

Abstract
A putative response element, GAGCCTC, was observed years ago in foot-printing analysis of the c-jun promoter and here we investigate its function in regulating c-jun expression and identify a protein complex that binds there. Electrophoretic mobility shift assays demonstrate a sequence-specific binding complex with this element in HEK293 cells. Additionally, unlabeled consensus AP-1 element DNA, but not a similar NF-jun element DNA, competes with complex formation. Mutations of this element decrease c-jun promoter reporter activity by nearly 5-fold in HEK293 cells. A new, two-step oligonucleotide trapping technique was developed to purify the element binding proteins. LC-Nanospray-ESI-MS/MS identification and western blotting show that the purified complex contains Ku80, and c-jun, which was further confirmed by antibody supershift, by immunoprecipitation with southwestern blot or with UV crosslinking analysis in vitro as well as chromatin immunoprecipitation in vivo. c-Jun promoter activity and c-jun expression were decreased by Ku80 siRNA introduction. A mutant Ku80 plasmid with normal amino acid sequence but immune to the siRNA recovers c-jun promoter activity from siRNA inhibition. Similarly, Ku70 wild type transfection can also up-regulate c-jun promoter activity. Thus , Ku80-c-jun activates c-jun expression by binding to this GAGCCTC element in the c-jun promoter and Ku70 may also serve a role.
Keywords: c-jun, promoter, Ku70, Ku80, transcription factor
INTRODUCTION
The c-jun protein is a component of the essential transcription factor Activator Protein-1 (AP-1). AP-1 is known to autoregulate c-jun expression by binding to AP-1 sites within the c-jun promoter (1). The c-jun gene, as a member of the early response gene family, is induced by serum, ionizing radiation, phorbol esters such as TPA (12-O-tetradecanoylphorbol-13-acetate), and tumor necrosis factor-α (2). After stimulation by serum, c-jun mRNA increased 7-8 fold, and it reached its maximal level (100-200 fold) one to two hours after stimulation by UV irradiation as a consequence of increasing transcription (3, 4).
AP-1 composition consists of either homodimers of c-jun, junB, or junD, or of heterodimers with members of the bZIP family, including c-jun, fos, ATF/CREB, Maf, CBP, NFAT, and c-Rel by physically interacting via a “leucine zipper” motif (5, 6). AP-1 regulates the expression of multiple genes essential for cell proliferation, differentiation and apoptosis by binding to the AP-1 sites on their promoters, and different AP-1 compositions execute distinct biological functions (2, 7). c-Jun-c-fos heterodimer binds in vitro with the TRE (TPA responsive element) sequence motif, TGAGTCA, which is an important element in the collagenase promoter (8). c-jun-ATF2 heterodimer has a high affinity for the asymmetric sequence of jun2TRE (TTACCTCA) in the c-jun promoter and functions as a main regulator for c-jun expression (9). It has been shown that different compositions of AP-1, c-jun-c-fos or c-jun-ATF2, affects transcriptional activity of collagenase or c-jun with or without stimulation by TPA and MEKK1-δ (7). The activity of AP-1 is strongly induced by numerous signals including growth factors, cytokines, and various extracellular stresses and is mediated, in part, by the jun N-terminal kinases (JNKs) phosphorylations of c-jun at sites on serines 63 and 73 (10, 11). Other kinases including CDK1, protein kinase C, casein kinase II, and pp44 MAPK also phosphorylate AP-1 (12).
The c-jun promoter has been reported to contain several binding sites for transcription factors by in vivo footprinting analysis, which includes two AP-1 sites, and a single SP-1, Mef-2 (RSRF), NF-jun, CTF, Smad3/Smad4 site and a putative element sequence (GAGCCTC) bound by an unknown factor (3, 4). The two AP-1 binding sites contain the proximal jun1TRE (-71 TGACATCA -64) and distal jun2TRE (-190 TTACCTCA-183). The jun1TRE has been reported to be a TPA response element (1). The jun2TRE, bound by c-jun-ATF2 heterodimer, is the decisive element in mediating c-jun induction by the 243 amino acid E1A product (9). UV and phorbol ester treatment of HeLa cells increases the binding of transcription factors to both elements (13). The Mef-2 site at −59 is an important element for EGF (epidermal growth factor), serum and TPA induction (14). The element sequence from −139 to −129 is bound by nuclear factor-jun (NF-jun) and also plays a role in inducing c-jun transcription (15). Recently, a positive regulatory factor in rat liver, different from NF-jun, has been reported to enhance transcription of c-jun by binding to the c-jun promoter region (nt -148 to −124) (16, 17). Until now, many cis-acting elements have been reported to be involved in the regulation of c-jun, but the exact mechanism of c-jun expression regulation is not clearly addressed under different physiological conditions and in different cell types. A putative element from -176 to -167 was identified by DNase I and MDS footprinting (3, 4). Interestingly, this region contains the sequence GAGCCTC with obvious dyad symmetry. Search of this sequence against all known elements reveals that this sequence is unknown as a transcription factor binding site (data not shown). This novel element was investigated here to further clarify the regulation of c-jun expression.
In this study, we purified and isolated the proteins binding to this putative element (-176-GAGCCTCcgc-167) in the c-jun promoter by a modified systematic oligonucleotide trapping approach, identified it by a combination of proteomics and immunological techniques, and confirmed its regulatory function in c-jun expression.
These investigations led to Ku80. Ku80, along with Ku70, are DNA end binding proteins, which dimerize bringing the ends together in the repair of double-stranded DNA breaks, although it is known to also serve other roles (18). Here, we found that Ku80 also interacts with c-jun to form a complex at the putative element which activates c-jun expression.
MATERIALS AND METHODS
Rabbit polyclonal antibodies: Lamin A/C (catalogue # H-110), c-jun (N), jun B (N-17), jun D (329), ATF1 (H-60), ATF2 (C-19), HMG-2 (FL-215), Tho4 (11G5), PARP1 (H-300) and mouse monoclonal antibodies: phospho-c-jun (KM-1), Ku70 (E-5), Ku80 (B-1), hnRNP A2 (DP3B3) and goat polyclonal antibody: hnRNP A1 (N-15) were obtained from Santa Cruz Biotechnology (California, USA). NF-45 antibody was from Aviva Systems Biology (San Diego, CA, USA).
Plasmid construction
Construction of a luciferase reporter plasmid for c-jun promoter wild type (WT), deletion (DEL) and mutation (MUT)
The c-jun promoter wild type (WT) (nt −200 to + 81) was amplified as described before (19) from pUC-19-c-jun plasmid using FP and RP primers listed in Table 1. c-Jun deletion (DEL) or mutation (MUT) were each prepared by an overlap extension technique. The first step was to amplify two fragments in separate reactions: using FP and Del 1 primers for one reaction and RP with Del 2 primers for another reaction for c-jun (DEL). The two PCR products have overlapping, complementary ends with the desired deletion. The products of the two reactions were combined, diluted 100-fold, and used as a template for a third PCR reaction using FP and RP primers to produce the complete sequence containing the deletion. Using the same method, c-jun mutation (MUT) was produced using Mut1 and Mut2 primers shown in Table 1. c-Jun (DEL) or c-jun (MUT) DNA was cloned into pUC19 for sequence confirmation and then subcloned to pMLuc-1 Luciferase vector (Promega, Madison, WI, USA) between the BamHI and the EcoRI restriction sites.
Table 1.
Oligonucleotide sequences
| Name | Oligonucleotide Sequence |
|---|---|
| FP | 5’-cgggatcccagcggagcattacctcatc-3’ |
| RP | 5’-cggaattcgctggctgtgtctgtctgtc-3’ |
| Mut 1 | 5’-tctgggcccgcgacgtgcaacgggatgaggt-3’ |
| Mut 2 | 5’-acctcatcccgttgcacgtcgcgggcccaga-3’ |
| Del 1 | 5’-tcttctctgggcccacgggatgaggtaa-3’ |
| Del 2 | 5’-ttacctcatcccgtgggcccagagaaga-‘3 |
| Putative element wild type (WT)* | Anneal 5’- ccgtgagcctccgcggg -3’ with 5’- cccgcggaggctcacgggtgtgtgtgt -3’ |
| Putative element mutation (MT) | 5’-ccgttgcacgtcgcggg -3’ duplex |
| AP-1 | 5’ -cgcttgatgactcagccggaa-3 duplex |
| NF-jun | 5’-tggagtctccatg-3’ duplex |
| GAPDH (ORF) primers | FP: 5’- acccagaagactgtggatgg-3’ |
| RP: 5’- ccctgttgctgtagccaaat-3’ | |
| c-jun (distal ORF) primers | FP: 5’- ttctatgacgatgccctcaa-3’ |
| RP: 5’- gttgctgaggtttgcgtaga-3’ | |
| Ku70 wild type (WT) | FP: 5’-cggctagcatgtcagggtgggagtcatattac-3’ |
| RP: 5’-cgggatccgtcctggaagtgcttggtgagg-3’ | |
| Ku80 mutant (MT) | FP1: 5’-cggctagcggcaacatggtgcggtcggggaataag-3’ |
| RP1: 5’-ctcgtgctgtataacgtccatgctcacgattagtgcatc-3’ | |
| FP2: 5’-atggacgttatacagcacgagacaataggaaagaagtttgag-3’ | |
| RP2: 5’-ctttttagctagagacatagagtcaatcaaagcatcaacagc-3’ | |
| FP3: 5’-tctatgtctctagctaaaaaggatgagaagacagacacccttg-3’ | |
| RP3: 5’-ctttaccaagacgcgaaaattttcagcaggattcacacttc-3’ | |
| FP4: 5’-aattttcgcgtcttggtaaagcagaagaaggccagctttgag-3’ | |
| RP4: 5’- cggaattctatcatgtccaataaatcgtc -3’ |
Putative element (WT) with a 3’ (GT)5 tail (shown in the table) was used for oligonucleotide trapping and the results in Fig. 5. For all other experiments, the WT element without a 3’ (GT)5 tail was used for the experiments shown although all experiments were also repeated with the tailed oligonucleotides with the same result (data not shown).
Construction of Ku80 mutant and Ku70 wild type plasmid
Total RNA was extracted from HEK293 cells using the RNeasy kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. cDNA synthesis was performed using T18 as primer according to the protocol provided (New England Biolabs, Ipswich, MA, USA). Ku70 full length (wild type, WT) was amplified by PCR using primers containing an NheI site at its 5’ end and a BamHI site at its 3’ end (Table 1), then it was cloned to pcDNA3.1 (-)/Myc-His A (Invitrogen, Carlsbad, California, USA) between the NheI and BamHI sites and after sequencing, a correct clone was used for eukaryotic expression in HEK293 cells. The full-length Ku80 (wild type) has three sites susceptible to the siRNA used (see Supplemental Data, Table S2, at 376-406, 1411-1431 and 1822-1842, accession number NM_021141). These were mutated by an overlap extension technique as described in the section of c-jun promoter mutant construction. The primers used are shown as Ku80 mutant in Table 1. The result is a Ku80 with the same amino acid sequence as wild-type but because of alternative codon usage cannot bind the siRNAs. The Ku80 mutant was cloned to pcDNA3.1 (-)/Myc-His A between NheI and EcoRI sites and sequenced. The way Ku80 mutant was initially cloned was not in correct reading frame to produce the myc tag, since this was not needed for other experiments not included here. To correct reading frame to produce also the myc tag, the Ku80 mutant pcDNA3.1 was cut at an EcoRI site, downstream of Ku80 but upstream of the myc tag, filled with Klenow fragment DNA polymerase, and re-ligated. This corrects reading frame so that Ku80 is produced as a fusion with in-frame myc antigen. The Ku70 or Ku80 plasmids with His/Myc tag, i.e. Ku70 (WT)-pcDNA3.1 or Ku80 (MT)-pcDNA3.1, was transfected into HEK293 cells and analyzed for myc tag as well as Ku70 or Ku80 expression by western blotting to confirm that Ku70 wild type or Ku80 mutant have been expressed correctly.
