

Abstract
The renin angiotensin system is activated in the early phase of two-kidney one-clip hypertension. The paraventricular nucleus (PVN) integrates inputs regulating sympathetic outflow. The PVN receives inputs from plasma angiotensin II via projections from circumventricular organs and from renal afferent nerves transmitted via the nucleus tractus solitarius. Nitric oxide within the PVN may exert a sympathoinhibitory effect. These studies tested whether decreasing endogenous nitric oxide by introducing dominant negative (DN) constructs for neuronal nitric oxide synthase (nNOS) into PVN chronically augments hypertension and/or modulates baroreflex function. Male six-week old Sprague Dawley rats underwent sham or right renal artery clipping and placement of radiotelemetry transmitters. One week later, the PVN was injected bilaterally with 250 nl artificial CSF containing 250 ng/μl of RSV β-galactosidase (β-gal); CMV wild type (WT nNOS); or RSV heme domain or RSV hemeRedF (DN nNOS). Hemodynamics were monitored for five weeks. Then left renal nerve electrodes were placed, and two days later the rats underwent baroreflex testing in the conscious state. The rise in MAP was significantly potentiated in the DN nNOS 2K-1C group beyond 15 days after PVN injection. By day 35, MAP in the 2K-1C groups was 152±6.3 (β-gal), 155.1±6.6 (WT nNOS) and 179±5.4 mmHg (DN nNOS, P<0.01 vs all other groups). Sham-clipped rats remained normotensive. All groups displayed progressive bradycardia over time that was attenuated in the DN nNOS 2K-1C group. Baroreflex curves shifted to higher pressures and baroreflex sensitivity of HR was diminished similarly in all groups of 2K-1C rats. The baroreflex response of RSNA was preserved. PVN tissue from DN nNOS rats had decreased dimerization of nNOS and generation of NOx. These findings indicate that chronic interference of nNOS dimerization required for generation of nitric oxide within the PVN potentiates the increase of blood pressure by modulating the sympathoexcitation that accompanies renovascular hypertension.
Keywords: arterial baroreflex, Goldblatt kidney, nitric oxide, paraventricular nucleus
Introduction
The brain plays a key role in the regulation of arterial blood pressure via baroreflex mechanisms and by altering chronic efferent sympathetic tone. In particular, alterations in renal sympathetic nerve activity (RSNA) that influence hormones affecting renal function and urinary salt and water excretion contribute to determining the long-term level of blood pressure (Hall et al., 1996). Several lines of evidence support the concept that sympathoexcitation (Weekley, 1992; Esler & Kaye, 1998) contributes to sustained hypertension and that baroreflex (Malpas, 2004; Thrasher, 2004) and baroreflex-independent (Osborn et al., 2005; Osborn et al., 2009) mechanisms are involved.
Activation of neurons within the hypothalamic paraventricular nucleus (PVN) can lead to increases in RSNA indirectly via a synapse in the rostral ventrolateral medulla (RVLM) or directly via a descending projection to spinal sympathetic preganglionic neurons (Tagawa & Dampney, 1999; Dampney et al., 2003). Importantly, in the two-kidney one-clip (2K-1C) model of renovascular hypertension (Bergamaschi et al., 1995) excitatory synaptic inputs to the RVLM are increased. The PVN is one source of these increased excitatory inputs (Stocker et al., 2004). In turn, the PVN receives projections from the circumventricular organs including the subfornical organ that lie outside the blood brain barrier and are richly endowed with angiotensin AT1 receptors (Giles et al., 2001). The subfornical organ appears to be crucial for the development of neurogenic hypertension resulting from systemic infusion of angiotensin II (Ang II) (Zimmerman et al., 2004). Notably, discrete lesions of the subfornical organ (Maliszewska-Scislo et al., 2008) consistently decrease systemic arterial pressure in 2K-1C hypertensive rats without decreasing circulating levels of Ang II, consistent with a neurally mediated component of the elevated arterial pressure. Circulating Ang II which is elevated in 2K-1C hypertension can activate a polysynaptic pathway from the circumventricular region to the PVN and then either via the RVLM or directly to the preganglionic spinal neurons to stimulate renal sympathetic nerves (Dampney et al., 2005; Guyenet, 2006).
Within the PVN itself, N-methyl-D-aspartate and AT1 receptors are among the primary mediators of the excitatory synaptic inputs (Bredt et al., 1990; Chen et al., 2003). These excitatory inputs are under tonic inhibition by gamma aminobutyric acid (GABA) (Yoshida et al., 1991). Several lines of evidence suggest that GABAergic inhibition of PVN sympathoexcitatory neurons is decreased in Ang II-dependent hypertensive rats (LaGrange et al., 2003). It is becoming increasingly recognized that nitric oxide enhances the GABAergic inhibition of excitatory PVN neurons that project to medullary centers (Zhang & Patel, 1998). Although a decrease in NADPH-diaphorase positive neurons (a marker of neuronal nitric oxide synthase [NOS] activity) within the PVN of spontaneously hypertensive rats has also been reported (DiCarlo et al., 2002), other studies in several models of hypertension (Krukoff et al., 1995; Ye et al., 1997; Lon et al., 1999), including the spontaneously hypertensive rat (Qadri et al., 2003) suggest the opposite, namely, that increased nitric oxide signaling occurs. This raises the possibility that nitric oxide within the PVN may play a pivotal role in buffering arterial pressure in hypertensive states.
As yet, no studies exist that directly assess the role of endogenous PVN nitric oxide to modulate chronic hypertension and influence the baroreflex in a model of endogenous high Ang II. Chronic inhibition of the generation of nitric oxide selectively within the PVN by a pharmacologic approach is far from ideal as it would block neuronal as well as endothelial sources. The dominant negative construct for neuronal nitric oxide synthase (nNOS) can very selectively decrease nitric oxide production in vitro by more than 90% (Phung & Black, 1999). These constructs have been expressed chronically in PVN after microinjection and functionally block nitric oxide suppression of the pressor response to injection of endothelin into the subfornical organ by 87% such that subsequent acute nonselective pharmacologic inhibition by Nω-nitro-L-arginine methyl ester (L-NAME) did not elicit any additional significant increase in pressure (Rossi et al., 2004).
Thus, we hypothesized that decreasing the generation of endogenous nitric oxide by introduction of a dominant negative construct for nNOS into the PVN of 2K-1C rats would result in a chronic and progressive augmentation of their hypertension whereas injection of wild type nNOS will exert a depressor effect. In addition to a shift of the baroreflex curve to higher pressures, we further postulated that, 2K-1C rats with impaired nitritergic signaling within the PVN would display decreased sensitivity of the baroreflex response of both heart rate and RSNA.
Materials and Methods
Male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN), were used for all experiments. The rats were housed under controlled conditions (21–23°C; lights on, 07:00–19:00 h) and were permitted free access to water and standard rat chow. The rats were cared for in accordance with the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols were approved by the Wayne State University Institutional Animal Care and Use Committee.
Renal artery clipping and telemetry transmitter placement
Five-week-old rats were anesthetized with combined ketamine 80 mg/kg and xylazine 5 mg/kg iv (Day −7). Using a right flank incision, a silver clip (0.2 mm) was surgically placed around the right renal artery (2K-1C group). Rats in the sham-clipped group underwent identical surgery but were not clipped. The flank incision was closed and a radiotelemetry transducer (TA11PA-C40, Data Sciences Intl) was implanted into each rat by exposing the femoral artery, temporarily occluding the proximal end, and inserting the gel-filled catheter attached to the transmitter device into the femoral artery with a 21-gauge needle. The catheter was advanced into the distal aorta and secured with medical adhesive and the transmitter placed subcutaneously and sutured to the underlying muscle. The skin was closed with surgical staples. At the end of surgery, each rat received butorphanol tartrate 0.075 mg sc for analgesia. Each rat was returned to its individual home cage with its receiver. Hemodynamic data was recorded continuously.
