

Abstract
We describe the use of a furanyl salicyl nitroxide derivative (“spin-labeled” compound), as a paramagnetic phosphotyrosine mimetic, to carry out a second-site screening by NMR against the PTPase YopH from Yersinia pestis. Using such a fragment-based screening approach we identified several small molecules targeting YopH that bind at sites adjacent to the spin-labeled compound. These second-site fragments were subsequently used to design and synthesize bidentate YopH inhibitors with sub-micromolar in vitro inhibition, selectivity against the human PTPase PTP1B, and cellular activity against Y. Pseudotuberculosis.
These initial compounds could result useful in elucidating the structural determinants necessary for YopH inhibition and may help in the design of even more active, selective and cell permeable compounds for the development of novel therapies against Yersiniae.
Keywords: Yersinia, NMR screening, spin label, YopH, Protein tyrosine phosphatase
Introduction
Yersiniae invade eukaryotic cells by transferring in their cytosol, through a type III secretion system, six effector proteins (1, 2). Once injected in mammalian cells, these effectors start to interfere with signaling pathways that control cytoskeletal dynamics and inflammation and thus they overcome the immune reaction of the contaminated organism (2–4). Among these effector proteins, YopH (Yersinia outer protein H) is a potent phosphotyrosine phosphatase (PTPase) able to dephosphorylate crucial protein substrates related to focal adhesion such as p130Cas and SKAP-HOM (5–7).
YopH is a protein composed of two domains; the N-terminal domain is essential for YopH translocation and for targeting it to phosphorylated substrates (8), the C-terminal domain contains the PTPase catalytic site (5). YopH phosphatase activity is fundamental for Yersiniae infections and this makes YopH an attractive target for the discovery of novel antibacterial agents (9).
It has been shown that Yersiniae lacking the YopH gene, or even strains with an inactivating C403S point-mutation in YopH, are essentially avirulent and can be successfully defeated by the immune system (10).
We recently reported the use of a furanyl salicyl derivative chemically linked to the “spin label” TEMPO (the 2,2,6,6-tetramethylpiperidine 1-oxyl) as a probe for NMR-based second-site screening in protein tyrosine phosphatases (11). Such technique, coupled with molecular docking studies and medicinal chemistry, is generally very useful for the design of selective and high affinity bi-dentate compounds for a given target (12–14), as we have recently demonstrated for the protein kinase JNK (C-Jun N-terminal protein kinase) (15).
Here we implement this method to screen a small library of chemical fragments against YopH from Yersinia pestis. Through this approach we have identified several small molecules that target pockets on the surface of YopH adjacent to the catalytic site. Further molecular modeling studies have helped us in the design and subsequent synthesis of a series of bidentate compounds. These compounds reveal to be potent inhibitors of YopH phosphatase activity with selectivity against the human PTPase PTP1B (16). The most active compounds can also revert Yersinia Pseudoturbeculosis induced cytopathology of human Hela cells.
Our work and the obtained bi-dentates can reveal structural determinants necessary for effective YopH inhibition and may help in the design of even more potent, selective and cell permeable compounds for the development of novel anti-Yersiniae treatments.
Methods and Materials
Reagents and Compounds
All anhydrous solvents were purchased from Sigma Aldrich and stored in Sure-seal bottles under nitrogen. All other reagents and solvents were purchased at the highest grade available and generally no further purification was implemented. Thin-layer chromatography (TLC) analysis of reaction mixtures was performed using Merck silica gel 60 F254 TLC plates, and visualized using ultraviolet light.
Compound 1 has been previously synthesized in our laboratories (11). Compounds 2 to 7 were synthesized in house using the synthetic procedures that are reported as Supplementary material. Compounds were analyzed by NMR spectroscopy and high resolution mass spectroscopy (See Supplementary Material). NMR spectra were recorded on a Bruker 600 MHz or a 300 MHz Varian instruments. High resolution ESI-TOF mass spectra were acquired at the Center for Mass Spectrometry, The Scripps Research Institute, La Jolla, CA. Compounds were all found to be in excess of 95% pure as established by LC-MS.
Protein expression procedures
The constructs used for GST-tagged full length YopH from Yersinia pestis (17) and for the His-tag containing N-terminal domain of YopH from Yersinia Pseudotuberculosis as well as their expression and purification procedures have been previously described (8, 18–20).