Genetic reporter assay
HEK293 cells were maintained in DMEM medium supplemented with 10% fetal bovine serum. For transient transfection assay, 2.0 μg pTK-Luciferase normalization reporter DNA (producing firefly luciferase) and 10 ng pMluc-1 test reporter DNA for Renilla luciferase activity, including empty vector pMLuc-1, pMLuc-1-c-jun (WT), pMLuc-1-c-jun (DEL) or pMLuc-1-c-jun (MUT), were mixed with jetPEI transfection reagent (6 μl) (Qbiogene Molecular Biology, CA, USA) and incubated at room temperature for 30 min, then the mixture was added to 2 × 105 HEK293 cells in a 6 well plate. Forty-eight hours later, firefly and Renilla luciferase activity was measured using the dual luciferase reporter assay system (Promega, Madison, WI, USA). The relative luciferase activity was calculated as the Renilla luciferase activity (pMLuc-1-c-jun) divided by the firefly activity (pTK-Luciferase). All transient transfection assays were carried out in triplicate with at least two independent experiments. The averages and standard deviations were calculated.
Preparation of HEK293 nuclear extract
HEK293 nuclear extracts were prepared as previously described (19, 20). Protein concentration was determined by the Bradford method (21).
Electrophoretic mobility shift assays (EMSA) and antibody blocking or supershift assay
Oligonucleotides were annealed and 5’ end labeled with [γ-32P] ATP using T4 polynucleotide kinase. HEK293 nuclear extract (2.5-5 μg) was mixed with 40 fmols labeled element (1.6 nM final concentration) in a total volume of 25 μl TE0.1 incubation buffer (10 mM Tris, pH 7.5, 1 mM EDTA, 0.1 M NaCl). After incubation at room temperature for 30 min, the complex was separated on a pre-electrophoresed 6% nondenaturing polyacrylamide gel in 0.25 × TBE buffer (22) and detected by autoradiography. In competition assays, unlabeled duplex putative element wild type (WT), mutation (MT) DNA, AP-1 or NF-jun DNA (Table 1) was also added. In some experiments, poly dI:dC, heparin, T18 and detergents were added to assess the optimal conditions for element binding as needed for subsequent oligonucleotide trapping (22). In EMSA antibody blockade or supershift assays, nuclear extract (5 μg) was incubated with antibody (0.4-1.6 μg) for 1 h on ice before the labeled DNA was mixed.
UV crosslinking and immunoprecipitation
UV crosslinking (23) was performed by incubating HEK293 nuclear extract (10 μg) with 1.6 nM labeled putative element (WT) in the presence of 20 ng/μl poly dI:dC in 25 μl TE0.1 incubation buffer. The mixture was incubated on ice for 30 min, and then UV irradiated (254 nm) for 30 min on ice. The crosslinked DNA-protein complex was separated on a 15% SDS-PAGE gel and exposed to autoradiography. For immunoprecipitation analysis, the UV crosslinked mixture was diluted 5-fold into E1A buffer (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 125 mM NaCl, 0.1% Triton X-100, 0.5 mM PMSF, 1 mM DTT). Antibody (1.5 μg) was then added and incubated at 4°C for 1 h, and mixed with Protein G-Sepharose beads (Sigma Aldrich Inc., St. Louis, MO, USA) at 4°C overnight. The proteins bound on the beads were eluted with SDS-PAGE sample buffer at 95°C for 5 min, analyzed by 15% SDS-PAGE and autoradiography (24).
Immunoprecipitation and southwestern blot analysis
HEK293 nuclear extract (50 μg, 10 μl), diluted 5-fold with E1A buffer and pre-cleared with Protein G-Sepharose, was mixed with 1.5 μg of antibody and incubated at 4°C for 1 h. Protein G-Sepharose (50 μl) was mixed at 4°C overnight. After centrifugation, the supernatant was retained to study the effect of immunodepletion. Proteins bound on the beads, after washing, were eluted with 50 μl 2 × SDS-PAGE sample buffer. The eluates were separated by 15% SDS-PAGE and electrophoretically transferred onto nitrocellulose membrane for southwestern blot analysis (17) at 4°C. The nitrocellulose-bound proteins were denatured in 50 ml denaturing buffer (6 M guanidine/HCl in binding buffer containing 10 mM HEPES, pH 7.9, 50 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% Triton X-100) to remove SDS for 10 min. The blot was then renatured by adding an equal volume of binding buffer sequentially to dilute the guanidine/HCl from 6 M to 3, 1.5, 0.75, 0.38 and 0.185 M with 5 min incubation after each addition. The membrane was then blocked 1 h in binding buffer containing 5% non-fat milk. The 5’ end labeled putative element (WT) (1.5 nM) was added to the blot in binding buffer containing 0.25% BSA and 10 μg/ml poly dI:dC and incubated overnight. The membranewas washed thrice for 10 min with binding buffer and detected by autoradiography.
Two-step oligonucleotide trapping chromatography
HEK293 nuclear extract was diluted 10-fold in TE0.1 buffer and passed over a 2 ml (AC)5-Sepharose column for pre-clearing. (AC)5-Sepharose beads were made by coupling (AC)5 oligonucleotide (5’-NH2-ACACACACAC-3’) to CNBr-preactivated Sepharose-4B (Sigma, St. Louis, MO, USA) as previously described (22). The flow through from pre-clearing was incubated at 4°C for 10 min with 8 μg/ml heparin, 15 μM T18 (TTTTTTTTTTTTTTTTTT), and 10 μg/ml poly dI:dC, and 0.1% octyl-glucopyranoside (OGP). The annealed element (WT) oligonucleotide with a 3’ (GT)5 tail was then added to a final concentration of 30 nM, and incubated for 30 min at 4°C to form the DNA-protein complex. The trapping mixture was applied to a fresh 2 ml (AC)5-Sepharose column, washed with 20-column volume of TE0.1, and the DNA-protein complex was eluted with TE buffer (10 mM Tris, pH7.5, 1 mM EDTA) containing 0.1% Tween-20 at 37°C. This treatment reverses annealing of the element DNA with the column and the DNA-protein complex elutes. NaCl (5 M) was added to the eluate to a final concentration of 0.1 M and the mixture was applied to a fresh 1 ml (AC)5-Sepharose column for the second trapping. After washing with 20 column volumes of TE0.1, the bound proteins on the column were then eluted with TE0.4 buffer (10 mM Tris, pH 7.5, 1 mM EDTA, 0.4 M NaCl). To ensure that elution was complete, a further wash with TE1.2 (10 mM Tris, pH 7.5, 1 mM EDTA, 1.2 M NaCl) was performed. Eluted fractions were concentrated by Microcon YM-10 (Amicon Inc., Beverly, MA, USA) or lyophilized following dialysis in 50 mM NH4HCO3. Concentrated samples were applied to a 15% SDS-PAGE gel, detected by silver staining, Coomassie brilliant blue staining or western blotting analysis.
Nano-LC-ESI-MS/MS analysis and database searching
The gel slices were destained with 1:1 acetonitrile (ACN): 50 mM NH4HCO3, reduced with 10 mM DTT at 56°C for 1 h and alkylated in the dark with 50 mM iodoacetamide at room temperature for 1 h. Then the gel plugs were lyophilized and immersed in 15-20 μL of 20 ng/μL trypsin (Promega, Madison, WI, USA) solution in 25 mM NH4HCO3 for digestion at 37°C overnight. Peptides were extracted twice with 50 μL 5% TFA in 50% ACN and the extracts combined. After lyophilization, the peptide mixture was analyzed by capillary HPLC-electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) using a Thermo Finnigan linear ion trap mass spectrometer (LTQ) equipped with a nano-ESI source. On-line HPLC separation of the digests was accomplished with an Eksigent NanoLC micro HPLC. The column was a PicoFrit™ (New Objective; 75 μm i.d.) packed to 10 cm with C18 adsorbent (Vydac 218MS, 5 μm, 300 Å). Mobile phase A was 0.5% acetic acid (HAc)/0.005% trifluoroacetic acid (TFA) and mobile phase B was 90% acetonitrile/0.5% HAc/0.005% TFA. The gradient was from 2 to 42% B in 30 min. The flow rate was 0.4 μl/min. MS conditions were a 2.9 kV ESI voltage, an isolation window for MS/MS of 3; 35% relative collision energy, with a scan strategy of a survey scan followed by acquisition of data dependent collision-induced dissociation (CID) spectra of the seven most intense ions in the survey scan above a set threshold. MS/MS spectra were searched against the human IPI protein database (version 3.07) using the SEQUEST algorithm of Bioworks software (version 3.1) for peptide and protein identifications. The proteins with two unique peptide matched were considered as high confidence.
Western blotting analysis
Gels were electroblotted onto 0.2 μm pore nitrocellulose membranes (BioRad, Hercules, CA, USA) as previously described (25). The dilution for primary antibodies including c-jun, Ku80, Ku70 and lamin A/C was 1:1,000. Immunoreactive proteins were visualized using 1:10,000 goat anti-rabbit or 1:5,000 goat anti-mouse secondary antibody-HRP conjugate (Santa Cruz Biotechnology, California, USA) as appropriate and detected by enhanced chemiluminescence (ECL).
RNA interference (siRNA) and expression plasmid transfection
HEK293 cells (2 × 105) in 1 ml of DMEM were transiently transfected with 2 μl of siRNA (final concentration 20 nM) or 2 ug of expression plasmid [Ku70 (WT)-pcDNA3.1 and Ku80 (MT)-pcDNA3.1] using 6 μl of transfection reagent for siRNA (Santa Cruz Biotechnology, California, USA) in a six-well plate and grown at 37°C in a CO2 incubator. After 5-7 h of growth, 1 ml of 20% FBS in DMEM was added to each well and growth continued. RNA was extracted 18 h after transfection. Cells were harvested 48 h after transfection and nuclear extract prepared for both western blotting (25 μg) and EMSA analysis (5 μg). In genetic reporter assay, 10 ng of pMLuc-1-c-jun (WT) reporter plasmid, 1 μg of pTK-Luc plasmid, 2 μl of siRNA, and/or 1.5 ug of expression plasmid(s) was transiently cotransfected into HEK293 cells as described above. The dual luciferase reporter assay was performed 48 h after incubation.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from siRNA transfected HEK293 cells and cDNA synthesis was performed as described above. For semi-quantitative RT-PCR analysis, GAPDH was used as a control for equal amounts of cDNA. PCR conditions were: 95°C for 5 min, then thermocycling 95°C 45 s, 56°C 45 s and 72°C 1 min, with 35 cycles for c-jun (Gene Bank Accession number J04111, using the c-jun downstream ORF primers to produce nt 1285-1785) or 17 cycles for GAPDH (Accession number NM002046, using the GAPDH ORF primers to produce nt 652-1072), and followed by 10 min at 72°C for extension. The primers for GAPDH and c-jun ORF amplification are shown in Table 1. The PCR products were electrophoresed, digitally photographed and quantified by Scion Image software. Real-time quantitative PCR was performed using LightCycler Master SYBER Green 1 (Roche Applied Science, IN, USA) following the protocol supplied with 400 nM primers. The primers were the same as for semi-quantitative PCR. A parallel reaction was done for each sample using GAPDH primers. Samples were amplified with a precycling hold at 95°C for 10 min, followed by 45 cycles of at 95°C for 10 s, 56°C for 10 s and 72°C for 25 s. The produced c-jun gene was normalized to GAPDH using the relative quantitation method.
