphosphate (PIP2). The cellular functions of PIP2 are numerous but the factors controlling targeting of PIP2 to specific proteins and organization of PIP2 in the inner leaflet of the plasma membrane remain poorly understood. To analyze the organization of PIP2 in a simplified planar system, we used Langmuir monolayers to study the effects of subphase conditions on monolayers of purified naturally derived PIP2 and other anionic or zwitterionic phospholipids. We report a significant molecular area expanding effect of subphase monovalent salts on PIP2 at biologically relevant surface densities. This effect is shown to be specific to PIP2 and independent of subphase pH. Chaotropic agents (e.g. salts, trehalose, urea, temperature) that disrupt water structure and the ability of water to mediate intermolecular hydrogen bonding also specifically expanded PIP2 monolayers. These results suggest a combination of water-mediated hydrogen bonding and headgroup charge in determining the organization of PIP2, and may provide an explanation for the unique functionality of PIP2 compared to other anionic phospholipids.
Introduction
Phosphatidyl inositol 4,5-bisphosphate (PI(4,5)P2 or PIP2) is uniquely import among membrane-bound lipids as a regulator of cell function. Despite its structural simplicity and relative scarcity in cells (<1% of all membrane lipids 1, 2), PIP2 is a critical mediator of a variety of cellular processes. The most widely recognized function of PIP2 is as a substrate for hydrolytic cleavage by phospholipase C (PLC) into diacyl glycerol (DAG) and inositol trisphosphate (IP3), which are effectors of protein kinase C and calcium signaling, respectively (reviewed in 3) and for phosphorylation by PI 3-kinase 4 to produce the signaling lipid PIP3. PIP2 itself has also signaling functions and is implicated in the regulation of proteins responsible for the maintenance and dynamics of the actin cytoskeleton 5, 6, attachment of these cytoskeletal structures to the actin cytoskeleton 7 regulation of membrane trafficking 8 and attachment 9, ion channel activity 10, and synaptic vesicle fusion 11.
How a small (~1kD) membrane-bound molecule such as PIP2 can have so many specific effects on a large number of structurally diverse binding partners is not known. Several lines of evidence suggest that control of PIP2 signaling comes not only from enzymatic regulation of its abundance, but also from regulation of its spatial organization. Some of the first evidence supporting this hypothesis was the finding that significant fractions of PIP2 in cell membranes were unavailable for PLC hydrolysis 12, 13, as well as the dependence of PLC activity in vitro on PIP2 concentration in monolayers 12. Detergent-resistant membrane fractions, the putative membrane-localized signaling complexes termed lipid rafts, were shown to be enriched in PIP2 14, 15. Imaging methods employing GFP-tagged PIP2 -binding domains 9, 16 and fluorescent anti- PIP2 antibodies 14, 17 have likewise confirmed the possibility of structurally distinct PIP2 fractions. Although the existence of these domains and their functional significance has been be disputed 18, 19, spatial segregation of PIP2 is a plausible for regulation of this critical lipid messenger.
Despite the mounting evidence for the existence of spatially distinct pools of PIP2, the mechanism for the formation of such domains has yet to be defined. Several studies demonstrate interaction between unstructured polybasic domains of proteins such as MARCKS and multiple PIP2 molecules, allowing concentration of this lipid through non-specific, electrostatic attraction 2, 14, 20–23 and shielding of the lipid from other potential cellular targets. This hypothesis views the interactions between neighboring PIP2 molecules as dominated by electrostatic repulsion between the charge-dense poly-anionic headgroups. On the other hand, recent experiments with liposomes containing PIP2 argue for the existence of PIP2 domains, independent of proteins, due to attractive interactions through hydrogen bonding 24, 25.
Here, we present results of experiments on monolayers of pure naturally-derived PIP2 that argue strongly for the existence of attractive interactions between adjacent PIP2 molecules that oppose the electrostatic repulsion of the anionic headgroups. Comparison of area-pressure isotherms of PIP2 with other acidic phospholipids over a range of subphase conditions reveal the extent to which electrostatics effects contribute to membrane surface pressure. The effects of several uncharged chaotropes preclude a strictly electrostatic interpretation and highlight the importance of hydrogen bonding or lipid hydration in maintaining the physical state of PIP2 in planar systems. Finally, the specificity of the observed effects over other anionic and inositol-based lipids suggests that PI(4,5)P2 may have unique ability to form hydrogen-bonded networks as a mechanism for its structural and functional sequestration.
