

Abstract
The bacterial RecA protein has been implicated as a bacterial drug target not as an antimicrobial target, but as an adjuvant target with the potential to suppress the mechanism by which bacteria gain drug resistance. In order to identify small molecules that inhibit RecA/ssDNA nucleoprotein filament formation, we have adapted the phosphomolybdate-blue ATPase assay for high throughput screening to determine RecA ATPase activity against a library of 33,600 compounds, which is a selected representation of diverse structure of 350,000. Four distinct chemotypes were represented among the 40 validated hits. SAR and further chemical synthesis is underway to optimize this set of inhibitors to be used as antimicrobial adjuvant agents.
Keywords: Antibiotic resistance, ATPase inhibition, high throughput screening, microbial drug resistance, RecA, SOS response.
INTRODUCTION
Antibiotic resistance is an innate problem involved in the treatment of bacterial infectious diseases and the incidence of these infections is escalating at an alarming rate [1-3]. Resistance not only renders current antibiotics useless against dangerous pathogens, but it is also a financial deterrent to efforts towards the discovery of novel antibiotics [4]. As a result, many pharmaceutical companies have reduced or eliminated their antibacterial drug discovery efforts [5], and since 1962 only three new classes of antibacterial agents have been introduced [6]. Genomics-driven discovery against targets considered essential has not yielded any clinically approved drugs [5], and one way to avert this looming public health crisis could be to replenish antibiotic drug pipelines with drugs against targets not considered essential for survival. In fact, two-thirds of Escherichia coli genes fall into this population [7], and targeting this previously ignored segment of the bacterial genome may offer possibilities for the discovery of non-traditional pharmaceutical agents that attenuate pathogenicity [8] or potentiate the pharmacologic effects of known antibacterial agents [9]. In this context, we have focused on the bacterial RecA protein as a prospective target in the treatment of bacterial infectious diseases. RecA’s activities are central to the repair of DNA damage and stalled replication, but are also important for adaptive mutagenesis and horizontal gene transfer. We hypothesize that small molecule inhibitors of RecA may sensitize bacteria to established antibacterial agents and prevent the development and acquisition of genes conferring drug resistance.
In Escherichia coli, a model system to study DNA damage, RecA’s expression is upregulated by events that challenge the integrity of the organism’s genome, and lead to the formation of single-stranded DNA (ssDNA), a hallmark of DNA damage [10]. In this event, RecA monomers respond by forming a homopolymer on the exposed ssDNA, initiating a series of programmed events that can be fundamentally associated with two major functions, de-repression of the SOS regulon and recombinational DNA strand exchange [11, 12]. In the first function, the SOS regulon is initiated when the RecA nucleoprotein filament comes into contact with the LexA repressor, which sits on promoters of up to 40 genes that are involved in repairing DNA damage [13]. Gene products expressed in the early stages of SOS repair the DNA damage by nucleotide excision (e.g., UvrABCD) or recombination (e.g., RecA), while gene products of the late stages of SOS are involved in error-prone DNA synthesis (e.g., PolIV and PolV) that systematically introduces mutations into the bacterial genome and facilitates adaptive evolution [14-17]. In the second function, RecA pairs ssDNA with homologous duplex DNA, and hydrolyzes ATP to induce the conformational rearrangements necessary to catalyze a strand exchange reaction that produces two new DNA molecules [18, 19]. This ATPase-dependent activity is necessary for recombinational DNA repair [20, 21] and horizontal gene transfer during bacterial transformation, transduction and conjugation [22].
Recently, the SOS and recombination processes mediated by RecA have been shown to be activated in response to antibiotic treatment [23-25]. The biological activities controlled by RecA likely enhance a bacterium’s tolerance to antibiotics and can result in the de novo development and horizontal transfer of antibiotic resistance genes. Because RecA and its functions in overcoming antibacterial chemotherapy are highly conserved among bacteria, small molecule modulators of RecA would be valuable for further exploration of its roles in bacterial pathogens and its potential exploitation as a therapeutic target. While ATP-competitive nucleotide analogs [26, 27], polysulfated naphthyl compounds [28], short alpha-helical peptides [29] and select transition metal cations [30] are effective inhibitors of RecA in vitro, none of these molecular classes have demonstrated RecA-specific biological activities in bacterial cell cultures.
