

Abstract
Aims
Rosiglitazone and fenofibrate, specific agonists of the peroxisome proliferator activated receptors-γ (PPARγ) and -α (PPARα), respectively, improve insulin sensitivity in diabetic animals and in patients with type 2 diabetes. Here we investigated how pre-diabetic Otsuka Long–Evans Tokushima Fatty (OLETF) rats fed with normal and high-fat diets respond to these PPAR agonists.
Main methods
Pre-diabetic OLETF rats were subjected to high-fat or standard diets with or without rosiglitazone or fenofibrate for 2 weeks. The metabolism of the rats and the levels of malonyl-CoA and activities of malonyl-CoA decarboxylase (MCD), acetyl-CoA carboxylase (ACC), and AMP-activated protein kinase (AMPK) in metabolic tissues were assessed.
Key findings
Rosiglitazone and fenofibrate significantly improved insulin sensitivity and reduced the levels of plasma triglycerides and free fatty acids in OLETF rats fed with a high-fat diet. Fenofibrate particularly reduced the body weight, fat, and total cholesterol in high fat diet OLETF rats. The highly elevated malonyl-CoA levels in the skeletal muscle and liver of OLETF rat were significantly reduced by rosiglitazone or fenofibrate due to, in part, the increased MCD activities and expression. On the other hand, ACC activities were unchanged in skeletal muscle and decreased in liver in high fat diet group. AMPK activities were dramatically decreased in OLETF rats and not affected by these agonists.
Significance
These results demonstrate that treatment of pre-diabetic OLETF rats–particularly those fed a high-fat diet–with rosiglitazone and fenofibrate significantly improves insulin sensitivity and fatty acid metabolism by increasing the activity of MCD and reducing malonyl-CoA levels in the liver and skeletal muscle.
Keywords: PPARγ, PPARα agonists, Rosiglitazone, Fenofibrate, Malonyl-CoA, Malonyl-CoA decarboxylase, Acetyl-CoA carboxylase
Introduction
Rosiglitazone and fenofibrate, specific agonists of peroxisome proliferator activated receptors-γ (PPARγ) and -α (PPARα), respectively, improve insulin sensitivity in diabetic animals (Guerre-Millo et al. 2000; Kramer et al. 2001; Wang et al. 2001), and in people with type 2 diabetes and insulin resistance syndrome (Mudaliar and Henry 2001; Saltiel and Olefsky 1996). PPARs are members of the nuclear hormone receptor superfamily and are activated by natural or synthetic fatty acids. They regulate transcription by forming heterodimers with the retinoid X receptor (RXR) and binding to specific PPAR-response elements (PPREs) in the promoter region of target genes (Blaschke et al. 2006; Jepsen and Rosenfeld 2002).
PPARγ is the key transcriptional regulator of adipogenesis and it plays a critical role in glucose homeostasis (Lehmann et al. 1995; Rosen et al. 1999). PPARα is critical for peroxisome proliferation (Lee et al. 1995) and it functions as fatty acid sensor important for the regulation of fatty acid metabolism and energy homeostasis (Blaschke et al. 2006). Although PPARγ and PPARα play specific and separate roles under physiological conditions, their agonists have similar effects on triglyceride levels in the blood of diabetic animals (Chaput et al. 2000). Malonyl-CoA is a potent inhibitor of mitochondrial carnitine palmitoyltransferase (CPT1), the key regulatory enzyme involved in the first committed step of fatty-acid oxidation in mitochondria (Rasmussen et al. 2002), and its levels are precisely regulated in various nutritional states (Chien et al. 2000). Inhibition of CPT1 by malonyl-CoA decreases the uptake of fatty acids into the mitochondria and slows mitochondrial fatty acid oxidation (Mcgarry 2002). Malonyl-CoA is synthesized by acetyl-CoA carboxylase (ACC) (Thampy 1989) and degraded to acetyl-CoA and carbon dioxide by malonyl-CoA decarboxylase (MCD) (Saha et al. 2000). It has been reported that exercise-induced activation of AMPK controls malonyl-CoA levels by coordinating the functions of MCD and ACC (Park et al. 2002).