Briefly, 15N labeled N-terminal domain of YopH was obtained by growing E. coli on M9 minimal medium containing 0.5 g/l of 15NH4Cl. Protein over-expression was induced overnight at 20°C at an OD600=0.6. The protein, which contains at the C-terminus a 6-His tag tail was purified on a Ni-column (Amersham) and extensively dialyzed in the following buffer (30 mM Tris, 150 mM NaCl pH=8.0). Upon purification the GST-attached full length YopH was instead dialyzed in 30 mM Tris, 150 mM NaCl, 1 mM DTT pH=6.5.
Enzyme Inhibition (Fluorescence based Assay with DiFMUP)
The enzyme inhibition assay relies on the phosphatase-catalyzed hydrolysis of 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP, INVITROGEN, Carlsbad, CA) in presence of the compounds at 25 °C (21). Enzyme inhibition was tested in a 96-well plate format with an assay volume of 100 μl and assay buffer: 30 mM Tris, 150 mM NaCl, 1 mM DTT, pH=6.5. Compounds were dissolved in DMSO (4.5% final concentration). Full length GST-YopH was used at a concentration of 3 nM. The concentration of substrate (DiFMup) was set at the Km value (50 μM).
Before addition of the substrate, enzyme and compounds were pre-incubated for 10 minutes at room temperature.
The initial reaction rate was measured using a fluorescence plate reader (VictorTM2 V, Perkin Elmer, Waltham, Massachusetts, USA) (excitation 358 nm, emission 455 nm). The non-enzymatic hydrolysis of the substrate was determined as control. The IC50 values were determined by plotting the percentage of inhibition versus the compound concentration (logarithmic scale) using the software GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). Screening of compounds against PTP1B was performed by using a DiFMUP concentration equal to the Km (57 μM) and PTP1B was purchased from BIOMOL Research Laboratories (Pennsylvania, USA). The complete IC50 curve for compound 6 against PTP1B is shown in the Supplementary Material, all other compounds have been tested at a single concentration (25 μM) and did not appreciably inhibited the enzyme under the given experimental conditions.
Modeling studies
The software GOLD (CCDC Software Ltd, Cambridge, UK) was implemented to carry out docking studies. Docking solutions were inspected with Sybyl 8.0 (Tripos, St. Louis) and molecular surfaces were created with MOLCAD (22). For these studies the crystal structure of YopH in complex with a phosphotyrosyl mimetic-containing hexapeptide (pdb code, 1QZ0 (5)) was used. Binding sites were generated by using as reference atoms the Cζ or Cδ of Arg205 and a radius of 25 Å. For docking of small fragments together with compound 1 (Table 1), a smaller radius of 8 Å was also applied. For each ligand ten solutions were generated and subsequently ranked according to Gold-Score (23). Docking studies with the phosphatase PTP1B were carried out with the crystal structure of the protein in complex with an inhibitor (pdb code: 2BGE (16)); the binding cavity was generated by using as a reference atom the Cα of Val49 and a radius of 25 Å.
Table 1.
Chemical structures, IC50 values and cell permeability data of furanyl salicylate derivatives.
| ID | STRUCTURE | IC50 (μM) YopH | IC50 (μM) PTP1B | PAMPA (logPe) |
|---|---|---|---|---|
| 1 [10] |  |
50.0 | >1000 | N.D. |
| 2 (76D12) | 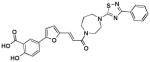 |
0.46 | >25 | −5.83 |
| 3 (76C12) | 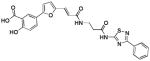 |
1.47 | >25 | −8.21 |
| 4 (76D2) |  |
0.54 | >25 | −7.59 |
| 5 (76D3) | 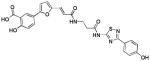 |
1.13 | >25 | −6.73 |
| 6 (76B12) | 0.29 | 21 | −5.81 |
PAMPA Assay
Cell permeability assays were conducted using a Parallel Artificial Membrane Permeation Assay (PAMPA), as we reported previously (24). The permeation of a compound through the artificial membrane layer is described by its logPe value. The logPe value of a given compound was calculated by considering the concentration of the compound in the acceptor compartment after 8 h and that of a reference well with the same concentration containing no membrane barrier.