Towards the goal of discovering potent cell-permeable inhibitors for the validation of RecA as an important and novel target for the chemotherapeutic treatment of bacterial infections, we carried out a two-tiered HTS campaign against RecA. As ssDNA-dependent ATP hydrolysis is a useful indicator that the RecA protein is active, we elected to screen the compounds using an inexpensive, yet robust and reliable phosphomolybdate blue ATPase assay to identify compounds that inhibit ATP hydrolysis. We initially screened the BRITE diversity set, which is comprised of approximately 10% of our proprietary collection of nearly 350K compounds selected to maximize chemical diversity. From this collection, we identified 40 hits that clustered into four chemotype classes which were then carried into dose-response confirmatory testing. We have also performed Tanimoto-based similarity searching in the entire BRITE library to identify related compounds that will be screened to provide initial insights into structure-activity relationships.
MATERIALS AND METHODOLOGY
RecA and ATPase Reagents
For the single-concentration screening, RecA protein was purified and stored as previously described by Singleton, et al. [31] and for additional screen of 8640 compounds, the enzyme was purchased from Epicentre Biotechnologies for the dose/response study. The commercially available enzyme has similar purity and specific activity as purified in-house, which is greater than 90% pure as judged by SDS-PAGE. Polydeoxythymidylic Acid (poly(dT)) single-strand DNA (average length = 300 nucleotides) was purchased from Midland Certified Reagent Company (Cat #P-2004, Lot 110607-403). Crystalline L-ascorbic acid and sulfuric acid were purchased from Fisher Scientific. Unless otherwise stated all other reagents were purchased from Sigma-Aldrich at the highest level of purity possible.
Compound Libraries and Handling
The general strategy for RecA screening was to test a small diverse library and then elaborate on the confirmed active compounds by similarity searching in the entire BRITE compound library. The BRITE diversity library screened in this HTS effort consists of 33,600 compounds selected for chemical diversity from the entire BRITE library, which was generated by combinatorial chemistry synthetic routes and was a gift from Biogen-Idec in 2005. All of the compound plates were stored in polypropylene deep-well blocks at 4 °C without humidity controls. The BRITE compound library is plated in 384-well Greiner V-bottom polypropylene plates in 100% DMSO in 10 mM and 1 mM aliquots. Plates are stored at 4 oC. Using the Biomek® NX and P20 tips, 0.5 µl of 1 mM compound in DMSO was spotted into columns 3 - 22 of Costar clear flat bottom 384-well assay plates (Corning # 3702) for single shot analysis. Columns 1, 2, 23, and 24 were spotted with 0.5 µl of DMSO with the Biomek® NX and P20 tips and used as positive and negative controls. This resulted in a final concentration of 10 µM for single-point screening.
For dose-response/IC50 curve analysis, 1 µl of the 10 mM stock of selected compounds was added to 4 µl of DMSO and mixed in an Axygen 384-well PCR plate for a 5-fold dilution and beginning concentration of 2 mM. This was performed with the Biomek® 3000 and P20 tips. A 10-point 2-fold serial dilution was then performed across the plate with the Biomek® 3000. The serially diluted compounds were then spotted to columns 3 – 22 of Costar clear flat bottom 384-well assay plates with the Biomek® NX and P20 tips. Columns 1, 2, 23, and 24 were spotted with 0.5 µl of DMSO with the Biomek® NX and P20 tips and used as positive and negative controls.
Phosphomolybdate-Blue ATPase Assay
A modified phosphomolbydate-blue ATPase assay (Unpublished data; Yeh L.A.) was used to quantify the amount of free phosphate as a measure of ATP hydrolysis, as an indirect measure of RecA nucleoprotein filament assembly [32]. The ATPase reactions were carried out in Costar clear flat-bottom 384-well assay plates (Corning, Lowell, MA) that the compounds were spotted in, and the final volume in each well was 30.5 µl, giving final concentrations of 17 µM for the library compound and 1.6% for DSMO. A 2.25 µM stock of ATP was prepared in H2O and 10 µl of this was added to all wells of the assay plates using a Thermo Mutidrop 384-well dispenser (Thermo Fisher, Waltham, MA), yielding a final ATP concentration of 0.75 µM. The reaction rate is linear at this low ATP concentration. The assay development, optimization and validation studies of known compounds were described in a recent publication by Wigle [33]. After assay assembly, the plates were then incubated at room temperature for 15 min. To columns 3-24 of the assay plates, using a Thermo Mutidrop 384-well dispenser, was added 20 µl of a cocktail of containing 0.5 µM RecA, 5 µM poly(dT) ssDNA, 10 mM Mg(OAc)2, 25 µM Tris·HOAc, and 5% v/v glycerol. For the negative control (minimum signal control), 20 µl of an identical solution omitting poly(dT)-DNA was added to columns 1 and 2 using a Thermo Mutidrop 384-well dispenser. The positive control (maximum signal control) in lanes 23 and 24 contained the full ATPase reaction reagents and 0.5 µl DMSO representing the full/uninhibited RecA activity. Compounds that exhibited inhibition greater than 50% in the primary screen were carried into the dose-response confirmation step.