Methods
Lipids and reagents
Natural lipids (bovine liver L-α-phosphatidylinositol, porcine brain L-α-phosphatidylinositol-4-phosphate, porcine brain L-α-phosphatidylserine, and porcine brain L-α-phosphatidylinositol-4,5-bisphosphate) were purchased as 1 mg/ml solutions from Avanti (Alabaster, AL) and stored at −20°C. Synthetic PIP2 analogs (dioleoyl phosphatidylinositol (x, y) bisphosphate) were purchased as dried 0.1 mg aliquots, dissolved in the supplied solvent and stored at −20°C. The concentrations of the lipid solutions were confirmed initially with phosphate analysis following acid digestion of organic components 26 and subsequently by comparing to the measured area per lipid molecule. Subphase reagents HEPES, EDTA, D-trehalose, and urea were purchased from Sigma (St. Louis, MO) and CsCl, NaCl, KCl, LiCl, MgCl2, CaCl2 were purchased from Fisher (Hampton, NH).
Pressure-area isotherms
Monolayer subphases were prepared with 10 mM HEPES, 0.1 mM EDTA, pH 7.4 dissolved in 18.2 MΩ ddH2O. For the low pH experiments, the buffer was 10mM sodium phosphate. 25–30 mL of subphase solution were filtered through a 0.2 μm syringe filter (Sigma) and introduced to a MicroTroughX Langmuir trough (Kibron Inc. Helsinki, Finland). Approximately 7 nmol of lipid was withdrawn through a septum from a container stored at −20°C to prevent solvent evaporation and deposited slowly on the subphase interface. After a 10 min stabilization of the monolayer, the lipids were compressed at 15 Å2/molecule/min by moving the barriers of the trough using a microstepping motor. The monolayer surface pressure was monitored with a surface probe using the Wilhelmy method 26 and the FilmWare software package (Kibron). Both the low amount of lipids and the slow deposition rate were critical parameters for reproducibility of monolayer isotherms. Monolayers of pure PIP2 could not be compressed past ~37 mN/m in our experiments because the Teflon coated barriers of the microtrough wetted at high surface PIP2 concentrations; hence the collapse pressure of the PIP2 monolayers could not be measured. Temperature of the subphase was maintained using a circulating water bath.
Time-course experiment
Approximately 0.01 nmol of PIP2 was deposited on the interface of 1 mL of filtered subphase added to a single well of a multiwell plate (Kibron). Lipid was added until the surface pressure increased to between 15–20 mN/m. The lipid was left to stabilize for ~30 min, until the surface pressure was stable (within 1 mN/m) for several minutes. 50 μL of 5 M NaCl were added to the subphase through an injection port and the change in surface pressure was measured as a function of time.
Results
Phase behavior of pure, natural PIP2
The relationship between the surface pressure (π) and molecular area of pure naturally-derived PIP2 was investigated by compressing monolayers of PIP2 from 250 to 50 Å2/molecule and observing the effect of compression on the surface pressure of the interface. Average isotherms for 10 separate trials are shown in Fig. 1a. As expected from the known composition of the acyl chains of pure PIP2 (~50% unsaturated, 33% arachadonic acid), these isotherms show a smooth, monotonic increase in surface pressure as the molecular area is decreased. No phase transitions were observed for monolayers of PIP2 under any of the conditions used in these experiments. The average area of PIP2 at a surface pressure corresponding to physiological conditions (~30 mN/m 27) was 73.1±3.0 Å2/molecule, somewhat larger than published values for SAPC (65 Å2) 28, which is to be expected from the added bulk of the sugar headgroup and electrostatic repulsions. Despite the size and relatively high charge density of the PIP2 headgroup at physiological pH, this molecule readily forms tightly compressed monolayers, as opposed to collapsing into aqueous micellar structures at higher surface pressures. Hysterysis of the monolayers due to loss of lipids through barrier leakage or monolayer collapse was minimal under all conditions, similar to control lipids (data not shown).
Figure 1.