Cell Based Assay
HeLa cells were first pretreated with the YopH inhibitor compounds for one hour, then infected with Y. pseudotuberculosis strain YPIII (multiplicity of infection =25) grown under conditions favoring Yop secretion. After infection for one hour, gentamicin was added and the cells were incubated an additional hour to allow cytopathology to progress. At this time, the fractions of cells retaining normal morphology and those with cytopathology were determined using our previously published procedure (25).
NMR Binding studies
A small library of ~500 compounds (26, 27) was used for the second site screening. Fragments binding to YopH, adjacent to the TEMPO containing compound 1, were identified through an NMR-based screening as previously described (11). Compounds were analyzed in mixtures (10 compounds per mixture) and T1ρ experiments were recorded with mixing times of 10 ms, 100 ms and 200 ms for the mixture of compounds alone, in presence of only GST-YopH and in samples containing both GST-YopH and the TEMPO labeled compound 1.
To investigate binding of the bidentate compounds 2 to 6 (Table 1) to the N-terminal domain of YopH, chemical shift perturbation studies (28, 29) with 15N labeled N-terminal domain of YopH (concentrations 70/153 μM) were carried out. 1H-15N HSQC spectra were acquired in presence and absence of each compound in a two or three fold excess with respect to the protein. NMR spectra were recorded on a 700 MHz Bruker AvanceIII spectrometer equipped with a TCI cryoprobe. Spectra were processed with Bruker software (Topspin version 2.0) and analyzed with NEASY (30) as implemented in Cara (http://www.nmr.ch/). Overlays of 2D spectra were generated with the program Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). An example of these chemical shift perturbation studies is reported in the Supplementary Material.
Results and discussion
We used a phosphotyrosine mimetic paramagnetic probe, made up of a furanyl salicyl derivative chemically linked to a TEMPO moiety (compound 1, Table 1), to carry out a second-site screening by NMR methods against the protein tyrosine phosphatase YopH from Yersinia pestis. Compound 1 was designed and synthesized in our laboratory (11) based on earlier findings demonstrating that furanyl salicylates were good phosphotyrosine mimetics and competitive YopH inhibitors (17). Hence the paramagnetic probe 1 was designed to occupy the highly conserved phosphotyrosine binding pocket of PTPases (P1), while leaving the TEMPO moiety reaching the edge of variable adjacent sub-pockets (P2, P3), as illustrated for YopH in Figure 1A (11).
Figure 1.

(A) Docked conformation of the TEMPO compound (number 1 in Table 1) in the YopH C-terminal domain (pdb code, 1QZ0). The surface of the protein is displayed in the cavity depth mode (yellow: deep, blue: shallow), the compound is shown in capped sticks. P1 represents the catalytic site, two close additional pockets are indicated as P2 and P3. (B) T1ρ spectra acquired with a mixing time of 200 ms of the small molecules f (YopH-binder) and i (control non-binder compound) at 1 mM concentration each (black); in presence of GST-YopH (10 μM) (green) and in simultaneous presence of GST-YopH and the TEMPO labeled compound 1 (500 μM) (red). Only the aromatic regions of the spectra are shown. Spectra were acquired on a 500 MHz Bruker Avance spectrometer; sample buffer consists of 30 mM Tris-d11, 150 mM NaCl pH=7.5 with 10% D2O. (C) Chemical structures of second-site binders identified through NMR screening. (D) Compound 1 and fragment f have been simultaneously docked in YopH. Two possible docking poses for the fragment f are shown.
Hence, the paramagnetic effect of the spin label can be effectively used to detect binding of chemical scaffolds in these sub-pockets in simple 1D 1H NMR experiments (11, 13–15). This approach, similar to the SAR by NMR (31–33), is useful to design high affinity bi-dentate compounds specific for a given target, as we recently demonstrated for the protein kinase JNK (15).
Here, in the search for second site binders for the phosphatase YopH, we have carried out a series of T1ρ 1D-1H NMR experiments to screen a small library of 500 low molecular weight compounds representing chemical fragments (Maybridge, Fisher Sci.). The description of the library can be found in our recent publication (26, 27).