For the IC50 determinations, the PMB ATPase assay was improved by the addition of 10 µg /mL BSA to reduce the immobilization of RecA in the assay plates, thereby increasing the amount of RecA in the well that can participate in nucleation and filament formation. It was also determined that the assay window was wider when the optical absorption was tabulated at 825 nm. Serial dilutions of compounds were performed in 100% DMSO with a two-fold dilution factor resulting in 10 final concentrations spanning 33 µM to 65 nM. The serially diluted compounds (0.5 µL) were dry spotted into assay plates and were assembled as described for the single-concentration screen. Four-parameter sigmoidal regressions were performed and IC50 values were determined using the XLfit software (XLFit software, IDBS, Guildford UK).
The assay plates were sealed by a Thermo Scientific microplate heat sealer and centrifuged at 1000 RPM for 1 min. The plates were then transferred to a 37 ºC air incubator and the ATPase reaction was allowed to proceed for 35 min. The plates were subsequently removed from incubator and 30 µl of freshly made phosphomolybdate blue dye (10% ascorbic acid and 2.5% ammonium molybdate at the ratio of 1:1 and adding 12 M sulfuric acid at 6.25% v/v) was added to all wells using a Thermo Mutidrop 384-well dispenser. After 30 min of incubation at room temperature, the plates were scanned for absorbance at 825 nm using a Spectramax-384 Plus (Molecular Devices, Sunnyvale, CA).
HTS Results and RecA Inhibitor Identification
In lieu of direct measurement of RecA nucleoprotein filament formation, we chose a more HTS-amenable endpoint of ATP hydrolysis in the presence of single-stranded DNA to identify RecA inhibitors. Here we report confirmed molecular structures for the most potent RecA inhibitors in each distinct class discovered in this screening effort. This screening approach successfully identified several chemotype classes and gave initial insight into the structure-activity relationship of small molecule inhibition of bacterial RecA.
RESULTS
The PMB ATPase screen of the BRITE diversity library resulted in 73 active compounds exhibiting RecA inhibition above a 50% threshold. RecA inhibitors were triaged by selecting the most potent single-point hits and were confirmed with dose-response. The average Z' for this screen was 0.83, with an overall hit rate of 0.22%. Of the 73 hits obtained, 71 of 73 compounds could be classified into one of four chemotype classes based on structural similarity (Fig. 1). The label for chemotype classes D and C are reversed in the lower portion of Fig. (1). “C - 9 compounds” should appear on the lower left, and “D – 2 compounds” should appear on the lower right.
Fig. (1).

Four chemotype classes of RecA inhibitors showing the number of compounds discovered for each classes in this PMB ATPase high thoughput screening effort.
The 73 hits identified in the single-point 10 µM screen were carried into dose-response confirmation with 40 compounds exhibiting dose-response behavior, yielding a 55% confirmation rate. A typical dose-response curve for RecA inhibitors is shown in Fig. (2), with characteristic steep dose-response curve likely due to multi-site inhibitor binding and well known positive cooperative binding kinetics [34, 35]. The structure and dose-response curve for the most potent RecA inhibitor identified in the BRITE diversity set screen, BRITE-338733, is shown in Fig. (2) and has been analyzed and exhibits a 4.7µM IC50 and a Hill slope of 7.6. The compound BRITE-345133 (chosen as representative of the 2-amino-4,6-diarylpyridine scaffold) has been analyzed by LC/MS and has a confirmed mass of 524.1 m/z (m+1 peak) and is 89% pure. The range of IC50 values for the 40 confirmed compounds is 4.7 µM to 39 µM and the average Hill slope is 7.4. The molecular structures and corresponding IC50 values are shown in Table 1. The 2-amino-4,6-diarylpyridine scaffold is the most potent as shown by BRITE-338733 having an IC50 of 4.7 µM. The 1,2,4-oxadiazole scaffold of BRITE-212537 followed by the quinaxolinone scaffold of BRITE-379089 and the benzimidazole diazepinone scaffold of BRITE-082776 produced IC50 values of 6.2, 11 and 32 µM respectively, and represent the most potent members of their respective classes.