Chromatin immunoprecipitation (ChIP) assays
Chromatin immunoprecipitation was performed using the ChIP kit (Upstate USA, Chicago, IL). First, the sonication procedure was optimized to generate 200-1000 bp fragments. HEK293 cells were crosslinked by 1% formaldehyde at 37°C for 10 min. After immunoprecipitation, protein-DNA crosslinks was reversed at 65°C for 4 h, and DNA was recovered using QIAquick PCR purification kit (Qiagen, Valencia, CA, USA). The primers for PCR amplification include c-jun promoter specific (FP and RP, nt -200 +81) and negative control primers, GAPDH ORF (open reading frame) and c-jun downstream ORF, are shown in Table 1. The experiment was replicated twice with similar results.
RESULTS
The putative element (nt-176-167) is a positive regulator of c-jun promoter transcription
To investigate if the putative element (nt −176 to -167) plays a role in c-jun promoter activity, the c-jun promoter wild type (WT), deletion (DEL) and mutation (MUT) reporter plasmids along with the thymidine kinase control reporter plasmid were co-transfected into HEK293 cells. Fig. 1B shows the result of the reporter assay. c-Jun promoter (Fig. 1A) wild type (WT) is composed of sequence between −200 and +81, the c-jun deletion (DEL) has 10 bp from -176 to -167 deleted, while the sequence of c-jun mutation (MUT) between -176 and -170 has been changed by randomly shuffling the bases (Fig. 1A). The c-jun promoter wild type (WT) has strong activity (77.6 ± 4.0, mean ± standard deviation) compared with empty vector control (0.039 ± 0.001). c-Jun deletion (DEL) (43.5 ± 7.2) and c-jun mutation (MUT) (16.1 ± 1.8) promoter reduces expression by 44% and 79%, respectively. The element is an important activator for c-jun promoter activity. Interestingly, mutation, which has no element but the same spacing has the largest effect.
FIG. 1.

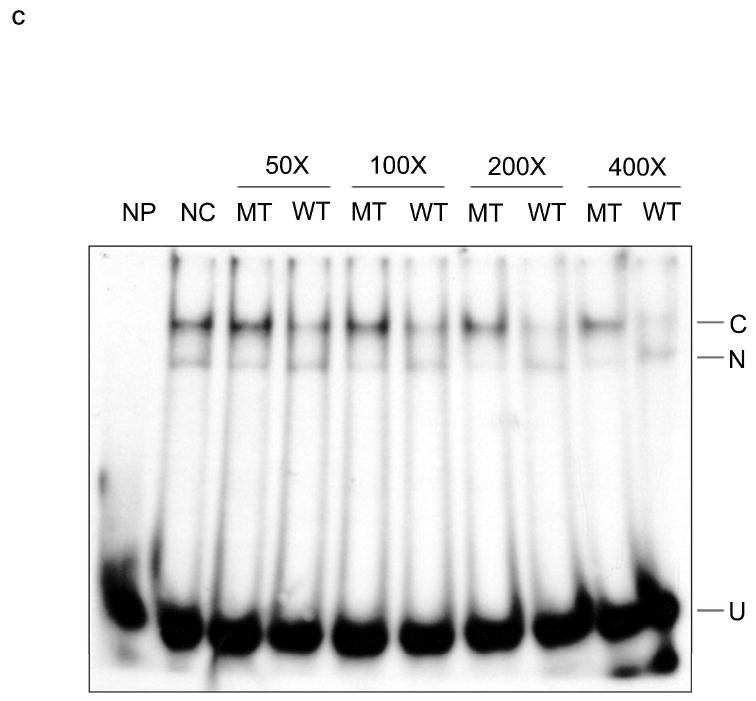
The GAGCCTC element is a positive regulator of c-jun promoter activity. A, The upper panel shows the wild-type promoter region of c-jun (nt −200 +81). The location of a novel, putative element (nt -176 -167) is in bold. The lower part of the panel shows mutations (in bold and italics) made by randomly scrambling the bases in this region (nt −176-170) or by deleting the entire region (nt -176-167). B, Genetic reporter assay. 60 ng of c -jun promoter wild type (WT) or its two mutants [deletion (DEL); mutation (MUT)], which were inserted in a Renilla luciferase expression vector (pMLuc-1) or the empty vector, was transiently co-transfected with a thymidine kinase control vector (pTK-firefly luciferase, 2 μg) in HEK293 cells. The promoter activity was detected by the dual luciferase assay. Relative activity was calculated as the ratio of c-jun driven Renilla luciferase activity divided by the thymidine kinase driven firefly luciferase activity. *, p < 0.05 vs. WT. C, EMSA identifies a specific element binding complex. 1.6 nM 5’ end labeled duplex WT element (Table 1) was incubated with 5 μg HEK293 nuclear extract. Unlabeled, duplex specific competitor, element wild type (WT), or unlabeled randomized element mutation (MT) (Table 1), were added individually at 50-400 fold molar excess of radiolabeled DNA. NP, no protein; NC, no competitor; C, specifically shifted complex; U, unshifted double stranded DNA. N, non-specific shifted complex.
The putative element has sequence-specific binding with proteins in HEK293 nuclear extract
Sequence-specific binding to the novel element was determined by an EMSA competition assay. Fig. 1C confirms binding proteins in the HEK293 nuclear extract. Two shifted bands are observed when nuclear extract is incubated with labeled wild-type (WT) element. The upper band (C) is decreased or abolished when 50-400 fold excess of unlabeled WT element DNA is added, while the mutant sequence (MT) could not compete away the band, indicating the upper band (C) represents the specific complex. In some EMSA experiments, the lower band (N) is not present. Thus, one or more proteins present in HEK nuclei binds specifically to the -176 to -167 footprint region of the c-jun promoter.
Molecular masses of the element binding proteins are determined by southwestern blot and DNA-protein crosslinking analysis
Southwestern blot analysis has the ability to estimate the molecular mass of proteins which bind the labeled element. As shown in Fig. 2A, four bands bind to the labeled element. Calculated from the molecular mass of the standards, the molecular sizes are approximately 115, 70, 45 and 37 kDa. Actually, there are three very closely spaced bands at 37 kDa. (data not shown), indicating the possibility of isoforms or modification of the same protein. In UV crosslinking experiments shown in Fig. 2B, the crosslinked complex gives rise to DNA-protein adducts of 125, 80, 47 and 32 kDa. in size. DNA of this size would add about 10 kDa. to the apparent mass of the complex, thus, these correspond to protein masses of about 115, 70, 37 and 22 kDa. The 47 kDa. DNA-protein complex contains three closely spaced bands, similar to those revealed by the southwestern blot for the corresponding 37 kDa. (Fig. 2A and data not shown) and these are probably the same protein(s). The 45 kDa. protein, seen on the southwestern blot (Fig. 2A) is not found upon crosslinking. This may be due to less crosslinking efficiency with DNA, suggesting that the 45 kDa. protein may not bind DNA tightly and indeed, it is not observed in all experiments (Fig. 2C). The 32 kDa. band, found upon DNA crosslinking, was not detected on the southwestern blot analysis; it may result from low efficiency of transferring or renaturing, or more likely represents DNA-DNA crosslinking of the radiolabeled probe.
FIG. 2.
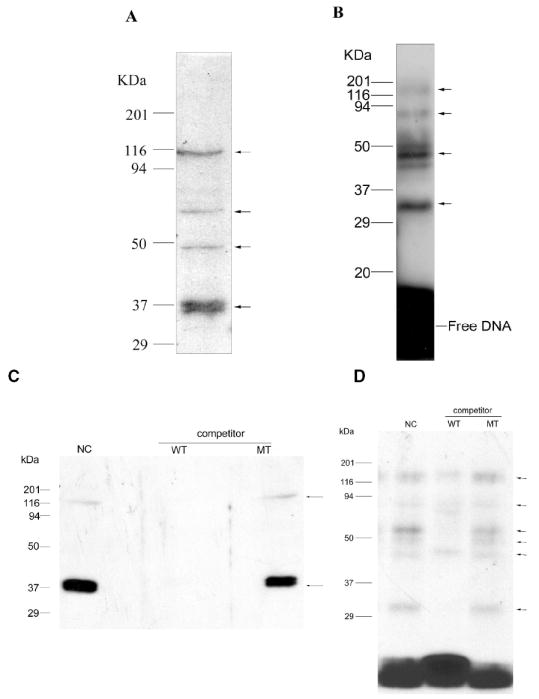
Southwestern blot and UV crosslinking of DNA-protein adducts determined the appropriate molecular masses of the proteins bound with the WT element. (A) Southwestern blot analysis. Nuclear extract (50 μg each) was separated by 15% SDS-PAGE and then transferred to nitrocellulose membrane, the proteins on the blots were denatured and renatured by serial dilution of guanidine/HCl. The membrane was probed with radiolabeled WT element (1.5 nM). The dried membrane was analyzed by autoradiography. (B) UV crosslinking of DNA-protein adducts was performed by incubating 10 μg nuclear extract with 1.6 nM radiolabeled element followed by UV crosslinking. The crosslinked complex was separated from free DNA by 15% SDS-PAGE. The molecular mass of the markers is shown on the left. Arrows indicate the bands bound with the element. (C) and (D) the same as in A and B, respectively, but showing either the effect of no competitor (NC) or the effect of adding a 100-fold excess of unlabeled wild type (WT) or mutant (MT) DNA to the radiolabeled wild type DNA used as the probe.
The 115, 70 and 37 kDa. proteins are confirmed by both UV crosslinking (after correcting for DNA mass) and southwestern blot analysis in Fig. 2, suggesting two possibilities: (1) The 70 kDa. protein is a homo- or heterodimer of the 37 kDa. proteins, or is a different protein of about 70 kDa.; and (2) The 115 kDa. protein is a heterodimer of 37 kDa. with another protein of about 80 kD, or possibly a trimer of the 37 kDa. protein. Another feature about the 125 kDa. (115 kDa. protein) band that may be important is that it sometimes appears as a closely spaced doublet in UV crosslinking, which is barely discernable in Fig. 2B but which is more easily seen later in Fig. 8E. Also, the intensity of the 115 and 70 kDa. protein bands vary in some southwestern blots and UV crosslinking experiments, as apparent from the experiments in Fig. 2C and D where the effect of competitor DNAs is shown. These experiments also show the effect on the 37 and 115 kDa. bands on a southwestern blot (Fig. 2C) when either unlabeled wild type or mutant DNA element were added to compete for binding with radiolabeled wild type DNA binding. Clearly, the 37 and 115 kDa. bands are competed by the wild type but not the mutant element. UV crosslinking in the presence of the unlabeled competitor DNAs provided more detail. While the mutant DNA competes very little, the same amount of the wild type element diminishes greatly the bands at approximately 125, 80, and 47 kDa. corresponding to 115, 70, and 37 kDa. protein masses. Other minor bands are also competed.