The screen consists of comparing the signal intensity of 1D-1H NMR experiments of test compounds (at 1 mM concentration) in presence of a substoichiometric amount of GST-YopH (10 μM) measured in presence or absence of the paramagnetic probe, compound 1 (500 μM). To enhance the relaxation induced by the paramagnetic probe, rotating frame relaxation filters of variable length have been introduced (10 ms, 100 ms or 200 ms) in the 1D-1H NMR experiments. Under these experimental conditions, if a given test compound binds in a second site, occupying for example one of the two pockets (P2, P3 Figures 1A) adjacent to P1, its signal intensity will dramatically decrease due to the proximity to the TEMPO labeled compound. This is illustrated in Figure 1B which shows an example of a typical T1ρ 1D-1H NMR experiment with a mixture of two compounds. The fragment f is a YopH binder while compound i is a control compound that doesn’t interact with YopH (Figure 1B). In presence of both compound 1 and GST-YopH the signals from f are nearly lost thus clearly indicating that f is a second site binder, while those of i remain unaffected.
From this screening, we identify a family of 8 related molecules (Figure 1C) interacting with YopH (Figure 1D).
Starting from these results we have designed in silico possible bi-dentate compounds linking furanyl salicylate derivates as phosphotyrosine mimetics, and one of the newly identified scaffolds or close analogues as second site binders (compounds 2–5 in Table 1). Docking studies (Figure 2 and Supplemental Figure 1) proved particularly useful in directing the medicinal chemistry efforts and especially in determining the length of the linker between the furanyl moiety and the second site binders.
Figure 2.

(Left) YopH surface in the “cavity depth” representation (yellow: deep, blue: shallow) with the docked structure of the bi-dentate compound 4. (Right) Curve showing the % of inhibition of the YopH phosphatase activity as function of the compound concentration.
Next, the ability of these compounds to inhibit YopH phosphatase activity has been monitored in an in vitro fluorescence based assay (See Materials and Methods section). The compounds resulted potent YopH inhibitors with IC50 values in the low- to sub-micromolar range (Table 1).
As mentioned, computational docking studies suggest that the furanyl salicylate group of these molecules invariantly occupy the P1 pocket while the second unit of each molecule including a thiazole or a thiadiazole ring tend to occupy the P3 pocket where it can make hydrogen-bond interactions with Arg205 (Figure 2 and Supplemental Figure 1). By further reducing the linker length, compounds can be tailored at occupying the sub-pocket P2, as suggested by molecular docking studies. Hence we designed and synthesized compound 6 (Table 1, Figure 3).
Figure 3.

(A). Docked pose of the bi-dentate compound 6. (B) IC50 curve for compound 6 determined with the DiFMUP assay (See Materials and Methods). (C) In vitro cell-based assay showing the inhibition of Yersinia-induced cytopathology by compound 2 and compound 6.
These compounds are selective for YopH as they are poor inhibitors of the human phosphatase PTP1B (16). This is not surprising due to the fact that while different PTPases have usually very similar active sites, the surfaces surrounding these sites are generally very different either in shape and/or in charge distribution, as we have recently surveyed (34). For example, molecular modeling studies suggest that while the furanyl salicylate group of compound 4 (Supplemental Figure 2, upper panel) can still occupy the P1 pocket of PTP1B, the surroundings of the active site appear clearly inappropriate to allow the thiazole part of the bi-dentate to engage in further intermolecular contacts (Supplemental Figure 2, upper panel). This observation proves that our screening method can be effective in producing potent and selective inhibitors of protein tyrosine phosphatases.
According to our docked structure, the furanyl salicylate group of compound 6 can still occupy the catalytic P1 site, while the nitro-phenyl ring fits mainly in the P2 pocket, where the nitro group can make a hydrogen bond with the backbone of the residue Gly442 (Figure 3A). Compound 6 is a very potent inhibitor of YopH phosphatase activity (IC50=0.3μM) (Figure 3B) while it can only inhibit PTP1B at very high concentrations (Table 1 and Supplemental Figure 3).