Stereotaxic injections
Exactly one week later (Day 0), rats from the 2K-1C and sham-clipped groups were randomly assigned by an individual blinded to the telemetry data to receive bilateral PVN injections of 250 nl saline containing plasmids (125 ng/μl DNA) of the constructs for one of the following: (a) RSV β galactosidase (β-gal); (b) CMV wild type nNOS (A333, WT nNOS), or (c) one of the dominant negative constructs: RSV heme domain nNOS (p725) or RSV hemeRedF nNOS (p739). Since the results from p725- and p739-injected rats were identical in all respects the data were pooled and reported as the dominant negative nNOS group (DN nNOS). The sequences of these constructs and their synthesis have been published by our laboratories (Phung & Black, 1999). The rats were anesthetized as before with ketamine and xylazine and secured in a stereotaxic apparatus with the skull leveled between the bregma and lambda. A cannula was inserted into the PVN using stereotaxic coordinates obtained in preliminary experiments to adjust for the younger age and smaller size of the animals: ±0.4 lateral to the midline, −1.3 dorsal to bregma, and −8.4 ventral to the skull surface. The injection was performed using a Hamilton syringe over a period of 1 min on each side. At the end of the injection, the cannula was left in place for 1 min prior to withdrawal. After the cannula was withdrawn, and the skin overlying the skull was closed with surgical staples. After recovery from the anesthetic, the rat was placed back into its home cage and telemetric monitoring of heart rate and arterial pressure was resumed.
Vascular catheters and nerve electrode placement
Five weeks later, each rat was conditioned daily for three days to remain for 180 min within a custom made Plexiglas study chamber that would be used during the experiment. The chamber allowed the rat to move forward and backward but not to turn around. They were returned to their home cage after each conditioning period.
On the day of surgery, the rats were anesthetized with pentobarbital sodium 50 mg/kg intraperitoneally. A midline ventral incision was made in the neck and catheters were inserted into the right carotid artery and jugular vein. The catheters were filled with heparinized saline (100 U/ml), secured, tunneled subcutaneously, and exteriorized at the base of the neck. The incision was sutured, and the rat was turned onto its right side. If needed, supplemental doses of sodium pentobarbital were given intravenously after placement of the catheters to maintain an adequate plane of anesthesia.
Electrodes were then implanted via a flank incision with a retroperitoneal approach to isolate the left renal nerve branch and carefully dissect it free. The nerve was placed on electrodes constructed of Teflon-coated silver wire (0.0055 in. diameter, A–M Systems) with the exposed ends wound into single loops. The nerve and electrodes were covered with silicone gel (Kwik-Sil, World Precision Instruments) which was allowed to harden before closure. A ground wire was sewn into the surrounding tissue. The electrodes and ground wire were also tunneled subcutaneously and exteriorized at the base of the neck. Each rat was returned to its home cage and permitted to recover. In all cases, the animals resumed normal grooming, eating and drinking and normal cage activity. Baroreflex function was assessed 24 hr later.
Baroreflex measurements
All baroreflex testing was performed in conscious animals. For these studies, arterial pressure was measured by connecting the arterial catheter to the pressure transducer (Gould P23 XL) which was coupled to an amplifier (Digi-Med BPA-200). Heart rate and MAP were derived by data-acquisition software (DasyLab, Biotech Products) using the arterial pressure pulse and averaged over 1-second intervals. Renal nerve activity was amplified (5,000–20,000 times) and filtered (100–1,000 Hz) with a Grass P511 differential preamplifier and a high-impedance probe (HIP511GB). Both the probe and animals were located inside a shielded Faraday cage. The amplified and filtered neurogram signal was channeled to an oscilloscope and Grass AM8 audiomonitor for visual and auditory evaluation, respectively. The amplified nerve activity was digitized, rectified, integrated, and averaged over 1-s intervals by the computer data acquisition software (DasyLab, Biotech Products). Background noise was determined at the end of experiment after administration of a bolus dose of the ganglionic blocker, trimethaphan camsylate, 20 mg/kg iv (Hoffman-La Roche). RSNA was defined as the amount of recorded nerve activity after subtraction of background noise.
Resting MAP and heart rate were measured in each of the sham-clipped and 2K-1C rats. Baroreflex responses of RSNA and heart rate were tested in all six groups of rats: sham-clipped β-gal; sham-clipped WT; sham-clipped DN nNOS; 2K-1C β-gal; 2K-1C WT; and 2K-1C DN nNOS. After achieving a stable resting baseline (~20 minutes), baroreceptor reflex curves were generated by producing ramp increases and decreases in arterial pressure using intravenous infusions of phenylephrine hydrochloride (200 μg/ml; Sigma RBI) and sodium nitroprusside (200 μg/ml; Ohmeda), respectively. Phenylephrine was infused in increasing rates of 5–50 μg/kg/min and nitroprusside at rates of 7.5–100 μg/kg/min. The ramp increase or decrease of MAP was completed in ~2 minutes. Fifteen to thirty minutes were allowed between the infusions to permit all parameters to return to baseline values. Infusions of phenylephrine or nitroprusside were performed randomly.
To assess the completeness of blockade of the generation of nitric oxide in vivo by the dominant negative constructs of nNOS, after completion of the baroreflex curves, we randomly chose two to three rats from each group and inserted chronic cannulae into the PVN. Two days after placement of the cannulae, 200 μg L-NG-nitroarginine methyl ester (L-NAME) was injected and the pressor response was recorded in the conscious animals. The brains of these rats were not used for subsequent assessment of nNOS dimers or nitric oxide generation by the PVN tissue in vitro.
When all experiments were completed, the rat were euthanized with sodium pentobarbital (100 mg/kg iv) and perfused transcardially with 0.9% saline followed by 10% neutral buffered formalin. The brains were then removed, dehydrated using an alcohol series, and embedded in paraffin. Coronal sections (40 μm) were stained with hematoxylin and eosin, and the site of injection verified. Rats with injection sites outside the PVN were eliminated from analysis.
At least four rats from each group were euthanized and the brains removed without fixation, frozen and stored at −70°C for analysis of nNOS expression, nNOS dimerization products and nitric oxide. These brains were submitted for evaluation to one of the authors (SMB) who was blinded to the treatment that the rat had received. Brains from β-galactosidase, p725 and p739 injected rats were randomly assigned to evaluation of nNOS or total nitric oxide (NOx = nitric oxide + nitrite + NOx).
Assay of nNOS protein and dimerization and total NOx levels
While still frozen, brain tissue for determination of nNOS protein and nitric oxide was obtained by cutting a thick coronal slice that extended rostrocaudally from −0.8 to −2.2 from bregma. The area of the PVN was then dissected and removed en bloc and process for nNOS and nitric oxide.
For determination of nNOS protein, the tissue block was homogenized in lysis buffer containing 50 mM Tris-HCl with 0.5% Triton X-100 and 20% glycerol at pH 7.6 and supplemented with protease inhibitors and clarified by centrifugation (20,000 × g for 20 min at 4°C). Prior to use, the protein concentration of the supernatant was determined by the Bradford Protein Assay.