FIG. 8.
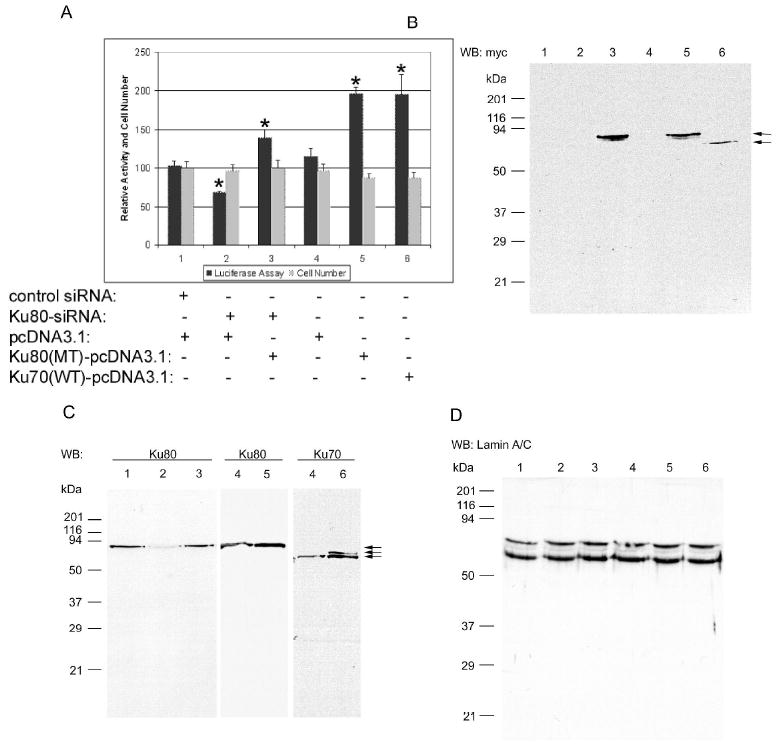
Ku80 and Ku70 introduction significantly increases c-jun promoter activity. A, Genetic reporter assay (black bars) and viable cell number counting (gray bars) indicates that Ku80 and Ku70 overexpression increases c-jun promoter activity without affecting cell proliferation. 10 ng of c-jun promoter (WT) plasmid, 1 μg normalization vector, 20 nM of siRNA and/or 1.5 μg of Ku80 or Ku70 plasmid was cotransfected into 2 × 105 HEK293 cells in 6-well plate as indicated in panel A, 48 hours later, cells were harvested for analysis by genetic reporter assay, western blotting as well as cell number counting as described in Fig. 7. 1, control siRNA+pcDNA3.1 (empty vector); 2, Ku80-siRNA+pcDNA3.1; 3, Ku80-siRNA+Ku80 (MT)-pcDNA3.1; 4, pcDNA3.1 (empty vector); 5, Ku80 (MT)-pcDNA3.1; 6, Ku70 (WT)-pcDNA3.1. The data are expressed as the means ± S.D. (n = 3). *, p < 0.05 vs. the control. B, C, D, The numbers shown are the same conditions as defined in panel A. Western blotting analysis of myc (B), Ku80 and Ku70 (C) and Lamin A/C (D). The arrow on the right indicates the Ku70 or Ku80 band and the molecular mass of standards are shown on the left.
c-Jun is a component of the element binding complex
Fig. 2 shows one or more proteins with molecular mass of about 37 kDa. binding to the element. Jun family members (c-jun, junB, junD) are approximate 39 kDa., which prompted us to explore if they play a role in forming the element-binding complex. Nuclear extract (5 μg) was incubated with antibodies (1.6 μg) against c-jun, junB and junD and then the complex was analyzed by EMSA using the WT duplex element. Fig. 3A shows that the c-jun antibody, but not jun-B, jun-D, or other antibodies, significantly blocks the specifically shifted complex (C), strongly suggesting c-jun binding. Similar results were found when lower amounts of each antibody (400 ng) was used (data not shown). Monoclonal antibody against c-jun phosphorylated on Ser63 (p-c-jun) blocks the specific complex somewhat but not as effectively as the c-jun polyclonal antibody, suggesting that the complex does not require this phosphorylation to any great extent (data not shown).
FIG. 3.
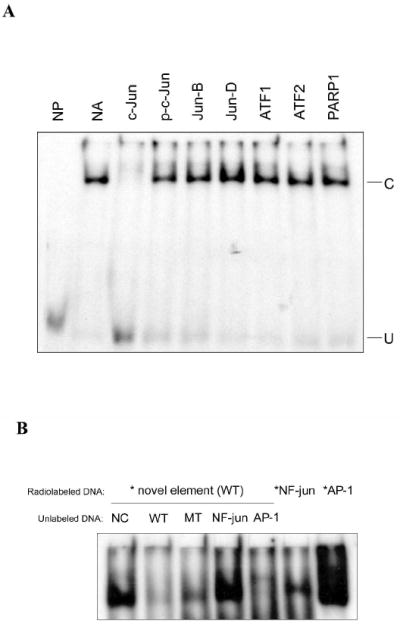
c-Jun is shown to be involved in the element-binding complex. (A) The c-jun antibody blocks the binding to the novel element. 1.6 μg of the indicated antibodies were incubated with nuclear extract (5 μg) on ice for one hour, followed by incubation with radiolabled WT element at room temperature for 30 min. NP, no protein; NA, no antibody; C, specific shifted complex; U, unshifted double stranded DNA. (B) EMSA competition assay. 200-fold excess of unlabeled DNA, including AP-1, NF-jun, element wild type (WT) and mutant (MT) (Table 1), were added to EMSA. Also shown are results from EMSA using radiolabeled AP-1 (*AP-1) and NF-jun (*NF-jun) are shown to the right. NC, no competitor.
To confirm that phosphorylation was not necessary, in supplemental data (Fig. S2B), we show that this same phospho-c-jun antibody does supershift the complex with the CREB element DNA, a complex known to involve an ATF2-phospho-c-jun dimer. In this experiment, the ATF2 antibody used was a blocking one, showing another case of a blocking antibody (Fig. S2B). Thus, the phospho-c-jun antibody does supershift complexes containing phospho-c-jun, and the results in Fig. 3A strongly suggests that the GAGCCTC complex does not require this c-jun phosphorylation.
It should be pointed out that the element sequence (tgagcctcc) shares considerable similarities with the recognition site of NF-jun (ggagtctcc) in the c-jun promoter (Fig. 1A) and consensus AP-1 binding sites (tgagctca). To determine if AP-1 components and NF-jun are possible candidates for binding, unlabeled NF-jun and AP-1 DNA as well as element wild type (WT) and mutant (MT), were added to the standard EMSA competition assay with the labeled element (WT). As shown in Fig. 3B, unlabeled AP-1 DNA competes with the specific complex (C) in a similar manner as unlabeled element wild type (WT), while unlabeled NF-jun at the same concentration does not. Moreover, there is a complex formed with labeled NF-jun (*NF-jun), but it is slower migrating than the WT element complex. Labeled AP-1 (*AP-1) reveals several complexes, as might be expected from the various protein compositions of AP-1, but shorter exposures show one is indeed the size of the complex under investigation (data not shown). The slight darkening at the bottom of the cropped image in Fig. 3B is from the non-specific binding discussed in Fig. 1C, and is not shown. We conclude that c-jun is a component of the element binding complex, and that the element represents a specialized kind of c-jun binding site. Since c-jun monomer and dimer would be the approximate size of the 37 and 70 kDa, bands in Fig. 2, this may account for these bands but the identity of the 115 kDa. band in Fig. 2 is still unclear.
The systematic optimization for oligonucleotide trapping is determined by EMSA
To further characterize the element-binding proteins, these were purified. The oligonucleotide trapping technique is used to purify transcription factors by their specific association with element DNA (22). EMSA can be used to estimate the DNA binding affinity, the concentration of transcription factor in nuclear extract, and determine the optimal concentration of element as well as additives that lessen non-specific binding prior to purification (22, 26). The results are shown in Fig. 4. The final conditions derived are: 10-fold dilution of nuclear extract (binding activity activity ≤ Kd), 30 nM element DNA (10 × Kd, and > Bmax to ensure nearly quantitative binding), 10μg/ml dI:dC, 15 μM T18, 8 μg/ml heparin, and 0.1% OGP (octyl-glucopyranoside) or Tween-20 in TE0.1 buffer with final elution in TE0.4 buffer.
FIG. 4.
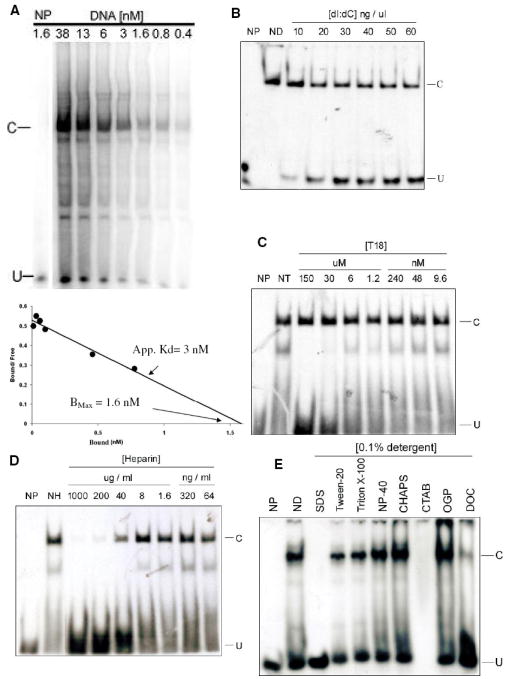
ESMA allows experimental determination of purification parameters for the element binding proteins. A, Determination of the apparent DNA-binding affinity and concentration of the transcription factor. No protein (NP) or an amount of 10-fold diluted nuclear extract was mixed with serial dilutions of radiolabeled WT element for the EMSA reaction. The density of the specifically shifted band (C), the unshifted DNA band (U), and the total density for each lane (T) are used to calculate the concentration of bound and free DNA for this nuclear extract dilution. Scatchard analysis (below) yields an estimate of Kd = 3 nM in this experiment for band C. The plot also shows the maximum binding (Bmax) of 1.6 nM for a 10-fold dilution, which provides an estimate of 16 nM binding protein in the undiluted nuclear extract. B, The optimal concentration of competitor poly dI:dC is determined. A serial dilution of poly dI:dC from 10 to 60 ng/μl shows the highest concentration of poly dI:dC that does not significantly interfere with complex formation is 10 ng/μl. ND, no poly dI:dC. C, The optimal concentration of competitor T18 is determined. Five-fold serial dilutions of T18 from 9.6 nM to 150 μM with poly dI:dC constant at 10 ng/μl. NT, no T18. Since T18 did not interfere throughout this concentration range, a cost-effective concentration of 15 μM was chosen for all subsequent experiments. D. The optimal concentration of the competitor heparin is determined. Five-fold serial dilutions of heparin from 64 ng/ml to 1 mg/ml with poly dI:dC constant at 10 ng/μl and T18 constant at 15 μM. Shown in this figure, the highest non-interfering concentration of heparin is 8 μg/ml, which was chosen for all subsequent experiments. NH, no heparin. E, The influence of different detergents (0.1%) on element binding is determined. The detergents indicated were included in the EMSA reaction with poly dI:dC constant at 10 ng/μl, T18 constant at 15 μM and heparin constant at 8 μg/ml. ND, no detergent. NP-40 (nonylphenyl-polyethylene glycol), CHAPS (3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate) and OGP (octyl-glucopyranoside), increase complex formation, while SDS (sodium dodecylsulfate), DOC (deoxycholic acid) and CTAB (hexadecyltrimethylammonium bromide) dramatically inhibit. Because OGP is monomeric and neutral, which will not interfere with mass spectrometry analysis, it was chosen for trapping purification. Thus, the conditions chosen for trapping are: 10-fold dilution of nuclear extract (binding activity ≤ Kd), 30 nM element DNA (10 × Kd, and > Bmax to ensure nearly quantitative binding), 10 ng/μl dI:dC, 15 μM T18, 8 μg/ml heparin, and 0.1% OGP in TE0.1 buffer. In experiments not shown, using EMSA, we also determined that the complex forms in TE0.1 (0.1 M NaCl), but is disrupted in TE0.4 (0.4 M NaCl) buffer.