Next, we investigated the cell permeability of these compounds using the PAMPA (Parallel Artificial Membrane Permeation Assay) method (Table 1 and Materials and methods section). While compounds 4 and 5 show limited cell permeability, compound 2 and 6 are predicted to have a medium permeability due probably to their less hydrophilic character. In order to determine whether P2 or P3 occupying bi-dentate YopH inhibitors elicit an effect in cell, we tested the ability of compounds 2 and 6 to reverse the phenotype that characterizes human cells when exposed to Yersiniae. We used an established (25) simple cell-based assay with human cervical cancer Hela cells in which the change of the cell morphology caused by Yersinia Pseudotubercolosis infection is monitored (Figure 3C). In this cell-based assay (25), greater than 90% of uninfected cells show typical HeLa cell morphology in confluent monolayers, with intact cellular junctions (Figure 3C). After infection with Yersinia Pseudotubercolosis, less than 5% of cells retain normal morphology. The remainder round up and lose cellular junctions. However, when pretreated with compounds 2 and 6 Hela cells present nearly normal morphology indicating that these inhibitors can effectively prevents YopH-mediated cytopathology at the concentrations used (Figure 3C). Finally, since the protein we have used in our in vitro fluorescence based assay is the full length YopH, made up of the N-terminal phosphotyrosine binding and C-terminal catalytic domains, we have also investigated if the inhibitory activity of the compounds could be related to their interaction with the YopH N-terminal domain. Thus, we have carried out chemical shifts perturbation studies by means of 2D [1H,15N] HSQC experiments (28, 29, 35) with 15N-labeled N-terminal domain of YopH from Yersinia pseudotbercolosis (99% sequence homology with the same domain from Yersinia pestis) (8). At the concentrations tested in our experiments (two or three fold excess of compound respect to the protein) none of the compounds produces chemical shifts variations in the spectra (Supplemental Figure 4). These NMR studies thus indicate that our compounds are targeting mainly the C-terminal domain of YopH.
Conclusions
We have reported on a series of novel bi-dentate compounds made up of a furanyl salicylate unit, targeting the phosphatase YopH catalytic site (P1) (Figure 1A, 1B), that is chemically linked to a more variable fragment (second-site binder) that can interact with one of two additional binding pockets P2 and P3 adjacent to P1 (Figure 1D). These compounds are able to inhibit in vitro the phosphatase YopH from Yersinia with IC50 values in the sub- to low-micromolar range. Compounds 2 and 6 (Table 1) are preliminarily also able to reduce Yersinia Pseudotubercolosis induced cytopathology (Figure 3C) in a cell based assay with human cervical cancer Hela cells (25).
The compounds do not possess appreciable inhibitory activity towards the human phosphatase PTP1B. This observation confirms again that the second-site screening techniques may be successful in generating bi-dentate inhibitors with increased affinity and selectivity for a given target. The reported compounds can be used to elucidate the role of YopH inhibition and may help in the design of novel antibacterial strategies.
Supplementary Material
Acknowledgments
Financial support was obtained thanks to NIH grants AI059572 and AI070494 to MP.
Abbreviations
- DiFMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- PTPs
Protein Tyrosine Phosphatases
- PAMPA
Parallel Artificial Membrane Permeation Assay
- TEMPO
2,2,6,6-tetramethylpiperidine 1-oxyl
- YopH
Yersinia outer protein H
Footnotes
Supplementary Material:
The following supplementary material is available for this article:
Synthetic procedures and compounds characterization by mass spectroscopy and NMR; Figure showing the results of docking studies with bi-dentate inhibitors and YopH; Docking studies with PTP1B; Chemical shift perturbation studies with compound 5; IC50 curve for the inhibition of the PTB1B phosphatase activity by compound 6.