LT-PAGE was used to distinguish dimeric and monomeric forms of nNOS by methods previously used in our laboratory (Phung & Black, 1999). Samples containing 40 μg of protein each were loaded on an 8% polyacrylamide gel immediately after the addition of Laemelli sample buffer. Running temperature was maintained below 10°C by keeping the electrophoresis unit on ice. Migrated proteins on gels were then transferred to a PVDF membrane and blocked with 5% milk in Tris buffered saline with 0.05% Tween before they were probed with anti-nNOS antibody (1:1000 dilution). Immunoblots were developed using the SuperSignal® West Femto Maximum Sensitivity Substrate Kit and imaged using the Kodak 440CF image station (Kodak, New Haven, CT, USA). The intensity of the reactive bands was quantified using the Kodak 1D software.
To measure total nitric oxide levels we utilized a modification of the chemiluminescence method used in nitric oxide determination in blood and reported by our laboratory (Tworetzky et al., 2000). Tissue samples were treated with cold ethanol for 1 hr at −20°C and then centrifuged at 20,000 × g to precipitate proteins that can interfere with nitric oxide measurements. Potassium iodide (KI)/acetic acid reagent was prepared fresh daily by dissolving 0.05 g of KI into 7 ml of acetic acid. This reagent was added to a septum sealed purge vessel and bubbled with nitrogen gas. The gas stream was connected via a trap containing 1N NaOH to a Sievers 280i Nitric Oxide Analyzer (GE). Samples were injected with a syringe through a silicone/Teflon septum. Results were analyzed by measuring the area under the curve of the chemiluminescence signal using the Liquid software (GE). The resultant NOx value represents total nitric oxide, nitrite and NOx. PVN tissue from each animal was assessed individually and in duplicate.
The time course of expression of the constructs was ascertained in the same preliminary experiments where we ascertained the stereotaxic coordinates for the younger animals. Both the histologic expression of co-administered β-galactosidase and nitric oxide generation were measured. Peak expression was observed at ~15 days post injection and persisted to about 80% of peak values at 40 days.
Analyses and statistics
Systolic, mean and diastolic pressures were recorded via telemetery. Heart rate was derived from the arterial pulse. The data represent the mean of ten-second averages of the heart rate and arterial pressure over a 5-day period.
For baroreflex studies, arterial pressure and heart rate were the one-second averages obtained via the arterial catheter attached to the pressure transducer. RSNA was normalized using resting nerve activity as the 100% value. MAP-RSNA and MAP-HR curves were constructed for each rat by fitting all individual data points to a four-parameter logistic function according to the equation: RSNA or HR = (P1−P2)/{1 + exp[P3(MAP − P4)]} + P2, where P1 is the upper plateau of the curve, P2 is the lower plateau, P3 is the slope coefficient, and P4 is MAP at the midpoint of the curve (BP50). The range of the baroreflex curve was defined as P1−P2, and the maximum gain was calculated as − (P1−P2)P3/4. For each individual animal’s curve, the four parameters (P1 to P4) were derived and then averaged across animals. The mean parameters were used to generate averaged baroreflex curves for each group of animals. Resting values recorded before phenylephrine or nitroprusside were averaged for each curve.
All data are presented as the mean ± SE. A two-way ANOVA was used to determine the effect of the PVN injections on baseline arterial pressure and heart rate over time as well as on baroreflex curve parameters. A P-value less than 0.05 was accepted as significant.
Results
Injection sites and gene expression
Figure 1 shows a typical microinjection site and the extent of expression with β-galactosidase in the PVN. The extent of expression of these constructs has been detailed in our previous study (Rossi et al., 2004) and is limited to the PVN with some areas of the ependyma of the 3rd ventricle also showing uptake. The distances caudal to bregma indicated on Figure 1 are given for the sites of expression verified in the PVN of the adult rats at the time of brain harvesting. The coordinates required to achieve these sites at the time of injection of the immature rats at 6 weeks of age are given in the Stereotaxic Injection section of the Methods.
Figure 1. Microinjection site into paraventricular nucleus.
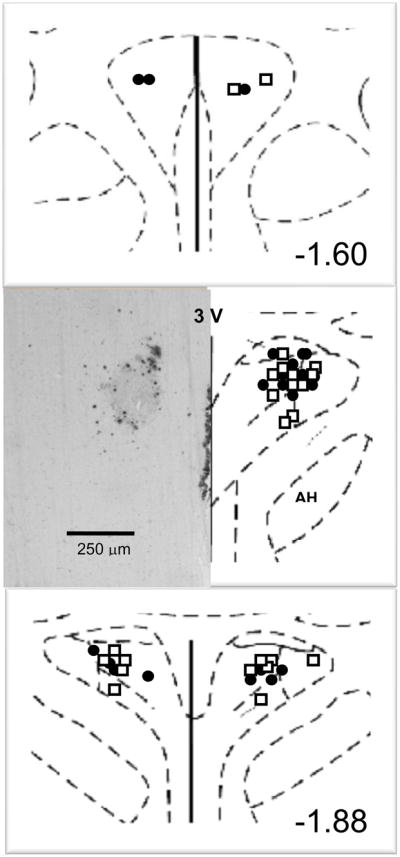
Typical microinjection site into paraventricular nucleus with β-galactosidase (left) and schematic diagram (right) of coronal section through the paraventricular nucleus are shown. The distance caudal to bregma indicated in the lower right corner is for the mature adult rat at the time of brain harvesting rather than the coordinates for microinjection used in the immature rat five weeks earlier to achieve the targeted area within the paraventricular nucleus. PVN, paraventricular nucleus; AH, anterior hypothalamus, 3V, third ventricle. •, injection sites for sham clipped groups; □, injection sites for two-kidney one-clip groups
Hemodynamic changes over time
The systemic pressures averaged over the previous 24 hr as measured by telemetry one week after renal artery clipping or sham clipping are shown in Table 1 for all six groups of rats in the awake freely moving state within their home cages prior to PVN injection. This day was designated Day 0 with respect to plasmid injection. Already at this time point, the systolic, diastolic and mean pressures were significantly different between the 2K-1C rats and their respective sham-clipped group. Pulse pressures did not differ among any of the groups (Table 1).
Table 1.
Systemic Pressures and Heart Rate on Days 0 and 35 in All Six Groups of Awake Rats
| Sham-Clipped | Two Kidney-One Clip | |||||
|---|---|---|---|---|---|---|
| β gal | WT nNOS | DN nNOS | β gal | WT nNOS | DN nNOS | |
| n | 8 | 5 | 7 | 5 | 5 | 9 |
| Day 0 | ||||||
| Systolic Pressure, mmHg | 124.0 ± 4.9 | 121.0 ± 2.5 | 122.3 ± 1.1 | 139.4 ± 4.0* | 141.9 ± 3.6* | 137.5 ± 1.7* |
| Diastolic Pressure, mmHg | 84.2 ± 4.0 | 84.1 ± 3.3 | 84.1 ± 1.3 | 104.1 ± 3.1 | 106.3 ± 3.2* | 104.7 ± 3.0* |
| Mean Pressure, mmHg | 97.5 ± 1.7 | 96.3 ± 2.4 | 96.8 ± 1.4 | 117.2 ± 3.1* | 118.2 ± 3.2* | 115.6 ± 2.9* |
| Pulse Pressure, mmHg | 39.7 ± 1.7 | 36.9 ± 1.7 | 38.2 ± 1.0 | 35.3 ± 1.9 | 35.7 ± 3.2 | 32.8 ± 1.8 |
| Heart Rate, bpm | 441 ± 5 | 452 ± 5 | 460 ± 4 | 426 ± 8* | 414 ± 8* | 418 ± 6* |
| Day 35 | ||||||
| Systolic Pressure, mmHg | 127.0 ± 5.9 | 120.0 ± 2.0 | 128.5 ± 1.9‡ | 185.1 ± 7.4*‡ | 185.8 ± 8.5*‡ | 218.8 ± 6.2*‡§◆ |
| Diastolic Pressure, mmHg | 88.6 ± 5.4 | 85.8 ± 3.0 | 89.2 ± 1.5‡ | 135.5 ± 5.7*‡ | 139.7 ± 6.1*‡ | 159.8 ± 5.0*‡§◆ |
| Mean Pressure, mmHg | 101.4 ± 5.6 | 97.2 ± 2.5 | 102.3 ± 1.7 | 152 ± 6.3*‡ | 155.1 ± 6.6*‡ | 179.5 ± 5.4*‡§◆ |
| Pulse Pressure, mmHg | 38.4 ± 1.0 | 34.2 ± 1.2 | 39.3 ± 1.0 | 49.6 ± 2.0‡ | 46.2 ± 3.0‡ | 59.0 ± 4.1*‡◆ |
| Heart Rate, bpm | 336 ± 16 | 353 ± 36 | 355 ± 6 | 366 ± 21 | 343 ± 11 | 374 ± 10 |
2K-1C, two kidney-one clip; β gal, β galactosidase; WT, wild type; DN, dominant negative; nNOS, neuronal nitric oxide synthase Values are mean ± SE; n is number of animals.