Two-step oligonucleotide trapping purifies the specific element-binding activity
Trapping normally involves DNA-protein complex formation in solution and complex trapping on a column. Here, we have extended this by adding a pre-clearing step and performed trapping elution in two different ways to lessen nonspecific binding proteins. First, nuclear extract was diluted 10-fold in TE0.1 buffer and passed through an (AC)5-Sepharose column for pre-cleaning to remove any proteins bound by that column. This approach was found useful in the purification of MafA (27). Then, the pre-cleared extract was mixed with 30 nM (GT)5-tailed element, T18 (15 μM), heparin (8 μg/ml), dI:dC (10 μg/ml) and the detergent OGP (0.1%), and the protein-DNA complex formed at 4°C was retained by the hybridization of the (GT)5 tail on the element with the (AC)5-Sepharose column. The element-column hybrid can be dissociated in low salt at 37°C and this is then used to elute the DNA-protein complex. The eluted complex was then made 0.1M NaCl, cooled to 4°C, and applied to a fresh (AC)5-Sepharose column where it again binds. The protein-DNA interaction was disrupted using 0.4 M NaCl, and the proteins were eluted, leaving the DNA still bound on the column.
Figure 5 shows the results of this trapping purification. Fig. 5A shows that during pre-clearing, the binding activity passes through the first (AC)5-Sepharose column unretained as expected.The temperature elution from the second column also behaves as expected (data not shown). On the third column, the binding activity is retained by the column since it is absent from the flow through (FT1) and eluted at 0.4 M NaCl (F1-3), also as expected. No further activity is observed when the column is washed with a higher salt concentration (TE1.2), showing that elution is complete (data not shown).
FIG. 5.
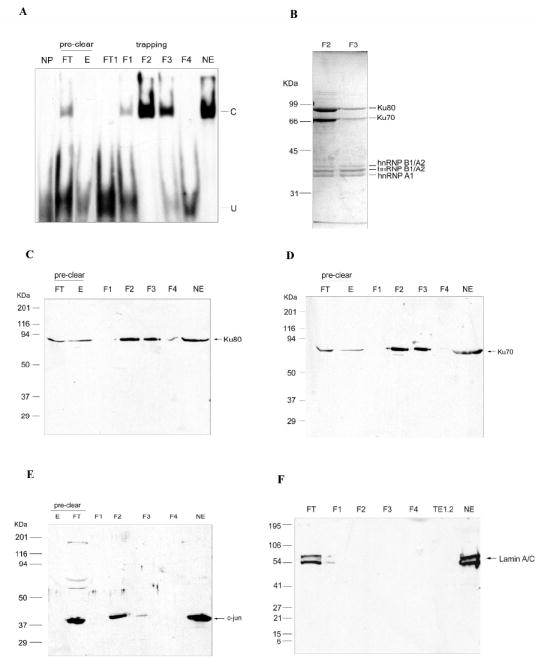
Purification and identification of element binding proteins. A, EMSA of WT element-binding activity during oligonucleotide trapping. Nuclear extract (3 ml, 15 mg) was diluted 10 fold to 30 ml with TE0.1 and passed over a 2 ml bed volume (AC)5-Sepharose column for pre-clearing, the column was washed with 20 column volumes of TE0.1 and “eluted” (E) with 5 ml TE0.4. The flow through (FT) from pre-clearing was incubated with 10 μg/ml poly dI:dC, 8 μg/ml heparin, 15 μM T18 and 0.1% OGP at 4°C for 10 min, then mixed to a final concentration of 30 nM with WT element DNA to form a complex at 4°C for 30 min. The mixture (30 ml) was applied to another 2 ml (AC)5-Sepharose column for the first trapping (not shown). The DNA-protein complex on the column was eluted with TE buffer (15 ml) containing 0.1% Tween-20 at 37°C, then the warm elution fraction was cooled and made 0.1 M NaCl for the second trapping (shown) on a fresh 1 ml (AC)5-Sephaose column at 4°C. The bound proteins were eluted with 2.5 ml TE0.4 (fractions F1-5, 0.5 ml/tube, only F1-4 are shown). Three μl of each fraction was used for EMSA. C, specifically shifted band; U, unshifted double stranded DNA. B, The elution fractions were dialyzed into 50 mM NH4HCO3, lyophilized, and dissolved in 0.1 ml water. 30 μl of lyophilized sample was electrophoresed on SDS-PAGE and stained with Coomassie brilliant blue. Five obvious bands (labeled) of fraction 2 (F2) were cut out, digested with trypsin and identified by LC-MS/MS analysis; the identified proteins are indicated on the right and the molecular mass of standard is shown on the left. C, D, E, Western blotting analysis of trapping fractions. The different fractions were resolved and analyzed by western blotting with antibodies against candidate proteins Ku80 (C) and Ku70 (D) as well as c-jun (E). F, Western blot analysis of lamin A/C. FT, flow through of pre-clearing; F1-4, elution fractions of second trapping in TE0.4 buffer; TE1.2, elution of second trapping with TE1.2 buffer. NE, HEK293 nuclear extract.
Most of the element binding activity is concentrated in fraction 2 (F2) and to a lesser extent fraction 3 (F3) (Fig. 5A). Two bands between 66-99 kDa. are more abundant in F2 than in F3 after Coomassie protein staining (Fig. 5B). LC-MS/MS analysis identified them to be Ku80 and Ku70 as shown in Fig. 5B and in Table S1 in Supplemental Data. Western blotting analysis confirms the presence of Ku80 and Ku70 in eluted fractions F2 and F3 (Fig. 5C and 5D) at the same mobility as the protein bands in Fig. 5B. As mentioned above, c-jun is a component of the element-binding complex, so we investigated c-jun in the purified fraction as well. Western blotting analysis indicates that c-jun is abundant in F2 and less so in F3 (Fig. 5E). Lamin A/C, an unrelated nuclear envelope protein, is not detected in the purified fractions (Fig. 5F), showing the specificity of trapping purification. Fig. 5B and Supplemental Data, Table S1 also show several other proteins which are identified and will be discussed later but for now, we will focus on c-jun and the Ku proteins.
Ku80 positively regulates c-jun expression by binding to the element
As demonstrated in the antibody supershift assay in Fig. 6A, the Ku80 antibody specifically supershifts the element complex (C), while other antibodies against Ku70, hnRNP A1, hnRNP A2 (proteins identified by mass spectrometry, Fig. 5B and Supplemental Table S1) and control IgG have no effect. In order to exclude the possibility that the complex is formed by Ku80 binding to the G-rich sequence of element wild type used, we then changed the downstream flanking sequences of element (ccgtgagcctccgcggg to ccgtgagcctccgcatc) and showed that the complex is still supershifted by the Ku80 antibody (data not shown). Then, we depleted Ku80 by incubating nuclear extract with Ku80 antibody followed by immunoprecipitation. The resulting supernatant, depleted of Ku80, shows less complex formation, while other antibodies (Ku70 and Tho4, also identified by mass spectrometry; or pre-immune IgG) do not affect EMSA complex formation (Fig. 6B). Additionally, the immunoprecipitated proteins were further investigated by southwestern blot analysis in Fig. 6C. Interestingly, an ≈115 kDa. protein that shows binding to WT element can be immunoprecipitated by Ku80 antibody, but no ≈80 kDa. band is observed. It is possible that immunoprecipitation has isolated a complex of two proteins, which remain tightly associated even during electrophoresis and bind to the element DNA. If immunopreciptitation with the Ku80 antibody has isolated a Ku80-c-jun complex (80 kDa. + 37 kDa. = 117 kDa.), these results are what might be expected. The c-jun antibody immunoprecipiated an approximately 70 kDa. protein binding to element, but does not reveal the 115 kDa. band, probably because its signal is so much less intense (compare the Ku80 and c-jun lanes in Fig. 6C). Fig. 6D shows that the UV crosslinked element-protein was immunoprecipitated by both c-jun and Ku80 antibodies. The c-jun antibody immunoprecipitated three protein at about 115 kDa., 70 kDa. and 37 kDa. (calculated by subtracting 10 kDa. for the crosslinked DNA mass), the darkest was approximately 70 kDa. This size is consistent with a c-jun-c-jun dimer but could also be a Ku70 bound singly. Similarly, the Ku80 antibody immunoprecipitated a band near 70-80 kDa. strongly, which may be Ku80 alone bound to DNA. More weakly, the antibody also precipitated a band that represents a 115 kDa. protein (more clearly shown in the rectangle below), which could be a Ku80-c-jun complex. The result of Fig. 6C and 6D suggests that Ku80-c-jun binds and that c-jun alone, probably as c-jun dimer (about 70 kDa.) or Ku70- or Ku80 alone may also binds to the element DNA.
FIG. 6.