Fig. (2).

Dose-response curve for the RecA inhibitor BRITE-338733, exhibiting an IC50 value of 4.7 ± 0.5 µM and characteristic steep Hill-slope (> 5).
Table 1.
Chemical Structures of Confirmed RecA Inhibitors
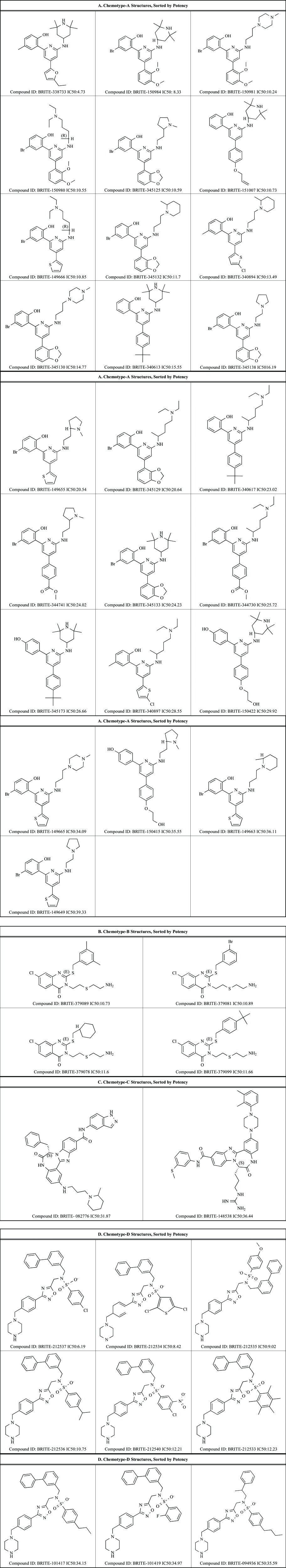
|
DISCUSSION
Here we present a focused high throughput screening effort that effectively identified several novel RecA inhibitors for molecular probes to investigate the impact of RecA inhibition on antibacterial drug resistance. Multiple chemical scaffolds were identified but the 2-amino-4,6-diarylpyridine scaffold was the most potent class of inhibitors. We have independently verified biological activity of this chemotype as described in Wigle, 2009 [33].
The PMB ATPase assay, while not a direct measure of nucleoprotein filament formation, provides a valuable means for the identification of RecA inhibitors with excellent screening statistics and normal confirmation rates (see Wigle, 2009) [33]. We found the assay to be very robust; however, the assay window and statistics could vary significantly from RecA protein batch to batch. The cause of this variation was determined to be residual free phosphate from the protein purification process involving phosphate-based buffers. We routinely performed min-max experiments in one 384-well plate (half-min, half-max) to investigate the assay window and signal-to-noise ratio for batch validation and would dialyze the RecA protein in the assay buffer to reduce the amount of free phosphate. Excellent assay statistics were achieved when the UV/Vis absorption assay window (max-minus-min) was above 0.2 for A650, and above 1.0 for A825. Two changes were made upon reflection on the initial BRITE-diversity set screen prior to the confirmation step; 1) shifting absorption wavelength to 825 nm and 2) with the addition of 10 µg/mL BSA, which resulted in an increase in Z' from 0.83 to 0.90.
Typical IC50 curves for RecA inhibition are steep, with fit Hill slopes typically above five. It is expected that positive cooperative binding (long known in the experimental RecA literature) would result in steep inhibitory Hill slopes. We have performed dynamic light scattering (DLS) experiments (Dynapro-384, Wyatt Tech. Santa Barbara, CA) to independently assess if the 2-amino-4,6-diarylpyridine scaffold (class A, most potent overall) was aggregating RecA protein in the absence of DNA. We found no indication of RecA aggregation in the presence of up to 10 µM BRITE-345133 (chosen as representative of the 2-amino-4,6-diarylpyridine scaffold) with DLS, which is approaching the solubility limit for the compound (data not shown). These DLS experiments confirm that chemotype-A compounds are genuine RecA inhibitors and the kinetics of inhibition adhere to a multi-site kinetic model. The DLS data will be published in a separate manuscript.