P < 0.05 vs corresponding sham clipped group, same day
P < 0.05 vs same group, day 0
P < 0.05 vs 2K-1C β gal, same day
P < 0.05 vs 2K-1C WT nNOS, same day
At the end of the next 5 weeks (Day 35), there was no difference in weight among the six groups of rats: sham-clipped β-gal, 382 ± 5 g; sham-clipped WT nNOS, 373 ± 10 g; sham-clipped DN nNOS, 385 ± 13 g; 2K-1C β-gal 369 ± 10 g; 2K-1C WT nNOS, 351 ± 9 g; and 2K-1C DN nNOS, 387 ± 14 g.
The changes in MAP and heart rate as measured by telemetry over the 5 weeks after plasmid injections into the PVN are depicted in Figure 2. Diurnal variation was maintained in all groups (data not shown). The change in MAP was greater and the change of heart rate was less from Day 15 onwards for each 2K-1C group compared with Day 0. In contrast, MAP did not change in any of the sham-clipped groups over time but heart rate decreased progressively and significantly from Day 5 onwards compared with Day 0.
Figure 2. Changes in mean arterial pressure (MAP) and heart rate (HR) as measured by telemetry after bilateral plasmid injection of the paraventricular nucleus.

On day 0, sham-clipped and two kidney-one clip (2K-1C) rats received bilateral injection with plasmids bearing constructs for β-galactosidase, wild type nNOS or dominant negative mutant nNOS in 250 nL artificial CSF. The groups are sham-clipped, β galactosidase (•, n = 8); sham-clipped, wild type nNOS (▲, n = 5); sham-clipped, dominant negative nNOS (■, n = 7); 2K-1C, β-galactosidase (○, n = 5), 2K-1C, wild type nNOS (△, n = 5); and dominant negative nNOS (□, n = 9). Values are mean ± SE. * P < 0.05 vs all other groups. § P < 0.05 for each 2K-1C group vs day 0 for the same group from days 15 through 35, inclusive. ‡ P < 0.05 for each sham-clipped group vs day 0 for the same group from days 5 through 35, inclusive. Brackets indicate P < 0.05 for MAP and HR in 2K-1C groups vs their respective sham-clipped groups from days 20 through 35, inclusive.
Notably, the change in MAP was significantly greater in the DN nNOS 2K-1C group compared not only with each of the sham-clipped groups but also with the β-gal and WT nNOS 2K-1C groups on Day 15 and all time points thereafter (Figure 2). The β-gal 2K-1C group and the WT nNOS 2K-1C groups displayed a significantly greater change in MAP than their respective sham-clipped group on Day 15 and thereafter.
The sham-clipped rats displayed faster absolute heart rate on Day 0 than the 2K-1C rats (Table 1). By Day 15, the decrease in heart rate was significantly greater in the β-gal and DN nNOS sham-clipped groups compared with their respective 2K-1C groups, whereas the decreases in heart rate in the WT nNOS sham-clipped and WT 2K-1C groups were similar (Figure 2). Thus, by Day 35 the absolute heart rate values did not differ among the groups (Table 1).
Baroreflex findings
Figure 3 depicts the average curves of the baroreflex responses of heart rate and RSNA by sham-clipped and 2K-1C groups. Resting MAP measured by the arterial catheter after conditioning in the three groups of 2K-1C rats were similar to those measured by telemetry whereas those in the sham-clipped animals tended to be higher by ~22 mmHg in each of the sham groups. Resting heart rate was elevated to a similar extent in all groups compared with telemetry values (compare Fig. 3 with Table 1). We also evaluated resting RSNA in terms of absolute μV. In β galactosidase sham-clipped and 2K-1C rats the values were 1.9 ± 0.4 and 3.6 ± 0.9 μV, respectively. A significant increase was observed 2K-1C rats injected with DN nNOS, 13.2 ± 3.5 μV (P < 0.05 vs 2K-1C β galactosidase 2K-1C). DN nNOS also resulted in an increase in the sham-clipped rats to 9.0 ± 3.2 μV, however this did not achieve significance compared with their β galactosidase sham-clipped rats.
Figure 3. Baroreflex curves of heart rate and renal sympathetic nerve activity (RSNA) responses.

Mean baroreflex curves of RSNA and heart rate (HR) responses are shown for β-galactosidase (— • —); wild type nNOS (--- ---); dominant negative nNOS (⋯○⋯) for sham-clipped (left panel) and two kidney-one clip (right panel) groups. Symbols on the curves represent resting values (mean ± SE). See Table 3 for complete statistics and numbers in each group.
As expected, MAP-RSNA and MAP-HR baroreflex curves were shifted to the higher values of arterial pressure in all 2K-1C groups. In addition, the lower plateau of the baroreflex curve of HR was significantly higher in all 2K-1C groups compared with sham-clipped rats consistent with an impaired bradycardic response to the rise in arterial pressure in hypertensive animals. Introducing DN or WT nNOS constructs into PVN had no effect on the baroreflex response of either HR or RSNA. In Figure 3, there appears to be a tendency to manifest a higher upper plateau of the baroreflex response of RSNA in the DN nNOS 2K-1C group but the response among the individual animals was highly variable and did not achieve significance (P = 0.16 vs 2K-1C β-galactosidase). In the two 2K-1C rats in which DN nNOS injections were located outside the PVN average resting MAP (144 mmHg) and HR (377 bpm) did not differ from that in rats in which the PVN was injected with β-galactosidase. Baroreflex parameters likewise were similar to the β-gal 2K-1C rats.
Intra-PVN administration of L-NAME at the end of the series of experiments (~ Day 38 ± 1) resulted in a rise in MAP of 15.9 ± 3.2 and 15.0 ± 2.5 mmHg in 2K-1C rats that had been previously injected with β-galactosidase or WT nNOS compared with 3.8 ± 1.0 mmHg in the DN nNOS treated 2K-1C rats (P < 0.02), consistent with functional blockade of roughly 76% of the nitric oxide at that locus. L-NAME increased MAP by 11.4 ± 1.2 mmHg in the β-gal sham-clipped group and 1.4 ± 1.2 mmHg in the DN nNOS sham-clipped group.