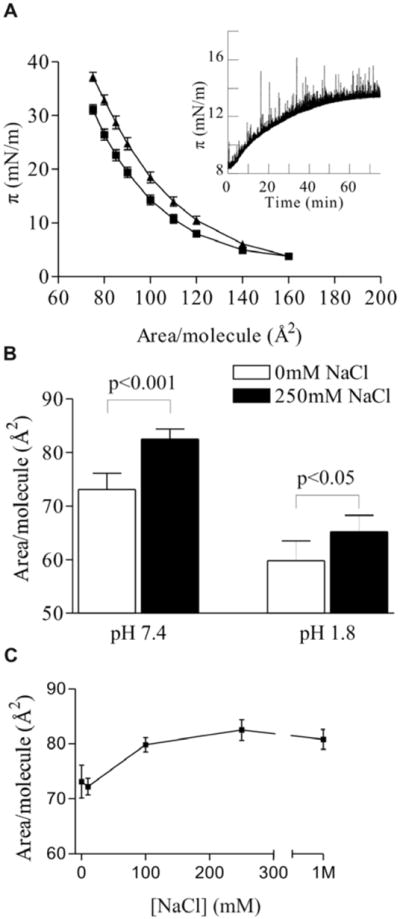
Expanding effect of NaCl on PIP2 monolayers. (A) π-A isotherms with 0 mM (squares) and 250 mM NaCl (triangles); (inset) change in surface pressure at constant area/molecule upon subphase injection of 250mM NaCl (at time = 0).
Expanding effect of increased ionic strength on monolayers of PIP2
To investigate the effect of ionic strength on the behavior of PIP2 monolayers, π-A isotherms were taken with varying concentrations of NaCl in the subphase. Addition of NaCl significantly expanded the monolayers at all surface pressures above 5 mN/m (Fig. 1a). This response was also observed upon addition of NaCl to the subphase of a preformed PIP2 monolayer. At constant molecular area, the surface pressure increased after addition of 250 mM NaCl with a magnitude commensurate to that observed in the isotherm experiments, on a diffusion-dependent time scale (Fig. 1a inset). At physiologically realistic surface pressure (π = 30 mN/m), the area per PIP2 molecule was increased by 13% to 82.5 Å2/molecule (Fig. 1b). Quantification of the dose response of this effect reveals that the effect saturates at approximately 200 mM NaCl and shows significant variation within the range of physiologically-relevant salt concentrations (Fig. 1c).
To rule out the possibility of an unexpected electrostatic mechanism (e.g. counter-ion cloud repulsion) causing the monolayer expansion, the effect of 250 mM NaCl was measured on another charged lipid (L-α PS) using the same conditions as employed in the PIP2 experiments. Monolayers of PS were not affected in the same way as those of PIP2, instead showing a very slight contraction in response to increased subphase ionic strength (Fig. 2a).
Figure 2.

Specificity of salt-expanding effect to PIP2. Area per molecule of (A) L-α PIP2 and L-α PS; and (B) L-α PIP2, L-α PI(4)P and L-α PI on HEPES-buffered subphase, pH 7.4, 30°C at π = 30mN/m. Mean ± SE, n=4.
Although the expanding effect of NaCl was specific for PIP2 over PS, this control did not rule out the possibility of the involvement of the acyl chains (those of natural PS are likely to be different from those of PIP2) or the inositol ring in inducing the expansion of the monolayer. To determine whether an inositol-based headgroup was the cause of the observed phenomenon, and at the same time control for acyl chain composition, the isotherm experiments above were repeated using two other inositol-based lipids, phosphatidyl inositol 4-phosphate (L-α PI(4)P) and phosphatidyl inositol (L- α PI). Because these molecules are precursors for enzymatic PIP2 production in cells, they have similar or identical fatty acid compositions as PIP2, and only differ in the degree of phosphate substitution on the inositol ring. In monolayer experiments, neither of these lipids showed a significant expansion in response to increased concentration of NaCl, although the monophosphate PI(4)P exhibited the same trend as the bisphosphate PIP2, suggesting a similar, although quantitatively smaller, effect (Fig. 2b). These data suggest that the mechanism involved in NaCl-induced expansion of PIP2 monolayers is specific to PIP2 over other anionic, as well as other inositol-based, lipids.
In addition to the specificity of the expanding effect of NaCl on PIP2 compared to other anionic phospholipids, the effect is also PIP2 isomer dependent. Quantification of the molecular areas of synthetic PIP2 analogs substituted at different positions on the inositol ring (3 and 5, 4 and 5, 3 and 4) shows that not only are the molecular areas dependent on the positions of the phosphate, but also that the magnitude of the NaCl-induced expansion is affected by the placement of the phosphomonoesters in the three different isomers (Fig. 5a). Direct comparison of this expansion reveals the greatest difference between 0 and 250 mM NaCl for PI(3,5)P2 (~22 Å2/PIP2), followed by PI(4,5)P2 (11 Å2/PIP2) and PI(3,4)P2 (5 Å2/PIP2), and that the differences between the specific PIP2 isomers is highly significant (p<0.001).