The preliminary structure-activity-relationship of the additional 8640 compounds selected from the rest of the 350,000 compound library for the active scaffolds shows a relatively flat profile, with most IC50 values hovering in the low micromolar range (data not shown). We believe that this PMB ATPase assay is not suitable for profiling the SAR because low micromolar concentrations are at the lower edge of the assay dynamic range. We are developing more sensitive assays to adequately profile the SAR for these compounds in the nano- to micromolar range.
CONCLUSION
We have screened 33,600 compounds selected from a library containing 350,000 compounds. Here we present 40 confirmed inhibitors of RecA ATPase activity. The 40 confirmed hits represented four novel scaffolds for RecA inhibition. These scaffolds will be used for future structure activity relation (SAR) analysis. Among the four scaffolds, the 2-amino-4,6-diarylpyridine compounds are the most potent with IC50 values in the single-digit micromolar range. We are in the process of developing new sensitive assays to further the SAR exploration. This high-throughput screening effort effectively identified a diverse set of active chemical scaffolds to serve as molecular probes for studying the effects of RecA in the development of antimicrobial resistance.
ACKNOWLEDGEMENTS
This work was supported in part by a grant to BRITE/NCCU from the Golden LEAF Foundation, a grant to S.F.S. from the National Institutes of Health (GM058114), and funds from the State of North Carolina.
REFERENCES
- 1.Diekema DJ, BootsMiller BJ, Vaughn TE, et al. Antimicrobial resistance trends and outbreak frequency in United States hospitals. Clin Infect Dis. 2004;38(1):78–85. doi: 10.1086/380457. [DOI] [PubMed] [Google Scholar]
- 2.Talbot GH, Bradley J, Edwards JE, Jr, Gilbert D, Scheld M, Bartlett JG. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis. 2006;42(5):657–68. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 3.Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 4.Power E. Impact of antibiotic restrictions: the pharmaceutical perspective. Clin Microbiol Infect. 2006;12(Suppl 5):25–34. doi: 10.1111/j.1469-0691.2006.01528.x. [DOI] [PubMed] [Google Scholar]
- 5.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6(1):29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 6.Lock RL, Harry EJ. Cell-division inhibitors: new insights for future antibiotics. Nat Rev Drug Discov. 2008;7(4):324–38. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- 7.Baba T, Ara T, Hasegawa M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2(2006):0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escaich S. Antivirulence as a new antibacterial approach for chemotherapy. Curr Opin Chem Biol. 2008;12(4):400–8. doi: 10.1016/j.cbpa.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 9.Cottarel G, Wierzbowski J. Combination drugs, an emerging option for antibacterial therapy. Trends Biotechnol. 2007;25(12):547–55. doi: 10.1016/j.tibtech.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Friedberg EC, Walker GC, Siede W. SOS responses and DNA damage tolerance in prokaryotes. DNA Repair and Mutagenesis. Washington, D.C: ASM Press; 1995. ; pp. 407–64. [Google Scholar]
- 11.Haruta N, Yu X, Yang S, Egelman EH, Cox MM. A DNA pairing-enhanced conformation of bacterial RecA proteins. J Biol Chem. 2003;278(52):52710–23. doi: 10.1074/jbc.M308563200. [DOI] [PubMed] [Google Scholar]
- 12.VanLoock MS, Yu X, Yang S, et al. ATP-mediated conformational changes in the RecA filament. Structure. 2003;11(2):187–96. doi: 10.1016/s0969-2126(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 13.Sassanfar M, Roberts JW. Nature of the SOS-inducing signal in Escherichia coli. The involvement of DNA replication. J Mol Biol. 1990;212(1):79–96. doi: 10.1016/0022-2836(90)90306-7. [DOI] [PubMed] [Google Scholar]
- 14.Kelley WL. Lex marks the spot: the virulent side of SOS and a closer look at the LexA regulon. Mol Microbiol. 2006;62(5):1228–38. doi: 10.1111/j.1365-2958.2006.05444.x. [DOI] [PubMed] [Google Scholar]