Although PPAR agonists markedly reduce blood triglyceride levels in diabetic animals (Chaput et al. 2000), to our knowledge their effect on the regulation of malonyl-CoA has not been investigated in the tissues of non-exercising pre-diabetic rats. In this study we investigated the effect of rosiglitazone and fenofibrate on insulin resistance and malonyl-CoA levels in the liver and skeletal muscles of Otsuka Long–Evans Tokushima Fatty (OLETF) rats. These rats spontaneously develop mild diabetes at 18 weeks of age and are pre-diabetic at 14 weeks of age with normal fasting glucose levels and high insulin levels (Kawano et al. 1992, 1994). We treated 14-week-old pre-diabetic OLETF rats fed with a normal or high-fat diet with rosiglitazone or fenofibrate for 2 weeks and found that these agonists stimulate MCD activity, decrease malonyl-CoA levels in the liver and skeletal muscles, and improve insulin sensitivity.
Materials and methods
Materials
[1, 3 14C]malonyl-CoA was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA), [14C]sodium bicarbonate from Moravek Biochemicals (Brea, CA, USA), [γ-32P]ATP from Amersham Biosciences (Piscataway, NJ, USA), and rat insulin RIA kit from Linco Research, Inc. (St. Charles, MO, USA). TaqMan EZ RT-PCR Core Reagents and the primers and probes of MCD and cyclophilin were purchased from Applied Biosystems (Foster City, CA, USA). The AMARAASAAALARRR (AMARA) peptide was synthesized by and purchased from Peptron Inc. (Taejeon, Korea). High-fat diet was purchased from BioGenomics, Inc. (Harlan, CA, USA). Whole blood glucose analyzer was obtained from Johnson & Johnson (Milpitas, CA, USA). Protease inhibitors, phosphatase inhibitors, and all other reagents were purchased from Sigma Chemical (St. Louis, MO, USA).
Animals and diet composition
Four-week-old male Long–Evans Tokushima Otsuka (LETO) rats (n=20) and Otsuka Long–Evans Tokushima Fatty (OLETF) rats (n=60) were donated by Otsuka Pharmaceuticals (Japan) and were group housed until the experiment was completed. At 14 weeks of age, the rats were randomly assigned to one of seven dietary regimens: 1) LETO (n=20) standard rat diet, 2) OLETF (n=10) standard rat diet, 3) OLETF (n=10) standard rat diet with rosiglitazone (4 mg/kg/d), 4) OLETF (n =10) standard rat diet with fenofibrate (100 mg/kg/d), 5) OLETF (n=10) high-fat diet, 6) OLETF (n=10) high-fat diet with rosiglitazone (4 mg/kg/d), 7) OLETF (n=10) high-fat diet with fenofibrate (100 mg/kg/d). The standard diet comprises approximately 3.6 kcal/g: 21% protein, 12.5% fat, and 66.5% carbohydrates and the high-fat diet comprises approximately 5.0 kcal/g: 21% protein, 66.5% fat, 12.5% carbohydrates as described (Bi et al. 2007). All rats were kept in the Department of Animal Experiment of Clinic Medical Research Center, Yonsei University Medical College, under controlled conditions of 23±1 °C and 12 h light: 12 h dark cycle, and given free access to food and water ad libitum. All rats were cared for as outline in the Guidelines of Animal Experiments recommended by Korean Academy Sciences.
Body weight and oral glucose tolerance test
Food consumed was checked every day, and body weight was checked every 3 days during the experiments. Oral glucose tolerance tests (OGTT) were conducted at the beginning and end of each experiment. Rats were given glucose orally (2 g/kg) and blood samples were collected from their tails 0, 30, 60, 90, 120 min later. Glucose levels were measured with a glucose analyzer (Milpitas, CA, USA).