Figure 5.

PIP2 isomer specificity of subphase NaCl expansion effect. (A) Area per molecule at π = 30 mN/m of DO- PIP2 isomers on HEPES-buffered subphase. Mean ± SE, n=7. (B) Difference in area per molecule of DO- PIP2 isomers between 250 mM NaCl and no subphase NaCl. The isomer dependence of the NaCl effect was measured to be significant to p = 0.0001 by two-way ANOVA. (C) Conceptual cartoon of the intermolecular interactions between PIP2 molecules. In absence of chaotropic agents (green ellipses), PIP2 molecules form water-mediated hydrogen-bonded networks. Upon addition of chaotropes, networks are broken, and electrostatic repulsion between charged phosphates induces expansion of the monolayer.
Effects of different counterions
To determine the ion specificity of the expanding effect of monovalent salts on PIP2 monolayers, the effects of other cationic counterions were tested. At 250 mM, all monovalent cations tested (i.e. Na+, K+, Li+, Cs+) showed similar, statistically significant expansion of the PIP2 monolayers, with the magnitude of the effect directly related to the charge density of the ion, i.e. Li+ > Na+ > K+ ~ Cs+ (Fig. 3a). These data show that the expanding effect of NaCl is not unique, but that the magnitude of the expansion depends on the charge density of the monovalent couterion. The charge-density dependence observed here differs from that reported for salt-induced expansion of less highly charged anionic phospholipid monolayers, where either no cation dependence or the opposite trend was observed 29. The magnitude of the expansion of PIP2, in contrast to PG 29, by the different cations appears to be directly related to the Hofmeister series describing the chaotropic nature of the ion (reviewed in 30). This observation suggests again that in addition to the purely electrostatic mechanism of headgroup protonation, these ions may also disrupt the structure of multi-molecular water-mediated hydrogen-bonded networks within the monolayer.
Figure 3.
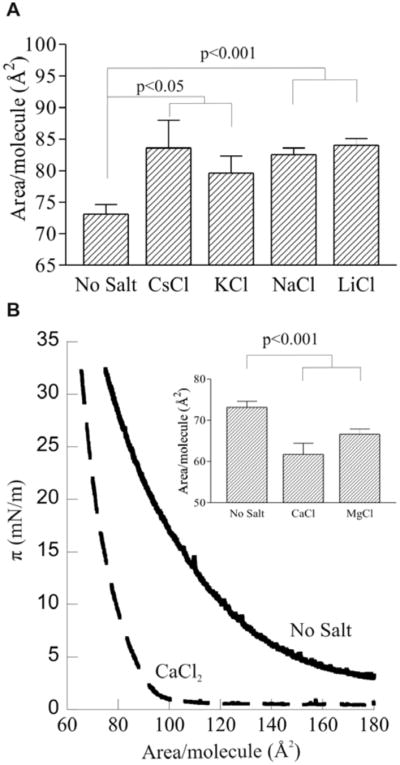
Effects of various counterions.
(A) Area per molecule at π = 30mN/m of L-α PIP2 on HEPES-buffered subphase with 250 mM salt; Mean ± SE, n=5.
(B) π-Area isotherms of L-α PIP2 HEPES-buffered subphase, pH 7.4, 30°C (solid line) and same conditions plus 250 mM CaCl2 (dashed line); (inset) quantification of the effects of 250 mM CaCl2 and MgCl2; mean ± SE, n=4.
Divalent counterions have a very different effect on PIP2 from any of the monovalent salts. Both CaCl2 and MgCl2 had a very significant compressing effect on pure PIP2 monolayers (Fig. 3b). The representative isotherms in Fig. 3b highlight these differences, both in the area per PIP2 at biologically relevant densities (π =30mN/m) and at lower surface pressures. The inset shows a quantification of the compressing effect of divalent cations and demonstrates that PIP2 monolayers with 250 mM Ca2+ and Mg2+ were compressed by 15% and 9% over control, respectively. These observations are consistent with the known ability of Ca2+ ions to act as PIP2 crosslinkers by binding and dehydrating multiple phosphates with high affinity 31, 32, neutralizing their charges, and bridging headgroups to form a tightly compressed monolayer33, even at low surface pressure.