- 15.Hastings PJ, Rosenberg SM, Slack A. Antibiotic-induced lateral transfer of antibiotic resistance. Trends Microbiol. 2004;12(9):401–4. doi: 10.1016/j.tim.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Schlacher K, Leslie K, Wyman C, Woodgate R, Cox MM, Goodman MF. DNA polymerase V and RecA protein, a minimal mutasome. Mol Cell. 2005;17(4):561–72. doi: 10.1016/j.molcel.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Schlacher K, Pham P, Cox MM, Goodman MF. Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation. Chem Rev. 2006;106(2):406–19. doi: 10.1021/cr0404951. [DOI] [PubMed] [Google Scholar]
- 18.Cox JM, Tsodikov OV, Cox MM. Organized unidirectional waves of ATP hydrolysis within a RecA filament. PLoS Biol. 2005;3(2):e52. doi: 10.1371/journal.pbio.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox MM. The bacterial RecA protein as a motor protein. Annu Rev Microbiol. 2003;57:551–77. doi: 10.1146/annurev.micro.57.030502.090953. [DOI] [PubMed] [Google Scholar]
- 20.Courcelle J, Ganesan AK, Hanawalt PC. Therefore, what are recombination proteins there for? BioEssays. 2001;23:463–70. doi: 10.1002/bies.1065. [DOI] [PubMed] [Google Scholar]
- 21.Roca AI, Cox MM. RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucl Acid Res Mol Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- 22.Radman M, Taddei F, Matic I. DNA repair systems and bacterial evolution. Cold Spring Harb Symp Quant Biol. 2000;65:11–9. doi: 10.1101/sqb.2000.65.11. [DOI] [PubMed] [Google Scholar]
- 23.Beaber JW, Hochhut B, Waldor MK. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature. 2004;427(6969):72–4. doi: 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- 24.Cirz RT, Jones MB, Gingles NA, et al. Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J Bacteriol. 2007;189(2):531–9. doi: 10.1128/JB.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130(5):797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 26.Lee AM, Ross CT, Zeng BB, Singleton SF. A molecular target for suppression of the evolution of antibiotic resistance: inhibition of the Escherichia coli RecA protein by N(6)-(1-naphthyl)-ADP. J Med Chem. 2005;48(17):5408–11. doi: 10.1021/jm050113z. [DOI] [PubMed] [Google Scholar]
- 27.Wigle TJ, Lee AM, Singleton SF. Conformationally selective binding of nucleotide analogues to Escherichia coli RecA: a ligand-based analysis of the RecA ATP binding site. Biochemistry. 2006;45(14):4502–13. doi: 10.1021/bi052298h. [DOI] [PubMed] [Google Scholar]
- 28.Wigle TJ, Singleton SF. Directed molecular screening for RecA ATPase inhibitors. Bioorg Med Chem Lett. 2007;17(12):3249–53. doi: 10.1016/j.bmcl.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cline DJ, Holt SL, Singleton SF. Inhibition of Escherichia coli RecA by rationally redesigned N-terminal helix. Org Biomol Chem. 2007;5(10):1525–8. doi: 10.1039/b703159a. [DOI] [PubMed] [Google Scholar]
- 30.Lee AM, Singleton SF. Inhibition of the Escherichia coli RecA protein: zinc(II), copper(II) and mercury(II) trap RecA as inactive aggregates. J Inorg Biochem. 2004;98(11):1981–6. doi: 10.1016/j.jinorgbio.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 31.Singleton SF, Simonette RA, Sharma NC, Roca AI. Intein-mediated affinity-fusion purification of the Escherichia coli RecA protein. Protein Expr Purif. 2002;26(3):476–88. doi: 10.1016/s1046-5928(02)00571-5. [DOI] [PubMed] [Google Scholar]
- 32.Lee AM, Wigle TJ, Singleton SF. A complementary pair of rapid molecular screening assays for RecA activities. Anal Biochem. 2007;367(2):247–58. doi: 10.1016/j.ab.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wigle TJ, Sexton JZ, Gromova AV, et al. Inhibitors of RecA activity discovered by high-throughput screening: cell-permeable small molecules attenuate the sos response in Escherichia coli. J Biomol Screen. 2009;14(9):1092–101. doi: 10.1177/1087057109342126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikawa T, Masui R, Kuramitsu S. RecA protein has extremely high cooperativity for substrate in its ATPase activity. J Biochem. 1998;123(3):450. doi: 10.1093/oxfordjournals.jbchem.a021958. [DOI] [PubMed] [Google Scholar]
- 35.Register J, Griffith J. The direction of RecA protein assembly onto single strand DNA is the same as the direction of strand assimilation during strand exchange. J Biol Chem. 1985;260(22):12308–12. [PubMed] [Google Scholar]