Assessment of nNOS monomers and dimers
Figure 4A shows that levels of monomeric nNOS from PVN tissue of 2K-1C rat were significantly increased in PVN from rats injected with either p725 (heme) or p739 (hemeRedF) dominant negative nNOS constructs compared with those injected with β-galactosidase. Despite the higher levels of monomers, the mutant nNOS constructs impaired dimerization of nNOS that is required for enzymatic activity (Figure 4B). The NOx values for p725 (n = 2) and p739 (n = 2) were virtually equivalent so the data were combined for comparison with β-galactosidase. NOx levels in the PVN from 2K-1C rats injected with the dominant negative constructs were ~44% of the value from β-galactosidase injected rats (Figure 5). Sham-clipped rats demonstrated the same pattern of changes with injection of β-galactosidase, WT or DN constructs for nNOS as 2K-1C rats (data not shown). NOx levels in PVN tissue were lower in the β-galactosidase treated sham-clipped group with 263 ± 90 pmol/mg protein compared with 779 ± 161 pmol/mg protein in the β galactosidase 2K-1C group (P < 0.05). DN nNOS injection also decreased NOx in the sham-clipped rats to 176 ± 112 pmol/mg protein but this value did not differ significantly from the β galactosidase treated group.
Figure 4.

Expression of nNOS protein and dimers in paraventricular nucleus (PVN) of 2K-1C rats. (A) Representative western blot analysis of total endogenous nNOS protein expression in PVN from 2K-1C rats microinjected 5–6 weeks earlier with plasmids delivering either β-galactosidase (β gal) or the dominant negative nNOS constructs p735 (heme; left panel) or p739 (hemeRedF; right panel). Results of densitometric analysis of 3 separate experiments are shown. For a description of constructs and their sequences see (Phung & Black, 1999). Values are mean ± SE; n = 3 for each group; * P < 0.05 vs β galactosidase. (B) Reduction of nNOS dimer formation in PVN. Densitometric analysis of the level of dimer to monomer levels expressed as percent of total nNOS from PVN tissue 5–6 weeks after injection as in (A). Values are mean ± SE; n = 3 for each group; * P < 0.05 vs β galactosidase.
Figure 5. Total nitric oxide (NOx) levels in paraventricular nucleus of 2K-1C rats.
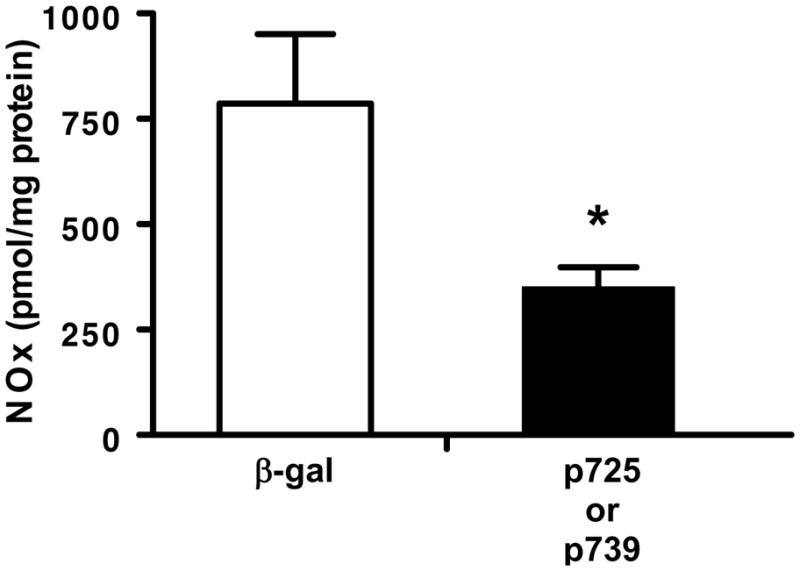
Total nitric oxide (NOx = nitric oxide + nitrite + NOx) in tissue from 2K-1C rats microinjection 5–6 weeks earlier with plasmids delivering either β-galactosidase (β-gal) or the dominant negative plasmids (p725 and p739). Tissue from each rat was analyzed separately. Results from the dominant negative plasmids were combined for analysis. Values are mean ± SE; n = 3 for β galactosidase and 4 for the combined dominant negative groups; * P < 0.05 vs β galactosidase.
Discussion
The present study assessed the role of nitric oxide within the PVN on the progressive increase in systemic arterial pressure in the 2K-1C model of renovascular hypertension. The major finding of this study was that systemic pressure increased at a more rapid rate and to higher absolute pressures in conscious, freely moving 2K-1C rats that had been injected with the dominant negative construct of nNOS into the PVN. As expected with hypertension, the baroreflex response curves were shifted to higher pressures in each of the 2K-1C groups compared with the sham-clipped groups; however, blockade of nitric oxide formation did not substantively alter any other parameters of the baroreflex responses of either heart rate or RSNA.
PVN nNOS and arterial pressure
By the second week after PVN injection, the 2K-1C group that had received the dominant negative constructs of nNOS already displayed a significantly higher pressure than the other 2K-1C groups and continued to display a steeper rise in arterial pressure and pulse pressure throughout the remaining period of observation. Contrary to our hypothesis, injection with a construct of the wild type nNOS construct did not prevent the rise in arterial pressure in 2K-1C rats at least during the timeframe of observation. Thus, it is possible that the ability to generate nitric oxide was already at or near maximal potential by three to five weeks after injection (i.e, four to six weeks after clipping; see below). Moreover, neither the wild type nor dominant negative nNOS constructs changed arterial pressure in the sham-clipped rats. Our constructs were not targeted specifically to either glia or neurons, so that the nitric oxide within the PVN could have been generated from either or both cell types. Together these findings indicate that generation of nitric oxide in PVN increases in 2K-1C hypertension and serves to dampen the rise in arterial pressure. Furthermore, normotensive animals do not appear to be as dependent on PVN nitritergic signaling as their hypertensive counterparts.
To our knowledge there are no reports of direct measurements of nNOS protein or its activity and NOx levels in the PVN of 2K-1C rats. The number of NADPH diaphorase positive neurons, an index of NOS activity, was decreased in spontaneously hypertensive rats (DiCarlo et al., 2002), however, cell positivity alone may not necessarily reflect ambient nitric oxide levels. The majority of NOS positive neurons in the hypothalamus are found in the PVN and supraoptic nuclei (Bredt et al., 1990; Vincent & Kimura, 1992; Krukoff et al., 1995), and Krukoff et al (1995) showed that nNOS gene expression in hypothalamic tissue which included the PVN from 2K-1C rats was decreased at three weeks and increased at six weeks after clipping. The only studies of nNOS protein in 2K-1C rats showed increased nNOS in the caudal medulla, but the PVN was not examined (Wang et al., 2002). In spontaneously hypertensive rats NOS activity was attenuated during the early phase of development of hypertension compared with normotensive Wistar-Kyoto rats but was clearly increased once hypertension was established (Qadri et al., 2003). Notably, in the spontaneously hypertensive rat, the elevated NOS activity in the hypothalamus was normalized by systemic inhibition of angiotensin converting enzyme activity or blockade of the angiotensin AT1 receptor (Qadri et al., 2003). This suggests that enhancement of sympathetic vasomotor tone by circulating Ang II may be mitigated by nitritergic signaling within the PVN. Conversely, the increase in basal nerve activity in the dominant negative nNOS treated sham-clipped and 2K-1C rats indicate that interference with dimerization of nNOS results in failure to dampen sympathetic outputs and that this exerts a greater regulatory influence in hypertensive animals.