Expanding effect of non-ionic chaotropes and temperature
The magnitude of the observed NaCl expansion and the dependence of the expansion of subphase cations suggest an unexpected mechanism for the intermolecular interactions between adjacent PIP2 molecules in a pure monolayer system. These effects, and their specificity for PIP2 over other inositol-based lipids, suggest adhesive interactions, possibly through hydrogen bonding, as a mechanism responsible for the compressed state of the PIP2 in the absence of salts. To validate the hypothesis that monolayers of naturally-derived PIP2 form PIP2 -bonded networks that allow these lipids to overcome the electrostatic repulsion expected from their high charge density, several non-ionic chaotropic factors were tested for their ability to disrupt these putative networks and induce monolayer expansion. Specifically, urea, a protein denaturant commonly used because of its chaotropic character, and trehalose, a non-reducing glucose dimer known for its cryoprotective properties which derive from its ability to disrupt water structure, were tested for their effect on PIP2 monolayers. Consistent with a role for hydrogen-bonding, both non-ionic chaotropes had a strong expanding effect on the monolayers. At π=30 mN/m, 5 M urea increased the area per PIP2 molecule by almost 25% to 90.9 Å2/molecule, the highest value observed for any of the conditions employed in these experiments (Fig. 4b). Similarly, 5 mM trehalose significantly increased the area of the PIP2 monolayer by 9%. These effects were specific to PIP2, as neither treatment had a significant effect on monolayers of PI.
Figure 4.

Evidence for water-mediated intermolecular hydrogen bonding. Area per molecule of L-α PIP2 and L-α PI at π = 30mN/m on HEPES buffered subphase, pH 7.4 (A) in presence of 5 mM trehalose and 5 M urea; and (B) as a function of the temperature of the subphase (circles = PIP2; squares = PI).
Finally, as confirmation of the hydrogen bonding hypothesis, the temperature-dependent behavior of PIP2 monolayers was tested. These monolayers showed a very significant contraction as the temperature of the subphase was decreased from 34 to 17°C, decreasing the area per molecule by almost 50% (Fig. 4a). In contrast, monolayers of PI were contracted by only ~10% over the same temperature range, consistent with a simple scaling of pressure with kBT. While some contraction is expected due to the decrease in kinetic energy of the lipids, the 50% difference observed for PIP2 strongly suggests an additional mechanism, such as the disruption of a hydrogen bonded network by increased thermal energy of the subphase. An additional potentially related finding was that pure PIP2 could not form compressed monolayers at subphase temperatures below ~15°C, instead exhibiting collapse at relatively low surface pressures (<10 mN/m; data not shown). This result could be relevant to understanding temperature-induced changes in cell structure, such as cold activation of platelets, a process during which changes in PIP2 organization at the plasma membrane trigger actin assembly 34.
Discussion
The results showing subphase ionic strength-induced expansion of the charged monolayers may seem to contradict a strictly electrostatic explanation because subphase ions would be expect to shield the anionic headgroups and allow tighter packing. However, the observed PIP2 monolayer expansion due to increased subphase cations can be explained partly by the dependence of the apparent pKa of the phosphate groups on ionic strength, previously shown for monolayers of lipids with phosphatidic acid headgroups 35. The plane of negative charge of the monolayer surface in the absence of counterions induces a high surface potential which in turn induces protonation of ionizable groups that would be deprotonated in the presence of subphase salts. This effect has been shown to be important in regulating the gel-liquid transition temperature of charged monolayers 36, although the measured magnitude of the expansion effect of subphase salts with other anionic lipids has been shown to be much smaller than the expansion observed here with PIP2 29.