Tissue sampling and measurement of fat mass
After 2 weeks of feeding with the normal or high-fat diet in the presence or absence of rosiglitazone or fenofibrate, animals were anesthetized with isoflurane and sacrificed for tissue sampling. Blood was collected by cardiac puncture for triglyceride (TG), total cholesterol (TC), free fatty acid (FFA), and insulin assays. Abdominal subcutaneous fat (SQ fat) and epididymal fat (epid fat) pads were surgically removed through a mid-abdominal incision and the weight of each dissected fat mass was recorded immediately. Skeletal muscles (gastrocneminus), livers, and pancreas were immediately isolated, freeze-clamped in liquid nitrogen, and stored at −80 °C until assays were performed.
Serum lipids and insulin, and pancreatic insulin content measurement
Triglyceride, total cholesterol, and free fatty acid levels were measured with infinity triglycerides reagent, infinity cholesterol reagent, and the ACS-ACOD enzyme method (NEFA ZYME-S, Aiken, Japan). To measure pancreatic insulin contents, pancreases were homogenized with an electric homogenizer in 500 μl of acid ethanol buffer (1.5 ml of 12 mol/L HCl in 100 ml of 70% ethanol) and extracted overnight at 4 °C. Insulin levels in the serum and pancreatic extract were measured with a rat RIA kit according to the manufacturer’s instructions.
Determination of malonyl-CoA levels
Malonyl-CoA was extracted from frozen (−80 °C) skeletal muscles and livers as previously described (King et al. 1988). The 6% perchloric acid extract was maintained at a pH of 2–3. Malonyl-CoA was measured with a modified high-performance liquid chromatography (HPLC) procedure described previously (King et al. 1988; Saddik et al. 1993) on a Beckman System Gold with a UV detector 167. Each sample (100 μl) was run through a pre-column cartridge (C18, size 3 cm, 7 μm) and a Microsorb short-one column (type C18, particle size 3 μm, size 4.6×100 mm). Absorbance was set at 254 nm and flow rate at 1 ml/min. A gradient was initiated using two buffers: buffer A consisted of 0.2 M NaH2PO4 (pH 5.0) and buffer B was a mixture of 0.25 M NaH2PO4 and acetonitrile (pH 5.0) in a ratio of 80:20 (v/v). Buffers were filtered using filter pure, Nylon-66 filter membrane (Pierce Chemical Co.). Initial conditions (97% buffer A, 3% buffer B) were maintained for 2.5 min and were changed thereafter to 18% buffer B over 5 min using Beckman’s curve 3. At 15 min the gradient was changed linearly to 37% buffer B over 3 min and subsequently 90% buffer B over 17 min. At 42 min the composition was returned linearly back to 3% buffer B over 0.5 min, and at 50 min column equilibration was complete. Peaks were integrated by a Beckman System Gold software package.
Determination of ACC activities
ACC activities were measured as described previously (Sakamoto et al. 2000). Briefly, approximately 200 mg of frozen tissues were homogenized using an electric homogenizer with a buffer containing Tris–HCl (10 mM), mannitol (200 mM), NaF (50 mM), EDTA (1 mM), 2-mercaptoethanol (10 mM), pH 7.5, and three proteolytic enzyme inhibitors (aprotinin, leupeptin, and antitrypsin, at a concentration of 5 mg/L). The homogenate was immediately centrifuged at 48,000 g for 30 min at 4 °C. The fraction containing ACC and AMPK was precipitated from the supernatant by addition of 144 mg ammonium sulfate/ml and by stirring for 60 min on ice. The precipitate was collected by centrifugation at 48,000 g for 30 min. The pellet was dissolved in 10% of the original volume of the homogenate buffer and centrifuged again to remove insoluble protein. The supernatant was assayed for ACC and AMPK activity.
ACC activity was measured using the CO2 fixation method, as described previously (Sakamoto et al. 2000). Briefly, 5 μl of the above supernatant containing 20 μg of total protein was added to a reaction mixture (final volume, 165 μl) containing tris acetate (60.6 mM), BSA (1 mg/ml), 2-mercaptoethanol (1.32 μmol/l), ATP (2.21 mM), acetyl-CoA (1.06 mM), magnesium acetate (5.0 mM), and NaHCO3 (18.08 mM). Samples were incubated at 37 °C for 10 min, and the reaction was stopped by adding 25 μl of 10% perchloric acid. Samples were centrifuged for 20 min at 3500 rpm, and 160 μl of supernatant was placed in minivials and dried in a fume hood overnight. H2O (100 μl), followed by scintillant, was added to the vials, and the vials were counted. ACC activity was expressed as the amount of malonyl-CoA produced/min/mg protein.