The present results with chronic interference of the generation of nitric oxide within the PVN are consistent with data from the acute pharmacologic studies of on et al (1999) who demonstrated that injection of a nitric oxide inhibitor into the left cerebral ventricle increased arterial pressure in mRen2(27) transgenic rats with renin dependent hypertension but not in normotensive Sprague Dawley rats. Together, these studies suggest that under conditions of an activated renin-angiotensin system nitric oxide serves to buffer the increase in systemic pressure, and that at least in the 2K-1C model the PVN is one site for such nitritergic modulation. The current data cannot unequivocally distinguish whether attenuation of nitritergic signaling within the PVN is important at some early critical time point during the Ang II-dependent sympathoexcitatory phase of 2K-1C hypertension (Martinez-Maldonado, 1991) or later as the model transitions to increased sympathetic output that is independent of Ang II (Kopp & Buckley-Bleiler, 1989; Martinez-Maldonado, 1991). Nonetheless, even at six weeks, we reported that circulating Ang II levels continue to be elevated up to three-fold in 2K-1C rats (Maliszewska-Scislo et al., 2008) and this Ang II may participate in generating neurogenic hypertension (Fink, 1997; Osborn et al., 2009). Blood for plasma Ang II was not obtained in these animals since the volume required may have compromised the assessment of the baroreflex and, conversely, assessment of the baroreflexes would have altered Ang II. In the dominant negative injected 2K-1C group, the increase in arterial pressure is significantly potentiated only after the third week post clipping. In light of earlier findings that nNOS expression and/or its activity is low prior to that time period (Krukoff et al., 1995), it is probable that nitric oxide within the PVN plays a more important role as blood pressure reaches a critical value. It is not likely that the delay occurs because of a longer time required for expression of the dominant negative protein since it has been shown that these constructs are expressed as early as four days after injection (Rossi et al., 2004). The small response to L-NAME in rats previously treated with the dominant negative nNOS construct also permit the reasonable inference that the primary source of this endogenous nitric oxide is from nNOS in PVN.
PVN nNOS and arterial baroreflex responses
Typical of hypertension in general (Guo et al., 1983; Head, 1995) and renovascular hypertension in particular (Moyses et al., 1994; Head & Burke, 2001; Vitela et al., 2005), the baroreflex responses of heart rate and RSNA were shifted rightward to higher pressures in each of the 2K-1C groups. Loading of the baroreceptors resulted in an attenuated bradycardic response in the hypertensive animals. These findings are consistent with those reported by Head and Burke (2001) in rabbits with 2K-1C hypertension.’
Although the dominant negative treated 2K-1C group showed a tendency toward a higher upper plateau, the responses showed considerable variation among the individual animals that may have been due to the degree to which the hypertension is dependent on Ang II at this six-week point (Yoshida et al., 1991). The baroreflex responses of heart rate or RSNA of wild type nNOS 2K-1C group were not different from the 2K-1C rats injected with the β-galactosidase construct. This observation is in line with a recent report that overexpression of eNOS in 2K-1C mice did not change baroreflex sensitivity despite decreasing arterial pressure (Gava et al., 2008). However, we cannot discount a potential effect of intervening surgical or environmental stress that may have obscured effects on the baroreflex especially as resting heart rate was higher in all groups compared with resting heart rate by telemetry in the home cage. The effect on RSNA is even harder to judge as telemetric nerve data for comparison are not available.
PVN nNOS and heart rate
Notably, prior to PVN injection and just one week after renal artery clipping (Day 0 in Table 1 and Figure 2), the 2K-1C groups displayed a significant bradycardia compared with their respective sham-clipped controls. Thus, early in the course of renovascular hypertension heart rate remains at least somewhat responsive to loading of the baroreceptors. The normotensive groups displayed a decrease in heart rate over time despite no change in systemic pressure. Jacob et al (2005) and others (Jaehne et al., 2008) have reported a similar decline in heart rate assessed by telemetry in normal Sprague Dawley rats over a comparable timeframe. We speculate that increased parasympathetic tone, decreased sympathetic tone, or a combination of both could account for this progressive bradycardia. However, neither of these earlier studies nor the present one directly addresses this mechanism. This decline in heart rate was diminished in the β-galactosidase 2K-1C group and 2K-1C rats that had received the dominant negative nNOS displayed the smallest decrease in heart rate despite having the highest arterial pressures. This response is consistent with the reduced bradycardic response observed with loading of the baroreceptors. The present findings are in keeping with studies showing a tachycardic response to inhibition of PVN nitric oxide generation by acute pharmacologic blockade of NOS activity (Zhang & Patel, 1998; Li et al., 2002; Li et al., 2003) as well as by antisense oligodeoxynucleotides directed at nNOS (Wang et al., 2005).
A concomitant decrease in parasympathetic tone in the 2K-1C groups cannot be excluded. In addition to projections to medullary loci regulating sympathetic output to the cardiovascular system, the PVN also contains separate neuronal populations that project to the dorsal vagal complex regulating parasympathetic autonomic function (Li et al., 2003). During β-adrenergic receptor blockade in anesthetized rats, Stauss et al (1997) unmasked a parasympathetic mediated decrease in heart rate in response to direct stimulation of the PVN with increasing frequencies. Not only does PVN nitric oxide inhibit neurons projecting to the rostral ventrolateral medulla, but PVN neurons projecting to the dorsal vagal complex also decrease their basal firing rate (Li et al., 2003). Under the current conditions of chronic nNOS inhibition, it appears that the inhibition of the sympathetic component overrides any inhibition of the parasympathetic output. The possibility that 2K-1C rats have lower overall vagal tone and/or that our injections encompassed a population of PVN neurons that are primarily directed at sympathetic autonomic targets cannot be excluded. The sham-clipped animals display similar bradycardia regardless of the type of nNOS construct they received which indicates that they may have more balanced sympathetic-parasympathetic outputs than their hypertensive counterparts.
Dominant negative nNOS injections
The current approach using plasmids designed to deliver gene constructs of nNOS into the PVN of rats with 2K-1C hypertension is a modification of adenoviral transfection to over-express nNOS in normal rats (Li et al., 2003) or antisense oligodeoxynucleotides to knockdown nNOS in PVN in rats with heart failure (Wang et al., 2005) to study neural regulation of cardiovascular function. The use of naked plasmid DNA has the advantage of introducing the recombinant DNA while minimizing transfection at distant sites without inducing an antibody response typically seen with viral vectors (Wolff & Budker, 2005). If the target cell is postmitotic or has long cell cycle, long term gene expression of the exogenous protein has been documented from up to 12 weeks (Herweijer et al., 2001) to more than one year (Sebestyen et al., 2007). Although the efficiency of gene transfer may be lower than with viral vectors, bolus injection results in uptake by neurons without attendant cellular damage. One caveat is that non-neuronal elements such as glial cells could also be transfected (Shi et al., 2007). Another advantage is that the central nervous system is more likely to be spared from an immune response to the expressed transgene.
The gene constructs of heme (p725) and hemeRedF (p739) interfere with wild type nNOS dimerization required for activity of the enzyme thereby resulting in less than 8% normal catalytic activity in COS-7 cells (Phung & Black, 1999). Studies in rats injected with plasmids bearing the same dominant negative nNOS gene resulted in potentiation of the acute pressor response to central endothelin (Rossi et al., 2004). Importantly, the PVN tissue from rats injected with either of the dominant negative constructs displayed an increase in monomeric nNOS but a significant decrease in dimer formation. This is consistent with upregulation of the endogenous nNOS gene in response to the dominant negatives, yet with a failure of dimerization needed for activity because of the presence of the dominant negative products. This decrease in nNOS activity is reflected in the significant decline in NOx directly measured in the PVN. Notably, the increase in monomeric nNOS and decrease in dimers is remarkably close to findings in COS-7 cells with these mutants (Phung & Black, 1999). Moreover, since two uniquely different dominant negative constructs decreased the level of dimer formation to very similar levels and also resulted in similar physiologic responses, the findings appear to be both reproducible and robust. Collectively, these findings demonstrate that the dominant negative nNOS plasmid injections were clearly able to interfere with the generation of nitric oxide over the period of observation.