To elucidate the complementary roles of subphase ionic strength (inducing protonation of headgroups and breaking hydrogen bonds) in expanding PIP2 monolayers, the purely electrostatic contribution was determined by modeling the system as a uniformly distributed plane of ionizable groups, the charge density of which is a function of both the pKa’s of the ionizable groups and the ionic strength of the subphase solution (see Supplementary/Cebers et al). The surface pressure due to electrostatic repulsion of the charged groups was then calculated by differentiating the thermodynamic potential with respect to the surface area. The results of the purely electrostatic model for the effects of subphase ionic strength on surface pressure correspond qualitatively with some of the observed experimental results. The high pressure observed with expanded monolayers (up to 150 Å2/molecule) at neutral pH can be explained by the repulsion of the highly charged headgroups. Additionally, both the crossing over between isotherms with low and high ionic strength and the expansion of the monolayer due to high ionic strength were confirmed with the electrostatic model at neutral pH (Fig. 1a and Supplementary Figure 4b). However, many of the experimentally observed results are not compatible with a purely electrostatic treatment. Specifically, the behavior of the PIP2 monolayers at non-neutral pH does not conform to that predicted by the model, neither in the magnitude of observed surface pressures at high pH, nor in the expanding effects of subphase salt at low pH (Fig. 1b and Supplementary figure 4). The expanding effect at low pH is particularly notable since theory predicts the charge on PIP2 at pH 1.8 to be between neutral and −1, i.e. approximately equal to the charge on PS and PI at neutral pH, for which no salt-dependent expansion was observed. Additionally, the varying effects of the monovalent salt series cannot be accounted for entirely by changes in subphase ionic strength. Finally, both the PIP2 isomer specificity of the NaCl-induced monolayer expansion and the effects of uncharged chaotropes and temperature point to a more complex molecular mechanism than the strictly electrostatic subphase ionic strength modulation of apparent headgroup pKa.
The results of the experiments described above highlight the importance of attractive interactions, probably mediated by hydrogen bonding that significantly counter the repulsive electrostatic interactions between PIP2 lipids in planar systems. These interactions can be disrupted by the introduction of chaotropic factors such as monovalent ions, trehalose, or urea. These findings are summarized in a qualitative model presented in Fig. 5c. In absence of disrupting agents, several PIP2 molecules are shown as interacting through a water-mediated hydrogen bonded network. When either ionic factors that disrupt water- PIP2 interactions or non-ionic chaotropes are present, hydrogen bonding is disrupted and electrostatic repulsion becomes the dominant mechanism of intermolecular interaction, causing an increase in molecular area. This model is supported by the magnitude of the expanding effect of monovalent cations on pure PIP2 monolayers, as well as data confirming that effect with urea and trehalose (strong non-ionic chaotropes), as well as high temperature. The calculated energy difference between the proposed hydrogen-bonded state and the chaotrope-disrupted expanded state (for 250 mM LiCl: ΔArea = 17.8 Å2/molecule at 35 mN/m = ~6 kJ/mol) is commensurate with the loss of approximately one hydrogen bond per PIP2 molecule. The possibility of intermolecular hydrogen bonding between PIP2 headgroups in mixed lipid systems has been shown both experimentally 24, 25 and in simulations 37, and the data presented here confirm that possibility through experiments showing hydrogen bonding to be an important factor in intermolecular PIP2 interactions.
An alternative explanation to electrostatics and hydrogen bonding for the observed effects of subphase salts involves the intercalation of the monovalent salts into the plane of the anionic headgroups to form a network lattice between the phosphates and cations. This explanation appears unlikely since the expansion is greatest with the smallest, most electropositive ion (Li+) and decreases with ion radius (Fig 3a). Also, while the formation of a rippled phase in the absence of salts could produce a more compressed monolayer, a phase transition from the liquid phase to the rippled phase was not observed with any of the isotherms (Fig. 1a). Additionally, the ripple phase would only be likely to form at high surface pressures, while the differences between the high and low salt states are apparent at pressure as low as 5 mN/m (Fig. 1a).
Two pieces of evidence argue for the importance of water in maintaining this network, as opposed to hydrogen bonding directly between adjacent PIP2 molecules. First, non-ionic factors not expected to interact with phosphate groups (i.e. urea and trehalose) were shown to have a strong expanding effect on PIP2 monolayers, likely as a result of their disruption of water structure and subsequent disturbance of the hydrogen-bonded network (Fig. 4a). Second, the significant reduction of the area per molecule of PIP2 induced by divalent cations (Ca2+ and Mg2+) confirms their ability to cross-link neighboring lipids through the dehydration of their phosphate groups, and suggests that although the PIP2 monolayers maintain a compressed state through their ability to hydrogen bond, they are not as tightly compressed as when directly crosslinked by divalent cations (Fig. 3b).