References
- 1.Persson C, Nordfelth R, Holmstrom A, Hakansson S, Rosqvist R, Wolf-Watz H. Cell-surface-bound Yersinia translocate the protein tyrosine phosphatase YopH by a polarized mechanism into the target cell. Mol Microbiol. 1995;18:135–50. doi: 10.1111/j.1365-2958.1995.mmi_18010135.x. [DOI] [PubMed] [Google Scholar]
- 2.Cheng LW, Schneewind O. Yersinia enterocolitica TyeA, an intracellular regulator of the type III machinery, is required for specific targeting of YopE, YopH, YopM, and YopN into the cytosol of eukaryotic cells. J Bacteriol. 2000;182:3183–90. doi: 10.1128/jb.182.11.3183-3190.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornelis GR. Type III secretion: a bacterial device for close combat with cells of their eukaryotic host. Philos Trans R Soc Lond B Biol Sci. 2000;355:681–93. doi: 10.1098/rstb.2000.0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornelis GR. Molecular and cell biology aspects of plague. Proc Natl Acad Sci U S A. 2000;97:8778–83. doi: 10.1073/pnas.97.16.8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phan J, Lee K, Cherry S, Tropea JE, Burke TR, Jr, Waugh DS. High-resolution structure of the Yersinia pestis protein tyrosine phosphatase YopH in complex with a phosphotyrosyl mimetic-containing hexapeptide. Biochemistry. 2003;42:13113–21. doi: 10.1021/bi030156m. [DOI] [PubMed] [Google Scholar]
- 6.Black DS, Marie-Cardine A, Schraven B, Bliska JB. The Yersinia tyrosine phosphatase YopH targets a novel adhesion-regulated signalling complex in macrophages. Cell Microbiol. 2000;2:401–14. doi: 10.1046/j.1462-5822.2000.00061.x. [DOI] [PubMed] [Google Scholar]
- 7.Black DS, Montagna LG, Zitsmann S, Bliska JB. Identification of an amino-terminal substrate-binding domain in the Yersinia tyrosine phosphatase that is required for efficient recognition of focal adhesion targets. Mol Microbiol. 1998;29:1263–74. doi: 10.1046/j.1365-2958.1998.01014.x. [DOI] [PubMed] [Google Scholar]
- 8.Khandelwal P, Keliikuli K, Smith CL, Saper MA, Zuiderweg ER. Solution structure and phosphopeptide binding to the N-terminal domain of Yersinia YopH: comparison with a crystal structure. Biochemistry. 2002;41:11425–37. doi: 10.1021/bi026333l. [DOI] [PubMed] [Google Scholar]
- 9.Guan KL, Dixon JE. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science. 1990;249:553–6. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- 10.Bubeck SS, Dube PH. Yersinia pestis CO92 delta yopH is a potent live, attenuated plague vaccine. Clin Vaccine Immunol. 2007;14:1235–8. doi: 10.1128/CVI.00137-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vazquez J, Tautz L, Ryan JJ, Vuori K, Mustelin T, Pellecchia M. Development of molecular probes for second-site screening and design of protein tyrosine phosphatase inhibitors. J Med Chem. 2007;50:2137–43. doi: 10.1021/jm061481l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jahnke W, Rudisser S, Zurini M. Spin label enhanced NMR screening. J Am Chem Soc. 2001;123:3149–50. doi: 10.1021/ja005836g. [DOI] [PubMed] [Google Scholar]
- 13.Jahnke W, Florsheimer A, Blommers MJ, Paris CG, Heim J, Nalin CM, et al. Second-site NMR screening and linker design. Curr Top Med Chem. 2003;3:69–80. doi: 10.2174/1568026033392778. [DOI] [PubMed] [Google Scholar]
- 14.Jahnke W. Spin labels as a tool to identify and characterize protein-ligand interactions by NMR spectroscopy. Chembiochem. 2002;3:167–73. doi: 10.1002/1439-7633(20020301)3:2/3<167::AID-CBIC167>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 15.Vazquez J, De SK, Chen LH, Riel-Mehan M, Emdadi A, Cellitti J, et al. Development of paramagnetic probes for molecular recognition studies in protein kinases. J Med Chem. 2008;51:3460–5. doi: 10.1021/jm800068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Black E, Breed J, Breeze AL, Embrey K, Garcia R, Gero TW, et al. Structure-based design of protein tyrosine phosphatase-1B inhibitors. Bioorg Med Chem Lett. 2005;15:2503–7. doi: 10.1016/j.bmcl.2005.03.068. [DOI] [PubMed] [Google Scholar]