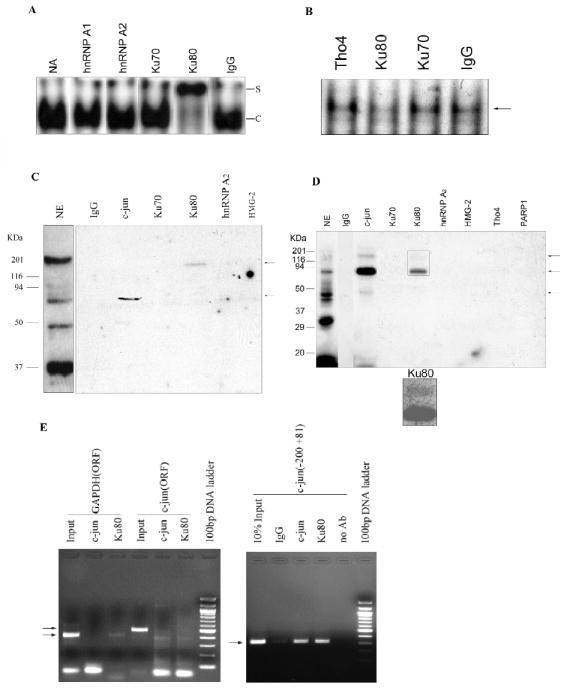
The presence of Ku80 and c-jun in the element-binding complex. A, Ku80 antibody supershifts the element binding complex. Nuclear extract (5 μg) was incubated with 1 μg of the indicated antibodies on ice for 1 h, radiolabeled WT element was then added and incubated at room temperature for 30 min and applied to EMSA. Only the shifted bands are shown for clarity. Lane five shows the specific band supershifted (S) by the Ku80 antibody. B, Immunodepletion and EMSA analysis. Nuclear extract pre-cleared with protein G-Sepharose (100 μg, 20 μl) was incubated with 1.5 μg of the indicated antibodies and then bound to Protein G-Sepharose beads in 100 μl E1A buffer for immunoprecipitation. The supernatants were used for EMSA and show that the Ku80, but not other antibodies, depletes the DNA-binding complex. C, Immunoprecipitation and southwestern blot analysis. The antibody-bound proteins on Protein G-Sepharose beads were eluted, separated on SDS-PAGE and transferred to nitrocellulose membrane for southwestern blot analysis. The immunoprecipitated proteins were probed with the radiolabeled WT element. Element-binding proteins are indicated with the arrows on the right. NE, southwestern blot analysis of nuclear extract without immunoprecipitation. D, UV crosslinking and immunoprecipitation analysis. Nuclear extract (10 μg) was incubated with 1.6 nM radiolabeled WT element, followed with UV cross-linking on ice for 30 min. The mixture was diluted five fold with E1A buffer and incubated with 1.5 μg of the indicated antibodies at 4°C for 2 h, the immunoprecipitated complex bound to Protein G-Sepharose beads was eluted and resolved by 15% SDS-PAGE, followed by autoradiography analysis. Cross-linked nuclear extract (NE) without immunoprecipitation is also shown. The UV cross-linked proteins immunoprecipitated are indicated with an arrow on the right. The molecular mass of standards is shown on the left. Below is an enlargement of a longer exposure of the area shown in the rectangle to show more clearly the 115 kDa. protein band. E, ChIP assay shows that Ku80 binds the c-jun promoter (nt −200+81) in vivo. Live HEK293 cells were crosslinked by 1% formaldehyde and sonicated to produce 200-1000 bp fragments, which were then immunoprecipitated by antibodies against c-jun and Ku80 as well as negative control, irrelevant IgG and no antibody (no Ab). Crosslinking was reversed, and the purified DNA was amplified by PCR using primers specific for c-jun promoter (nt −200+81) and negative control primers for GAPDH ORF and c-jun distal ORF. The PCR products of the correct size are indicated with arrows on the left.
The purified protein was also used for southwestern blotting and UV crosslink and the data are included in Supplemental Data Fig. S1A and B. The results are quite similar to the similar experiments for nuclear extract already shown in Fig. 2 showing that purification has indeed enriched the DNA-binding proteins of interest.
To further investigate whether the Ku70 antibody can supershift any Ku70 containing complex, in the Supplementary Data (Fig. S2A), we show an experiment in which we synthesized a DNA end binding oligonucleotide duplex known to bind the Ku70-Ku80 heterodimer (28) and used it for an EMSA assay testing both the Ku70 and Ku80 antibodies for their ability to supershift this complex. The result is that the Ku80 antibody causes a supershift but the Ku70 antibody has no effect. Thus, the inability of the Ku70 antibody to supershift or block the GAGCCTC-protein complex in Fig. 6A does not rule out Ku70 involvement and the other uses of the antibody in Fig. 6 are of questionable value. We also investigated two other Ku70 antibodies (Santa Cruz catalogue # 9033 and H-308) but neither of these supershift or block DNA complexes or immunoprecipitate effectively (data not shown) We conclude that Ku70 may be a part of the Ku80-c-jun complex but this is not conclusively shown by the data at this point.
c-Jun and Ku80 bind the c-jun promoter in vivo
In order to confirm that Ku80 binds to c-jun promoter in vivo, formaldehyde crosslinking of live HEK293 cells, lysis, sonication (to produce DNA fragments in the 200-1000 bp size range) and chromatin immunoprecipitation was performed. For the ChIP assay, the PCR primers were designed for the c-jun gene promoter (nt −200+81) that contains the novel element as well as AP-1 binding sites (right panel), while the primers for GAPDH ORF and distal c-jun ORF (a region 1.5 Kb away from promoter sequence) were used as negative controls (left panel). As shown in Fig. 6E, input chromatin resulted in the PCR products using primers for c-jun promoter (nt −200+81), GAPDH ORF and distal c-jun ORF. Immunoprecipitation using control IgG or no antibody (no Ab) gave little PCR product of the expected size. c-Jun antibody, used as positive control, specifically pulled down the c-jun gene promoter as expected since it contains not only the novel element but also two AP-1 binding sites. However, the c-jun antibody did not bind the negative control sequences (GAPDH and distal c-jun ORF). Ku80 antibody ChIP gave the same results as the c-jun antibody and provides the first evidence that Ku80 binds to c-jun promoter in vivo. The band seen at the very bottom of the gel (left panel, below the size of the smallest size marker) of some wells is an artifact caused by using too much primer, often referred to as “primer dimers”, and can be ignored.
Ku80 and Ku70 involvement is also implicated by HEK293 cell transfections
To confirm a role for Ku80 in c-jun expression, HEK293 cells were transfected with Ku80-siRNA to diminish Ku80 expression. Western blotting (Fig. 7A) and densitometry shows that Ku80-siRNA transfection (+) decreases the Ku80 protein by 81% ± 10% (n = 3, p < 0.01) compared with control siRNA (-), and c-jun expression was decreased by 35% ± 18% (n = 5, p < 0.05), confirming that Ku80 is activating c-jun expression. The same blot was stripped and re-probed for lamin A/C, showing equal loading in each well. Using semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR), c-jun mRNA expression was decreased 82% ± 6% (n=2, p < 0.05) by Ku80-siRNA (Fig. 7B) while the GAPDH expression was unchanged and confirms equal amounts of cDNA. Real-time RT-PCR (data not shown) confirms a 48% decrease of c-jun mRNA by Ku80-siRNA. Moreover, the effect of Ku80-siRNA on the c-jun promoter reporter assay is shown in Fig. 7C. The c-jun promoter activity of Ku80-siRNA transfected HEK293 cells (+) significantly (P < 0.05) decreased 28% as compared with control siRNA transfected cells (−). EMSA (Fig. 7D) shows that Ku80 siRNA decreases the formation of the specific element complex; the complex decreased 70% ± 12% (n = 3, p < 0.05) by densitometry. By UV crosslinking analysis, Fig. 7E shows that the 125 kDa. DNA-protein adduct (115 kDa. of which is protein) is dramatically decreased in Ku80-siRNA (+) transfected HEK293 cells. Interestingly, the 80 kDa. band is also diminished. These results are consistent with a model in which the 115 kDa. protein is a heterodimer of c-jun-Ku80 (37+80 kDa.) while the 80 kDa. band may well represent the Ku80 monomer binding DNA directly or this band may be the c-jun dimer (37+37) diminished because of diminished c-jun expression.
FIG. 7.

Ku80-siRNA introduction decreases Ku80 and c-jun expression. A, Western blotting analysis of Ku80, c-jun and lamin A/C show the down-regulation of Ku80 and c-jun protein in HEK293 cells after Ku80-siRNA transfection. Ku80-siRNA (20 nM, +) or control siRNA (20 nM, -) was transiently transfected into 2 × 105 HEK293 cells and after 48 h, nuclei were isolated. Nuclear extract (50 μg) was used for western blotting analysis with antibodies against Ku80, c-jun and lamin A/C. B , Ku80-siRNA affects c-jun mRNA expression by RT-PCR analysis. Eighteen hours after Ku80-siRNA was transfected into HEK293 cells, the total RNA was extracted and reverse transcribed, the synthesized cDNA was then used for semi-quantitative PCR using primers for c-jun and GAPDH ORFs. c-Jun was amplified for 35 cycles, while GAPDH was thermocycled for 17 cycles and serves as a control, showing approximately equal cDNA in each sample. The PCR product of the correct size is indicated by the arrow on the right. C, Ku80 plays a positive role in c -jun promoter activity by genetic reporter assay using Ku80-siRNA. HEK293 (2 × 105) cells were transiently cotransfected with the 10 ng of pMLuc-1-c-jun reporter plasmid, 2 μg of normalization plasmid (pTK-Luc) and 20 nM of Ku80 (+) or control siRNA (−). After 48 h, the cells were lysed and analyzed by dual luciferase assay. The data are expressed as the mean ± S.D. (n = 4). *, p < 0.05 vs. the control. D, EMSA analysis confirms the presence of Ku80 in the element binding complex using Ku80-siRNA transfection. Nuclear extract (5 μg) from Ku80-siRNA (+) or control siRNA (-) transfected HEK293 cells was incubated with labeled WT element for EMSA analysis. C, specifically shifted band; U, unshifted double stranded DNA. E, UV cross-linking analysis of DNA-protein adducts formed in Ku80-siRNA transfected HEK293 cells. The DNA-protein complex as in Fig. 7D was UV cross-linked on ice for 30 min. The arrows on the right indicates the 125 kDa. complex (115 kDa. of which is protein) and the 80 kDa. band. The molecular mass of standards are shown on the left.
In order to exclude that the inhibition of c-jun expression by Ku80 is caused by siRNA toxicity, a Ku80 mutant plasmid, which was constructed by mutating the three Ku80 sites targeted by the siRNA but having the wild-type amino acid sequence, was introduced into HEK293 to overexpress Ku80. Fig. 8A strongly suggests Ku80 mutant plasmid overexpression can recover the c-jun promoter activity inhibited by Ku80-siRNA (black bars) without influencing the number of viable cells (gray bars). The cells remain viable and cell morphology (data not shown) was also indistinguishable. As shown in Fig. 8A, Ku80-siRNA introduction significantly (indicated by “*”, p < 0.05) inhibits c-jun promoter activity by about 34% (Bar 2) compared with control (bar 1), the same result as shown in Fig. 7C. Co-transfection of Ku80-siRNA and Ku80 mutant plasmid increases c-jun promoter activity by about 40% (bar 3) compared with Ku80-siRNA transfection alone (bar 2). Similarly, introduction of Ku80 mutant plasmid [Ku80 (MT)-pcDNA3.1] significantly up-regulates c-jun promoter activity by about 70% (bar 5) compared with empty vector transfection (bar 4, pcDNA3.1). Interestingly, introduction of Ku70 wild type plasmid [Ku70 (WT)-pcDNA3.1] also significantly increases c-jun promoter by about 70% (bar 6). Western blotting results confirm the overexpression of Ku70 wild type and Ku80 mutant in HEK293 cells. Fig. 8B suggests that both plasmids, Ku70 (WT)-pcDNA3.1 and Ku80 (MT)-pcDNA3.1, can correctly express the fusion protein with the myc tag. In Fig. 8C, transfection of Ku80 mutant plasmid or Ku70 wild type increases the Ku80 or Ku70 expression in HEK293 cells, compared with empty vector. Ku80 mutant plasmid up-regulates Ku80 protein by about 60% (Fig. 8C), which was caused by a Ku80 fusion protein with myc tag (Fig. 8B), and Ku70 wild type plasmid produces a Ku70 fusion protein with a detectably higher molecular mass compared with endogenous Ku70 as shown in Fig. 8C (Lane 6, upper band). Ku80-siRNA significantly decreased Ku80 expression as shown in Fig. 7A and 7C, and the addition of Ku80 mutant recovers the expression of Ku80 (Fig. 8C), indicating that Ku80 siRNA can not block the overexpression induced by Ku80 mutant introduction. Western blotting for Lamin A/C (Fig. 8D) shows equal sample loading throughout. Clearly, Fig. 7 and Fig. 8 strongly suggest that Ku80 is required for efficient protein-binding to the element DNA in vivo and in vitro and for c-jun promoter activity. Also, Figs. 7 and 8A show for the first time that Ku70 is also important to c-jun expression.