Perspectives
In summary, the present study provides evidence that nitric oxide generated by nNOS within the PVN chronically buffers the rise in arterial pressure that accompanies 2K-1C hypertension. This effect becomes apparent ~3 weeks post clipping and is sustained throughout the course of observation. This timeframe is pivotal in the development of 2K-1C hypertension since it coincides with the timeframe that nNOS message and activity transition from low to high levels and when the model moves toward greater dependence on sympathoexcitation. Overexpression of nNOS does not appear to alter the responses and may well indicate that nitric oxide signaling may already be maximized. Formal baroreflex testing revealed that PVN nitric oxide does not substantively modulate the reflex responses of either heart rate or RSNA. However, the telemetry data show that despite having the highest arterial pressures, the dominant negative nNOS 2K-1C group exhibited a significantly attenuated bradycardia. The specific reason for this discrepancy is not readily apparent, however, intervening anesthesia and surgery for electrode placement may well have contributed even though the rats were tested in the conscious state. The telemetry data suggest that PVN nitric oxide may modulate the baroreflex response of heart rate in this model. This will require further evaluation of the baroreflexes using technologies permitting both heart rate and nerve recordings by telemetry.
Acknowledgments
This study was supported by the National Institutes of Health grants HL-079102 to NF Rossi and HD-039110 to SM Black.
References
- Bergamaschi C, Campos RR, Schor N, Lopes OU. Role of the rostral ventrolateral medulla in maintenance of blood pressure in rats with Goldblatt hypertension. Hypertension. 1995;26:1117–1120. doi: 10.1161/01.hyp.26.6.1117. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- Chen QH, Haywood JR, Toney GM. Sympathoexcitation by PVN-injected bicuculline requires activation of excitatory amino acid receptors. Hypertension. 2003;42:725–731. doi: 10.1161/01.HYP.0000085197.20043.44. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Horiuchi J, Killinger S, Sheriff MJ, Tan PS, McDowall LM. Long-term regulation of arterial blood pressure by hypothalamic nuclei: some critical questions. Clin Exp Pharmacol Physiol. 2005;32:419–425. doi: 10.1111/j.1440-1681.2005.04205.x. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Horiuchi J, Tagawa T, Fontes MA, Potts PD, Polson JW. Medullary and supramedullary mechanisms regulating sympathetic vasomotor tone. Acta Physiol Scand. 2003;177:209–218. doi: 10.1046/j.1365-201X.2003.01070.x. [DOI] [PubMed] [Google Scholar]
- DiCarlo SE, Zheng H, Collins HL, Rodenbaugh DW, Patel KP. Daily exercise normalizes the number of diaphorase (NOS) positive neurons in the hypothalamus of hypertensive rats. Brain Res. 2002;955:153–160. doi: 10.1016/s0006-8993(02)03400-5. [DOI] [PubMed] [Google Scholar]
- Esler M, Kaye D. Increased sympathetic nervous system activity and its therapeutic reduction in arterial hypertension, portal hypertension and heart failure. J Auton Nerv Syst. 1998;72:210–219. doi: 10.1016/s0165-1838(98)00107-6. [DOI] [PubMed] [Google Scholar]
- Fink GD. Long-term sympatho-excitatory effect of angiotensin II: a mechanism of spontaneous and renovascular hypertension. Clin Exp Pharmacol Physiol. 1997;24:91–95. doi: 10.1111/j.1440-1681.1997.tb01789.x. [DOI] [PubMed] [Google Scholar]
- Gava AL, Peotta VA, Cabral AM, Vasquez EC, Meyrelles SS. Overexpression of eNOS prevents the development of renovascular hypertension in mice. Can J Physiol Pharmacol. 2008;86:458–464. doi: 10.1139/y08-044. [DOI] [PubMed] [Google Scholar]
- Giles ME, Sly DJ, McKinley MJ, Oldfield BJ. Neurons in the lamina terminalis which project polysynaptically to the kidney express angiotensin AT1A receptor. Brain Res. 2001;898:9–12. doi: 10.1016/s0006-8993(01)02113-8. [DOI] [PubMed] [Google Scholar]
- Guo GB, Thames MD, Abboud FM. Arterial baroreflexes in renal hypertensive rabbits. Selectivity and redundancy of baroreceptor influence on heart rate, vascular resistance, and lumbar sympathetic nerve activity. Circ Res. 1983;53:223–234. doi: 10.1161/01.res.53.2.223. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Hall JE, Guyton AC, Brands MW. Pressure-volume regulation in hypertension. Kidney Int Suppl. 1996;55:S35–41. [PubMed] [Google Scholar]
- Head GA. Baroreflexes and cardiovascular regulation in hypertension. J Cardiovasc Pharmacol. 1995;26(Suppl 2):S7–16. [PubMed] [Google Scholar]
- Head GA, Burke SL. Renal and cardiac sympathetic baroreflexes in hypertensive rabbits. Clin Exp Pharmacol Physiol. 2001;28:972–975. doi: 10.1046/j.1440-1681.2001.03567.x. [DOI] [PubMed] [Google Scholar]
- Herweijer H, Zhang G, Subbotin VM, Budker V, Williams P, Wolff JA. Time course of gene expression after plasmid DNA gene transfer to the liver. J Gene Med. 2001;3:280–291. doi: 10.1002/jgm.178. [DOI] [PubMed] [Google Scholar]
- Jacob F, Clark LA, Guzman PA, Osborn JW. Role of renal nerves in development of hypertension in DOCA-salt model in rats: a telemetric approach. Am J Physiol Heart Circ Physiol. 2005;289:H1519–1529. doi: 10.1152/ajpheart.00206.2005. [DOI] [PubMed] [Google Scholar]
- Jaehne EJ, Salem A, Irvine RJ. The effect of long-term repeated exposure to 3,4-methylenedioxymethamphetamine on cardiovascular and thermoregulatory changes. Psychopharmacology (Berl) 2008;201:161–170. doi: 10.1007/s00213-008-1258-9. [DOI] [PubMed] [Google Scholar]
- Kopp UC, Buckley-Bleiler RL. Impaired renorenal reflexes in two-kidney, one clip hypertensive rats. Hypertension. 1989;14:445–452. doi: 10.1161/01.hyp.14.4.445. [DOI] [PubMed] [Google Scholar]
- Krukoff TL, Gehlen F, Ganten D, Wagner J. Gene expression of brain nitric oxide synthase and soluble guanylyl cyclase in hypothalamus and medulla of two-kidney, one clip hypertensive rats. Hypertension. 1995;26:171–176. doi: 10.1161/01.hyp.26.1.171. [DOI] [PubMed] [Google Scholar]
- LaGrange LP, Toney GM, Bishop VS. Effect of intravenous angiotensin II infusion on responses to hypothalamic PVN injection of bicuculline. Hypertension. 2003;42:1124–1129. doi: 10.1161/01.HYP.0000102181.83892.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang W, Stern JE. Nitric oxide inhibits the firing activity of hypothalamic paraventricular neurons that innervate the medulla oblongata: role of GABA. Neuroscience. 2003;118:585–601. doi: 10.1016/s0306-4522(03)00042-3. [DOI] [PubMed] [Google Scholar]