- 17.Tautz L, Bruckner S, Sareth S, Alonso A, Bogetz J, Bottini N, et al. Inhibition of Yersinia tyrosine phosphatase by furanyl salicylate compounds. J Biol Chem. 2005;280:9400–8. doi: 10.1074/jbc.M413122200. [DOI] [PubMed] [Google Scholar]
- 18.Tautz L, Mustelin T. Strategies for developing protein tyrosine phosphatase inhibitors. Methods. 2007;42:250–60. doi: 10.1016/j.ymeth.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 19.Khandelwal P, Keliikuli K, Smit CL, Saper MA, Zuiderweg ER. 1H, 15N and 13C assignments of the N-terminal domain of Yersinia outer protein H in its apo form and in complex with a phosphotyrosine peptide. J Biomol NMR. 2001;21:69–70. doi: 10.1023/a:1011971202626. [DOI] [PubMed] [Google Scholar]
- 20.Alonso A, Bottini N, Bruckner S, Rahmouni S, Williams S, Schoenberger SP, et al. Lck dephosphorylation at Tyr-394 and inhibition of T cell antigen receptor signaling by Yersinia phosphatase YopH. J Biol Chem. 2004;279:4922–8. doi: 10.1074/jbc.M308978200. [DOI] [PubMed] [Google Scholar]
- 21.Wu S, Vossius S, Rahmouni S, Miletic AV, Vang T, Vazquez-Rodriguez J, et al. Multidentate small-molecule inhibitors of vaccinia H1-related (VHR) phosphatase decrease proliferation of cervix cancer cells. J Med Chem. 2009;52:6716–23. doi: 10.1021/jm901016k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teschner M, Henn C, Vollhardt H, Reiling S, Brickmann J. Texture mapping: a new tool for molecular graphics. J Mol Graph. 1994;12:98–105. doi: 10.1016/0263-7855(94)80074-x. [DOI] [PubMed] [Google Scholar]
- 23.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–45. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 24.Wu B, Rega MF, Wei J, Yuan H, Dahl R, Zhang Z, et al. Discovery and binding studies on a series of novel Pin1 ligands. Chem Biol Drug Des. 2009;73:369–79. doi: 10.1111/j.1747-0285.2009.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Browne SH, Lesnick ML, Guiney DG. Genetic requirements for salmonella-induced cytopathology in human monocyte-derived macrophages. Infect Immun. 2002;70:7126–35. doi: 10.1128/IAI.70.12.7126-7135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becattini B, Culmsee C, Leone M, Zhai D, Zhang X, Crowell KJ, et al. Structure-activity relationships by interligand NOE-based design and synthesis of antiapoptotic compounds targeting Bid. Proc Natl Acad Sci U S A. 2006;103:12602–6. doi: 10.1073/pnas.0603460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becattini B, Pellecchia M. SAR by ILOEs: an NMR-based approach to reverse chemical genetics. Chemistry. 2006;12:2658–62. doi: 10.1002/chem.200500636. [DOI] [PubMed] [Google Scholar]
- 28.Pellecchia M, Becattini B, Crowell KJ, Fattorusso R, Forino M, Fragai M, et al. NMR-based techniques in the hit identification and optimisation processes. Expert Opin Ther Targets. 2004;8:597–611. doi: 10.1517/14728222.8.6.597. [DOI] [PubMed] [Google Scholar]
- 29.Pellecchia M. Solution nuclear magnetic resonance spectroscopy techniques for probing intermolecular interactions. Chem Biol. 2005;12:961–71. doi: 10.1016/j.chembiol.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 31.Pellecchia M, Bertini I, Cowburn D, Dalvit C, Giralt E, Jahnke W, et al. Perspectives on NMR in drug discovery: a technique comes of age. Nat Rev Drug Discov. 2008;7:738–45. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hajduk PJ, Dinges J, Miknis GF, Merlock M, Middleton T, Kempf DJ, et al. NMR-based discovery of lead inhibitors that block DNA binding of the human papillomavirus E2 protein. J Med Chem. 1997;40:3144–50. doi: 10.1021/jm9703404. [DOI] [PubMed] [Google Scholar]
- 33.Hajduk PJ. SAR by NMR: putting the pieces together. Mol Interv. 2006;6:266–72. doi: 10.1124/mi.6.5.8. [DOI] [PubMed] [Google Scholar]
- 34.Tautz L, Pellecchia M, Mustelin T. Targeting the PTPome in human disease. Expert Opin Ther Targets. 2006;10:157–77. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- 35.Pellecchia M, Sem DS, Wuthrich K. NMR in drug discovery. Nat Rev Drug Discov. 2002;1:211–9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.