The specificity of these observed effects for PIP2 over structurally similar lipids, such as PI(4)P, could provide an insight into the physicochemical explanation for the relative biological importance of PIP2 over the monophosphorylated inositol-based lipids in regulating cell function. Although the possibility of modification by phosphate substitution at multiple sites makes inositol-based phospholipids a versatile platform for signaling, the monophosphorylated PIPs provide the same versatility as PIP2 as kinase and phosphatase substrates. However, while PIP2 is known to bind dozens of unique proteins and function as a critical second messenger and regulator of numerous cellular processes, the monophosphorylated PIPs are not implicated in nearly as many signaling pathways. The results presented here suggest that perhaps PIP2’s unique role in cell function is due to the ability of this molecule to form stable multi-molecular aggregates, thereby providing a hierarchal mechanism for regulation of signaling. In addition to enzymatic control of PIP2 abundance, the presentation and availability of this molecule could be modulated by a variety of chaotropic and kosmotropic factors that could affect its aggregation state and consequent signaling properties.
Many experiments suggest that there are at least two distinct modes of interaction for the many cellular binding partners of PIP2. Some proteins (e.g. those containing PH domains) have a specific binding site for individual PIP2 molecules 38–40, whereas others contain unstructured polybasic domains thought to bind several PIP2 molecules simultaneously through non-specific electrostatic attraction (e.g. MARCKS 23, 41). It is interesting to consider the possibility that a cell could regulate PIP2-mediated signaling by influencing the balance between hydrogen-bonding and electrostatic repulsion, thereby moderating the pools of PIP2 available for single-lipid binding protein domains versus those that bind multi-molecular assemblies.
Footnotes
SUPPORTING INFORMATION: Submitted manuscript including the theoretical calculations for electrostatic surface pressure in monolayers of polyanionic lipids.
References
- 1.Ferrell JE, Jr, Huestis WH. J Cell Biol. 1984;98:1992. doi: 10.1083/jcb.98.6.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLaughlin S, Wang J, Gambhir A, Murray D. Annu Rev Biophys Biomol Struct. 2002;31:151–75. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 3.Berridge MJ, Irvine RF. Nature. 1984;312:315. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- 4.Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, Cantley LC, Roberts TM, Vogt PK. Science. 1997;276:1848–50. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 5.Yin HL, Janmey PA. Annu Rev Physiol. 2003;65:761–89. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 6.Janmey PA, Lindberg U. Nat Rev Mol Cell Biol. 2004;5:658–66. doi: 10.1038/nrm1434. [DOI] [PubMed] [Google Scholar]
- 7.Gilmore AP, Burridge K. Nature. 1996;381:531–5. doi: 10.1038/381531a0. [DOI] [PubMed] [Google Scholar]
- 8.Martin TFJ. Curr Op Cell Biol. 2001;13:493. doi: 10.1016/s0955-0674(00)00241-6. [DOI] [PubMed] [Google Scholar]
- 9.Varnai P, Lin X, Lee SB, Tuymetova G, Bondeva T, Spat A, Rhee SG, Hajnoczky G, Balla T. J Biol Chem. 2002;277:27412–22. doi: 10.1074/jbc.M109672200. [DOI] [PubMed] [Google Scholar]
- 10.Hilgemann DW, Feng S, Nasuhoglu C. Sci STKE. 2001;2001:RE19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 11.Cremona O, De Camilli P. J Cell Sci. 2001;114:1041–52. doi: 10.1242/jcs.114.6.1041. [DOI] [PubMed] [Google Scholar]
- 12.James SR, Paterson A, Harden TK, Demel RA, Downes CP. Biochemistry. 1997;36:848–55. doi: 10.1021/bi962108q. [DOI] [PubMed] [Google Scholar]
- 13.Boguslavsky V, Rebecchi M, Morris AJ, Jhon DY, Rhee SG, McLaughlin S. Biochemistry. 1994;33:3032–7. doi: 10.1021/bi00176a036. [DOI] [PubMed] [Google Scholar]