- Li YF, Roy SK, Channon KM, Zucker IH, Patel KP. Effect of in vivo gene transfer of nNOS in the PVN on renal nerve discharge in rats. Am J Physiol Heart Circ Physiol. 2002;282:H594–601. doi: 10.1152/ajpheart.00503.2001. [DOI] [PubMed] [Google Scholar]
- Lon S, Szczepanska-Sadowska E, Paczwa P, Ganten D. Enhanced blood pressure buffering role of the brain nitrergic system in renin transgenic rats. Brain Res. 1999;842:384–391. doi: 10.1016/s0006-8993(99)01857-0. [DOI] [PubMed] [Google Scholar]
- Maliszewska-Scislo M, Chen H, Augustyniak RA, Seth D, Rossi NF. Subfornical organ differentially modulates baroreflex function in normotensive and two-kidney, one-clip hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R741–750. doi: 10.1152/ajpregu.00157.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpas SC. What sets the long-term level of sympathetic nerve activity: is there a role for arterial baroreceptors? Am J Physiol Regul Integr Comp Physiol. 2004;286:R1–R12. doi: 10.1152/ajpregu.00496.2003. [DOI] [PubMed] [Google Scholar]
- Martinez-Maldonado M. Pathophysiology of renovascular hypertension. Hypertension. 1991;17:707–719. doi: 10.1161/01.hyp.17.5.707. [DOI] [PubMed] [Google Scholar]
- Moyses MR, Cabral AM, Marcal D, Vasquez EC. Sigmoidal curve-fitting of baroreceptor sensitivity in renovascular 2K1C hypertensive rats. Braz J Med Biol Res. 1994;27:1419–1424. [PubMed] [Google Scholar]
- Osborn JW, Averina VA, Fink GD. Current computational models do not reveal the importance of the nervous system in long-term control of arterial pressure. Exp Physiol. 2009;94:389–396. doi: 10.1113/expphysiol.2008.043281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn JW, Jacob F, Guzman P. A neural set point for the long-term control of arterial pressure: beyond the arterial baroreceptor reflex. Am J Physiol Regul Integr Comp Physiol. 2005;288:R846–855. doi: 10.1152/ajpregu.00474.2004. [DOI] [PubMed] [Google Scholar]
- Phung YT, Black SM. Use of chimeric forms of neuronal nitric-oxide synthase as dominant negative mutants. IUBMB Life. 1999;48:333–338. doi: 10.1080/713803520. [DOI] [PubMed] [Google Scholar]
- Qadri F, Arens T, Schwarz EC, Hauser W, Dendorfer A, Dominiak P. Brain nitric oxide synthase activity in spontaneously hypertensive rats during the development of hypertension. J Hypertens. 2003;21:1687–1694. doi: 10.1097/00004872-200309000-00018. [DOI] [PubMed] [Google Scholar]
- Rossi NF, Black SM, Telemaque-Potts S, Chen H. Neuronal nitric oxide synthase activity in the paraventricular nucleus buffers central endothelin-1-induced pressor response and vasopressin secretion. J Cardiovasc Pharmacol. 2004;44(Suppl 1):S283–288. doi: 10.1097/01.fjc.0000166275.32421.c6. [DOI] [PubMed] [Google Scholar]
- Sebestyen MG, Hegge JO, Noble MA, Lewis DL, Herweijer H, Wolff JA. Progress toward a nonviral gene therapy protocol for the treatment of anemia. Hum Gene Ther. 2007;18:269–285. doi: 10.1089/hum.2006.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Gounko NV, Wang X, Ronken E, Hoekstra D. In situ entry of oligonucleotides into brain cells can occur through a nucleic acid channel. Oligonucleotides. 2007;17:122–133. doi: 10.1089/oli.2007.0034. [DOI] [PubMed] [Google Scholar]
- Stauss HM, Persson PB, Johnson AK, Kregel KC. Frequency-response characteristics of autonomic nervous system function in conscious rats. Am J Physiol. 1997;273:H786–795. doi: 10.1152/ajpheart.1997.273.2.H786. [DOI] [PubMed] [Google Scholar]
- Stocker SD, Keith KJ, Toney GM. Acute inhibition of the hypothalamic paraventricular nucleus decreases renal sympathetic nerve activity and arterial blood pressure in water-deprived rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R719–725. doi: 10.1152/ajpregu.00494.2003. [DOI] [PubMed] [Google Scholar]
- Tagawa T, Dampney RA. AT(1) receptors mediate excitatory inputs to rostral ventrolateral medulla pressor neurons from hypothalamus. Hypertension. 1999;34:1301–1307. doi: 10.1161/01.hyp.34.6.1301. [DOI] [PubMed] [Google Scholar]
- Thrasher TN. Baroreceptors and the long-term control of blood pressure. Exp Physiol. 2004;89:331–335. doi: 10.1113/expphysiol.2004.027441. [DOI] [PubMed] [Google Scholar]
- Tworetzky W, Moore P, Bekker JM, Bristow J, Black SM, Fineman JR. Pulmonary blood flow alters nitric oxide production in patients undergoing device closure of atrial septal defects. J Am Coll Cardiol. 2000;35:463–467. doi: 10.1016/s0735-1097(99)00576-8. [DOI] [PubMed] [Google Scholar]
- Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience. 1992;46:755–784. doi: 10.1016/0306-4522(92)90184-4. [DOI] [PubMed] [Google Scholar]
- Vitela M, Herrera-Rosales M, Haywood JR, Mifflin SW. Baroreflex regulation of renal sympathetic nerve activity and heart rate in renal wrap hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R856–862. doi: 10.1152/ajpregu.00620.2004. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen ZJ, Luo CQ, Pan JY. The expression of neuronal nitric oxide synthase in caudal medulla of two-kidney one clip Goldblatt hypertension rat. Sheng Li Xue Bao. 2002;54:175–178. [PubMed] [Google Scholar]
- Wang Y, Liu XF, Cornish KG, Zucker IH, Patel KP. Effects of nNOS antisense in the paraventricular nucleus on blood pressure and heart rate in rats with heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H205–213. doi: 10.1152/ajpheart.00497.2004. [DOI] [PubMed] [Google Scholar]
- Weekley LB. Renal renin secretion rate and norepinephrine secretion rate in response to centrally administered angiotensin-II: role of the medial basal forebrain. Clin Exp Hypertens A. 1992;14:923–945. doi: 10.3109/10641969209036227. [DOI] [PubMed] [Google Scholar]
- Wolff JA, Budker V. The mechanism of naked DNA uptake and expression. Adv Genet. 2005;54:3–20. doi: 10.1016/S0065-2660(05)54001-X. [DOI] [PubMed] [Google Scholar]
- Ye S, Nosrati S, Campese VM. Nitric oxide (NO) modulates the neurogenic control of blood pressure in rats with chronic renal failure (CRF) J Clin Invest. 1997;99:540–548. doi: 10.1172/JCI119191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Takata M, Ueno H, Tomoda F, Yasumoto K, Iida H, Sasayama S. Sympathetic and renin-angiotensin system in conscious rabbits with two-kidney, one clip hypertension. Nippon Jinzo Gakkai Shi. 1991;33:879–884. [PubMed] [Google Scholar]
- Zhang K, Patel KP. Effect of nitric oxide within the paraventricular nucleus on renal sympathetic nerve discharge: role of GABA. Am J Physiol. 1998;275:R728–734. doi: 10.1152/ajpregu.1998.275.3.R728. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]