In Fig. 5B and Supplemental Table S1 are shown several other proteins identified by MS in the proteins purified by oligonucleotide trapping. Supplemental Table S1 has a column “Confirm” which shows results from using various commercially available antibodies to test if these affected the EMSA complex (either blocking or supershifting). Since all these antibodies, with the exception of Ku80 and c-jun, were negative, the data is not shown except for the data shown in Fig. 6.
DISCUSSION
The GAGCCTC element in the c-jun promoter was clearly visible in footprinting analysis from over a decade ago (4) but remained uninvestigated. EMSA (Fig. 1C, 4, and 6D) shows it binds a specific complex. Genetic reporter assays (Fig. 1B, 6C, 7A) clearly show that it plays a significant role in regulating c-jun expression, perhaps contributing as much as a five-fold enhancement of expression (Fig. 1B). Identifying the proteins regulating this expression is thus important to the understanding of c-jun promoter function and the regulation of c-jun expression. Here, we have presented evidence that both c-jun itself and Ku proteins play important roles.
There can be little doubt that both Ku80 and c-jun are involved in binding the GAGCCTC element. (1) The c-jun antibody blocks the specifically shifted complex (Fig. 3A) while the Ku80 antibody supershifts the complex (Fig. 6A); (2) Western blotting analysis detects both Ku80 and c-jun in the oligonucleotide trapping purification product (Fig. 5C and E); (3) The specific element complex can be competed away by the AP-1 consensus sequence, DNA known to bind c-jun (Fig. 3B) and is decreased by Ku80-siRNA (Fig. 7E); (4) The protein complex immunoprecipitated by the c-jun or Ku80 antibodies bind to the element DNA (Fig. 6C); (5) c-jun or Ku80 antibodies immunoprecipitate the UV crosslinked element-protein complex (Fig. 6D); and (6) southwestern blotting and UV crosslinking give molecular weights consistent with the known size of c-jun, its homodimer, and its heterodimer with Ku80 (Fig. 2, 5C and D).
Ku80 was identified by MS analysis. Several additional lines of evidence suggest that Ku80 plays an important role in binding the GAGCCTC element. (1) Immuno-depletion of Ku80 diminishes complex formation (Fig. 6B); (2) ChIP assay confirmed the binding of Ku80 to c-jun promoter in vivo; and (3) Ku80-siRNA diminishes Ku80 protein and c-jun expression (Fig. 7A and 6B) and the formation of the specific complex measured by EMSA (Fig. 7D and 6E). How it participates in element-complex formation is, however, a separate question.
It is curious that c-jun was not identified in the mass spectral analysis, however, this has been previously observed in other studies and may be related to the high abundance of other nuclear proteins and the relatively low abundance of c-jun. In trapping fractions, we identified heterogeneous nuclear RNA binding protein (hnRNP) family members with molecular mass from 37-39 kDa., i.e. hnRNP A2/B1, hnRNP A1 and hnRNP A3. We found no evidence these are directly involved in element-binding (Supplemental Table S1, Fig. 6, and data not shown), however, they may interfere with c-jun detection. This is the same approximate mass as c-jun and the more abundant hnRNP proteins may be masking lower abundance c-jun during LC-MS/MS identification. Samuel et al used DNA affinity chromatography to purify AP-1 element binding proteins. By nanospray MS/MS microsequencing, they identified YB-1 (Y-box-binding protein-1), but failed to find other AP-1 component proteins, probably because YB-1 is much more abundant than other AP-1 proteins (29). This is likely the same situation as in our study.
Ku (Ku80/Ku70 heterodimer) has been reported to bind to free double-stranded DNA ends independent of the exact structure of the ends. However, it is unlikely that Ku80 participates here because of this end-binding property. The end-binding affinity (Kd) for Ku is between 0.2-4.0 ×10−10 M (30, 31). EMSA analysis shows an apparent Kd of 3 nM (Fig. 4) for the GAGCCTC element in our study, nearly two-order of magnitude different. In addition, the reporter assays are performed with circular DNA lacking ends and ChIP assay is carried out to verify the binding of Ku80 to intact chromatin containing c-jun promoter sequence in vivo. Nuclear DNA has infrequent ends which are distant from the c-jun promoter. EMSA competition assays show that the Ku80-containing complex is specific for the wild-type GAGCCTC element sequence and cannot be competed away by large excesses of the mutant sequence (Fig. 1C) or NF-jun element oligonucleotide (Fig. 3B), all of which have free double-stranded ends. Interestingly, there is only one base pair difference between NF-jun and element consensus sequence, i.e., -c- in the middle of element (-gagcctc-), while -t- in the middle of NF-jun (-gagtctc-). This shows how sensitive complex formation is for this particular DNA sequence motif. Moreover, the element complex can be supershifted and immunoprecipitated by Ku80 antibody, but not by other antibodies except c-jun. Ku80 was detected by UV crosslinking assays but varies somewhat from experiment to experiment (compare 115 and 70 kDa. protein bands in the nuclear extracts in Fig. 2B, 5D and 6E) suggesting that its association with DNA may not be in as close contact as the c-jun. This is consistent with other studies, showing that Ku80 crosslinking to DNA is variable (32).
Many studies show that Ku80 has specific sequence binding activity and participates in transcription regulation. Ku recruits DNA-PK (DNA-dependent protein kinase) by binding to negative regulatory element 1 (NRE1) with high affinity (Kd = 0.84 ± 0.24 nM) (33). Rosenfeld and colleagues demonstrated by ChIP analysis the specific recruitment of Ku80, Ku70 and DNA-PK to the pS2 promoter upon induction in vivo, and the activation of pS2 promoter was regulated by transient double strand DNA breaks and DNA repair (34). Ku also functions as a repressor of human immunodeficiency virus (HIV-1) transcription by binding to the Ku-specific binding sites within the negative regulatory region of HIV-1 long terminal repeat (35). By competing with HSF1 (heat-inducible heat shock factor), Ku proteins suppress the promoter activity of hsp70 (heat shock protein) through binding with the consensus sequence (-ngaan-) of heat shock element (HSE) (36). Ku protein, as an integral part of CTC box binding factor (CTCBF), shows specific binding to the CTC box in the human collagen type IV gene promoter (37). Intriguingly, the GAGCCTC element contains this CCTC box motif. Moreover, Ku80 has been suggested to activate expression of p50 (an NF-κB subunit) and NF-κB activity. The binding element for Ku80 in the p50 promoter is –gcttc- (38). The GAGCCTC element in the c-jun promoter contains this same motif with a one base difference. In our study, Ku80 is a component of the GAGCCTC element complex and positively regulates c-jun expression.
The question remains whether the binding of c-jun and Ku80 to the element DNA may be independent events to independent sites on the DNA. This is unlikely given the small size of the oligonucleotide (17-mer) used in EMSA. Assays with specific antibodies confirm that both c-jun (Fig. 3A) and Ku80 (Fig. 6A) are present in the specific complex with this oligonucleotide. Also, a single base change in the GAGCCTC motif in the NF-jun oligonucleotide (Fig. 3B and Table 1) was ineffective to compete for complex formation. In other experiments described in the text, we also altered the downstream flanking sequences of this element without affecting complex formation. The results of these studies constrict binding to a small region of an already short oligonucleotide making it unlikely that the two proteins bind distinct sites. Ku80 siRNA inhibits formation of this complex (Fig. 7D) and inhibits formation of the 125 kDa. (of which 115 kDa. is protein) band upon crosslinking whose size is consistent with the c-jun-Ku80 heterodimer (37 + 80 kDa.) that we conclude binds to this element. Other data would suggest that either c-jun dimer or Ku80 monomer may also be able to bind to the element independent of the other. c-Jun dimer (37 + 37 = 74 kDa.) and Ku80 monomer are too similar in size to be able to reliably separate on SDS-PAGE and either could account for the ~70 kDa. band observed in the southwestern blots (Fig. 2A and 5C) or the ~80 kDa. band observed upon UV crosslinking (Fig. 2B, 5D, and 6E). However, since c-jun and Ku80 antibodies both have a nearly complete effect (either blocking or supershifting) on complex formation observed by EMSA (Fig. 3A and 5A), the binding of either protein alone must not be the predominant binding event.
How Ku80 and c-jun interact with one another and the precise details of how the complex binds DNA are unknown. c-Jun is a bZIP motif transcription factor which normally binds DNA by a basic sequence motif (amino acids 257-272) and dimerizes with other proteins by way of a leucine zipper (from Leu274-Leu308) motif, composed of a heptad repeat of leucines, in this case six leucines. A Prosite search of Ku80 reveals that it also contains a leucine zipper motif (from Leu138-Leu165) of five leucines. This may be how the two proteins interact to form a complex but this will require independent confirmation. Ku80, however, lacks the signature basic sequence motif in c-jun which binds DNA.
Until now, we are unaware of any report of a direct role of Ku80 on c-jun promoter activity and expression. In this study, Ku80 is found to bind, along with c-jun itself, to the GAGCCTC element in the c-jun promoter and activate c-jun expression. We speculate there are two possibilities for Ku80 to regulate c-jun expression: (1) Ku80 as a transcription factor binds directly to the element DNA or (2) Ku80 binds to c-jun protein as a co-activator. The variable UV-crosslinking of Ku80 to DNA (including the c-jun-Ku80 heterodimer of 115 kDa. protein and the ~70 kDa. protein band which probably results from both monomer Ku80-DNA and dimer c-jun -DNA binding) in some experiments favors the second possibility and suggests that Ku80 binds c-jun, and that it is c-jun which interacts directly with the DNA. This co-activator model is supported by a report that Ku is a novel transcriptional recycling coactivator of the androgen receptor by direct interaction with androgen receptor (39). It is also possible that both c-jun and Ku80 bind directly to DNA. Regardless of the exact mechanism, a Ku80-c-jun complex is what is binding the novel GAGCCTC element and regulating c-jun expression.