- 14.Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. J Cell Biol. 2000;149:1455–72. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pike LJ, Miller JM. J Biol Chem. 1998;273:22298. doi: 10.1074/jbc.273.35.22298. [DOI] [PubMed] [Google Scholar]
- 16.Huang S, Lifshitz L, Patki-Kamath V, Tuft R, Fogarty K, Czech MP. Mol Cell Biol. 2004;24:9102. doi: 10.1128/MCB.24.20.9102-9123.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran D, Gascard P, Berthon B, Fukami K, Takenawa T, Giraud F, Claret M. Cell Signal. 1993;5:565–81. doi: 10.1016/0898-6568(93)90052-n. [DOI] [PubMed] [Google Scholar]
- 18.van Rheenen J, Jalink K. Mol Biol Cell. 2002;13:3257–67. doi: 10.1091/mbc.E02-04-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Rheenen J, Achame EM, Janssen H, Calafat J, Jalink K. EMBO J. 2005;24:1664–73. doi: 10.1038/sj.emboj.7600655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Gambhir A, Hangyas-Mihalyne G, Murray D, Golebiewska U, McLaughlin S. J Biol Chem. 2002;277:34401–12. doi: 10.1074/jbc.M203954200. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Gambhir A, McLaughlin S, Murray D. Biophys J. 2004;86:1969–86. doi: 10.1016/S0006-3495(04)74260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Crocker E, McLaughlin S, Smith SO. J Biol Chem. 2003;278:21459–66. doi: 10.1074/jbc.M301652200. [DOI] [PubMed] [Google Scholar]
- 23.Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S. Biophys J. 2004;86:2188–207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redfern DA, Gericke A. Biophys J. 2004;86:2980–92. doi: 10.1016/S0006-3495(04)74348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redfern DA, Gericke A. J Lipid Res. 2005;46:504–15. doi: 10.1194/jlr.M400367-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Kates M. Techniques of Lipidology. 2. Elsevier Science Publishers B.V; Amsterdam: 1986. [Google Scholar]
- 27.Demel RA, Geurts van Kessel WS, Zwaal RF, Roelofsen B, van Deenen LL. Biochim Biophys Acta. 1975;406:97–107. doi: 10.1016/0005-2736(75)90045-0. [DOI] [PubMed] [Google Scholar]
- 28.Brockman HL, Applegate KR, Momsen MM, King WC, Glomset JA. Biophys J. 2003;85:2384–96. doi: 10.1016/s0006-3495(03)74662-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sacre MM, Tocanne JF. Chem Phys Lipids. 1977;18:334–54. doi: 10.1016/0009-3084(77)90019-6. [DOI] [PubMed] [Google Scholar]
- 30.Cacace M, Landau E, Ramsden J. Quar Rev Biophys. 1997;30:241–277. doi: 10.1017/s0033583597003363. [DOI] [PubMed] [Google Scholar]
- 31.Flanagan LA, Cunningham CC, Chen J, Prestwich GD, Kosik KS, Janmey PA. Biophys J. 1997;73:1440–7. doi: 10.1016/S0006-3495(97)78176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah DO, Schulman JH. J Lipid Res. 1965;6:341. [PubMed] [Google Scholar]
- 33.Papahadjopoulos D. Biochim Biophys Acta. 1968;163:240–54. doi: 10.1016/0005-2736(68)90103-x. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmeister KM, Falet H, Toker A, Barkalow KL, Stossel TP, Hartwig JH. J Biol Chem. 2001;276:24751. doi: 10.1074/jbc.M011642200. [DOI] [PubMed] [Google Scholar]
- 35.Trauble H. Membrane Electrostatics. In: Abrahamsson S, Pascher I, editors. Structure of Biological Membranes. Plenum Press; New York London: 1977. pp. 509–550. [Google Scholar]
- 36.Trauble H, Eibl H. Proc Natl Acad Sci U S A. 1974;71:214–9. doi: 10.1073/pnas.71.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liepina I, Czaplewski C, Janmey P, Liwo A. Biopolymers. 2003;71:49–70. doi: 10.1002/bip.10375. [DOI] [PubMed] [Google Scholar]
- 38.Harlan JE, Hajduk PJ, Yoon HS, Fesik SW. Nature. 1994;371:168–70. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 39.Harlan JE, Yoon HS, Hajduk PJ, Fesik SW. Biochemistry. 1995;34:9859–64. doi: 10.1021/bi00031a006. [DOI] [PubMed] [Google Scholar]
- 40.Lemmon MA, Ferguson KM, O’Brien R, Sigler PB, Schlessinger J. Proc Natl Acad Sci U S A. 1995;92:10472–6. doi: 10.1073/pnas.92.23.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rauch ME, Ferguson CG, Prestwich GD, Cafiso DS. J Biol Chem. 2002;277:14068. doi: 10.1074/jbc.M109572200. [DOI] [PubMed] [Google Scholar]