The role of Ku70 in complex formation is less definite. Ku70 over-expression in Fig. 8, lane 6 suggests strongly that it is somehow involved in c-jun promoter activity. In fact, this experiment contradicted our original expectation. If Ku80 alone was involved in complex formation, Ku70 overexpression would force predominance of Ku80-Ku70 dimer, depleting Ku80 monomer and have a “dominant negative” inhibitory effect. Obviously, the opposite is true and Ku70, like Ku80, enhances c-jun expression. The UV-crosslinked complex and southwestern blots (Fig. 2, 5C, 5D) is not consistent with a complex as large as Ku70-Ku80-c-jun (~ 190 kDa.). However, since single crosslinks with DNA would be more likely than double or triple crosslinks, Ku70 could be a member of the complex but give infrequent crosslinks to DNA because it is not in close contact with DNA or simply that this is not the most favored crosslink to form. In southwestern blots, only complexes which can persist on SDS-PAGE and renature will be revealed and Ku70 may simply not persist in the complex under these conditions or not renature properly. Thus, Ku70 may bind too loosely to be detected by these techniques, or alternatively, it may be binding elsewhere (other than the GAGCCTC motif) in the c-jun promoter to have its effect in the reporter assays. Thus, while we have presented considerable positive data showing the Ku80-c-Jun binding to the GAGCCTC motif, the data on Ku70 is at this point less complete.
Regardless, this is the first report of a complex involving the Ku proteins and c-jun, binding as an activator to a novel element, GAGCCTC, in the c-jun promoter. Ku proteins are thought to function in DNA repair but obviously also serve a role in transcriptional regulation. Whether this new role for Ku proteins will extend to other promoters is an exciting area for future investigation.
Supplementary Material
Acknowledgments
This work was supported by NIH grant 5R01GM43609. We thank Ms. Magda Loranc and Maria Macias for their excellent technical assistance and our labmates Linda Nagore, Markandeswar Panda, Shoulei Jiang, and Yongmin Xiong for their discussion and encouragement. For mass spectrometry analysis and interpretation, we are indebted to Dr. Susan Weintraub and the Institutional Mass Spectrometry Laboratory of the Univ. TX Health Science Center in San Antonio.
Footnotes
SUPPORTING INFORMATION AVAILABLE Two tables and two figures of supporting information are provided online. Table S1 gives the complete list of proteins identified by LC-MS analysis of the purified element binding complex while Table S2 gives the sequences of the Ku80 siRNAs used. Fig. S1A shows a southwestern blot of the purified proteins while Fig. S1B shows UV crosslinking results for the purified proteins. Fig. S2 shows the effect of Ku80, c-jun and other antibodies on the EMSA assay of an oligonucleotide known to bind Ku70-Ku80 dimers and a different oligonucleotide known to bind phospho-c-jun.
References
- 1.Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 2.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 3.Rozek D, Pfeifer GP. In vivo protein-DNA interactions at the c-jun promoter: preformed complexes mediate the UV response. Mol Cell Biol. 1993;13:5490–9. doi: 10.1128/mcb.13.9.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rozek D, Pfeifer GP. In vivo protein-DNA interactions at the c-jun promoter in quiescent and serum-stimulated fibroblasts. J Cell Biochem. 1995;57:479–87. doi: 10.1002/jcb.240570313. [DOI] [PubMed] [Google Scholar]
- 5.Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- 6.Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- 7.Steinmuller L, Cibelli G, Moll JR, Vinson C, Thiel G. Regulation and composition of the activator protein-1 (AP-1) transcription factors controlling collagenase and c-jun promoter activities. Biochem J. 2001;360:599–607. doi: 10.1042/0264-6021:3600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angel PIM, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–39. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- 9.van Dam H, Duyndam M, Rottier R, Bosch A, de Vries-Smits L, Herrlich P, Zatema A, Angel P, van der Eb AJ. Heterodimer formation of c-Jun and ATF-2 is responsible for induction of c-jun by the 243 amino acid E1A protein. EMBO J. 1993;12:479–87. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis RJ. Signal Transduction by the JNK Group of MAP Kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 11.Dunn C, Wiltshire C, MacLaren A, Gillespie DA. Molecular mechanism and biological functions of c-Jun N-terminal kinase signalling via the c-Jun transcription factor. Cell Signal. 2002;14:585–93. doi: 10.1016/s0898-6568(01)00275-3. [DOI] [PubMed] [Google Scholar]
- 12.Baker SJ, Kerppola TK, Luk D, Vandenberg MT, Marshak DR, Curran T, Abate C. Jun is phosphorylated by several protein kinases at the same sites that are modified in serum-stimulated fibroblasts. Mol Cell Biol. 1992;12:4694–705. doi: 10.1128/mcb.12.10.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein B, Angel P, van Dam H, Ponta H, Herrlich P, van der Eb A, Rahmsdorf HJ. Ultraviolet-radiation induced c-jun gene transcription: two AP-1 like binding sites mediate the response. Photochem Photobiol. 1992;55:409–15. doi: 10.1111/j.1751-1097.1992.tb04255.x. [DOI] [PubMed] [Google Scholar]
- 14.Han TH, Lamph WW, Prywes R. Mapping of epidermal growth factor-, serum-, and phorbol ester-responsive sequence elements in the c-jun promoter. Mol Cell Biol. 1992;12:4472–4477. doi: 10.1128/mcb.12.10.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brach MA, H F, Yamada H, Bäuerle PA, Kufe DW. Identification of NF-jun, a novel inducible transcription factor that regulates c-jun gene transcription. EMBO J. 1992;11:1479–1486. doi: 10.1002/j.1460-2075.1992.tb05192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohri S, Sharma D, Dixit A. Interaction of an approximately 40 kDa protein from regenerating rat liver with the -148 to -124 region of c-jun complexed with RLjunRP coincides with enhanced c-jun expression in proliferating rat liver. Eur J Biochem. 2004;271:4892–902. doi: 10.1111/j.1432-1033.2004.04458.x. [DOI] [PubMed] [Google Scholar]
- 17.Sharma D, Ohri S, Dixit A. The -148 to -124 region of c-jun interacts with a positive regulatory factor in rat liver and enhances transcription. Eur J Biochem. 2003;270:181–9. doi: 10.1046/j.1432-1033.2003.03369.x. [DOI] [PubMed] [Google Scholar]
- 18.Tuteja R, Tuteja N. Ku autoantigen: a multifunctional DNA-binding protein. Crit Rev Biochem Mol Biol. 2000;35:1–33. doi: 10.1080/10409230091169177. [DOI] [PubMed] [Google Scholar]
- 19.Jiang D, Moxley RA, Jarrett HW. Promoter trapping of c-jun promoter-binding transcription factors. J Chromatogr A. 2006;1133:83–94. doi: 10.1016/j.chroma.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Abmayr S, Workman J. Preparation on Nuclear and Cytoplasmic Extracts from Mammalian Cells, in Current Protocols in Molecular Biology. John Wiley & Sons. 1993:12.1.1–12.1.9. doi: 10.1002/0471142727.mb1201s75. [DOI] [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Moxley RA, Jarrett HW. Oligonucleotide trapping method for transcription factor purification systematic optimization using electrophoretic mobility shift assay. J of Chromatography A. 2005;1070:23–34. doi: 10.1016/j.chroma.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 23.Molnar G, OL N, Pardee AB, Bradley DW. Quantification of DNA-protein interaction by UV crosslinking. Nucleic Acids Res. 1995;23:3318–3326. doi: 10.1093/nar/23.16.3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Dam H, D M, Rottier R, Bosch A, de Vries-Smits L, Herrlich P, Zantema A, Angel P, van der Eb AJ. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12:479–487. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gadgil H, Jarrett HW. Oligonucleotide trapping method for purification of transcription factors. J Chromatogr A. 2002;966:99–110. doi: 10.1016/s0021-9673(02)00738-0. [DOI] [PubMed] [Google Scholar]
- 27.Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol. 2003;23:6049–62. doi: 10.1128/MCB.23.17.6049-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deriano L, Guipaud O, Merle-B H, et al. Binet JL, Ricoul M, Potocki-Veronese G, Favaudon V, Maciorowski Z, Muller C, Salles B, Sabatier L, Delic J. Human chronic lymphocytic leukemia B cells can escape DNA damage-induced apoptosis through the nonhomologous end-joining DNA repair pathway. Blood. 2005;105:4776–4783. doi: 10.1182/blood-2004-07-2888. [DOI] [PubMed] [Google Scholar]
- 29.Samuel S, Twizere JC, Bernstein LR. YB-1 represses AP1-dependent gene transactivation and interacts with an AP-1 DNA sequence. Biochem J. 2005;388:921–8. doi: 10.1042/BJ20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falzon M, Fewell JW, Kuff EL. EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single- to double-strand transitions in DNA. J Biol Chem. 1993;268:10546–52. [PubMed] [Google Scholar]
- 31.Blier PR, Griffith AJ, Craft J, Hardin JA. Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J Biol Chem. 1993;268:7594–601. [PubMed] [Google Scholar]
- 32.Dynan WS, Yoo S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998;26:1551–9. doi: 10.1093/nar/26.7.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giffin W, Kwast-Welfeld J, Rodda DJ, Prefontaine GG, Traykova-Andonova M, Zhang Y, Weigel NL, Lefebvre YA, Hache RJ. Sequence-specific DNA binding and transcription factor phosphorylation by Ku Autoantigen/DNA-dependent protein kinase. Phosphorylation of Ser-527 of the rat glucocorticoid receptor. J Biol Chem. 1997;272:5647–58. doi: 10.1074/jbc.272.9.5647. [DOI] [PubMed] [Google Scholar]
- 34.Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 35.Jeanson L, Mouscadet JF. Ku represses the HIV-1 transcription: identification of a putative Ku binding site homologous to the mouse mammary tumor virus NRE1 sequence in the HIV-1 long terminal repeat. J Biol Chem. 2002;277:4918–24. doi: 10.1074/jbc.M110830200. [DOI] [PubMed] [Google Scholar]
- 36.Kim D, Ouyang H, Yang SH, Nussenzweig A, Burgman P, Li GC. A constitutive heat shock element-binding factor is immunologically identical to the Ku autoantigen. J Biol Chem. 1995;270:15277–84. doi: 10.1074/jbc.270.25.15277. [DOI] [PubMed] [Google Scholar]
- 37.Genersch E, Eckerskorn C, Lottspeich F, Herzog C, Kuhn K, Poschl E. Purification of the sequence-specific transcription factor CTCBF, involved in the control of human collagen IV genes: subunits with homology to Ku antigen. Embo J. 1995;14:791–800. doi: 10.1002/j.1460-2075.1995.tb07057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim JW, Kim H, Kim KH. The Ku antigen-recombination signal-binding protein Jkappa complex binds to the nuclear factor-kappaB p50 promoter and acts as a positive regulator of p50 expression in human gastric cancer cells. J Biol Chem. 2004;279:231–7. doi: 10.1074/jbc.M308609200. [DOI] [PubMed] [Google Scholar]
- 39.Mayeur GL, Kung WJ, Martinez A, Izumiya C, Chen DJ, Kung HJ. Ku is a novel transcriptional recycling coactivator of the androgen receptor in prostate cancer cells. J Biol Chem. 2005;280:10827–33. doi: 10.1074/jbc.M413336200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.