Determination of AMPK activities
AMPK activity was measured as previously described (Sakamoto et al. 2000). Briefly, 2 μl of the supernatant prepared as described above for the determination of ACC activity was added to a reaction mixture composed of HEPES-NaOH (40 mM), NaCI (80 mM), glycerol (8% wt/vol), EDTA (0.8 mM), AMARAASAAALARRR (AMARA) peptide (200 μmol/L), dithiothreitol (DTT) (0.8 mM), [γ-32P]ATP (200 μmol/l), MgCl2 (5 mM), and 0.18% Triton X-100. Samples were incubated in the presence or absence of 200 μM AMP for 3 min at 30 °C. 15 μl of this mixture was spotted on 1 cm2 phosphocellulose paper and the paper was washed four times for 10 min each with 150 mM phosphoric acid, followed by a 5 min acetone wash. The papers were then dried and counted for radioactivity. AMPK activity was expressed as nmol of 32P incorporated into the AMARA peptide/min/mg protein.
Determination of MCD activities
Approximately 300 mg of frozen tissues was homogenized in a buffer of 0.1 M Tris–HCl (pH 8.0), 2 mM PMSF, 5 μM aprotinin, 5 μM leupeptin, and 5 μM pepstatin A. 40 mM β-glycerophosphate, 40 mM NaF, 4 mM NaPPi, and 1 mM Na3VO4 were added to inhibit phosphatase activity. The homogenates were then centrifuged at 500 g for 10 min. Powdered (NH4)2SO4 was slowly added to the supernatant with stirring until 40% (243 g/L of ammonium sulfate) saturation was achieved. The mixture was stirred for 1 h on ice and centrifuged at 14,000 g for 10 min. The supernatant from this spin was treated with additional (NH4)2SO4 until 55% (351 g/L of ammonium sulfate) saturation was achieved. The mixture was recentrifuged at 14,000 g. The resultant pellet fraction was dissolved in 0.1 M Tris–HCl (pH 8.0) and MCD activity was determined as described below.
The enzyme activity was assayed by measuring the amount of 14CO2 generated from [3-14C]malonyl-CoA, as previously described (Goodwin and Taegtmeyer 1999) with the following modification. Briefly, a reaction mixture of 10 μmol of Tris–HCl buffer (pH 8.0), 0.01 μmol of dithioerythritol (DTE), 0.02 μmol of [3-14C]malonyl-CoA was incubated with enzyme in a total volume of 0.1 ml for 10 min at 37 °C. The 14CO2 generated by [3-14C] malonyl-CoA was trapped in filter papers soaked with 2 N KOH and assayed by liquid scintillation spectrometry. Enzyme activity is expressed as nmol/min/mg protein.
RNA extraction and real-time RT-PCR
Total RNA was extracted from frozen (−80 °C) tissues with TRIzol Reagent (GIBCO BRL) according to the manufacturer’s instructions. Real-time RT-PCR was performed using an ABI PRISM 7700 Sequence Detection System instrument and software (PE Applied Biosystems). Primers and probes for MCD and cyclophilin were used as described previously (Young et al. 2001). MCD primer/probe: forward 5′-CGGCACCTTCCTCATAAAGC-3′, reverse 5′-GGGTATAGGTGACAGGCTGGA-3′, probe 5′-FAM-AGTGGTCAAGGA GCTGCAGAAGGAGTTT-TAMRA-3′. Cyclophilin primer/probe: forward 5′-CTGA TGGCGAGCCCTTG-3′, reverse 5′-TCTGCTGTCTTTGGAACTTTGTC-3′, probe 5′-FAM-CGCGTCTGCTTCGAG-CTGTTTGCA-TAMRA-3′. The level of transcripts for the constitutive housekeeping gene product cyclophilin was measured in triplicate in each sample to adjust for sample-to-sample differences in RNA concentration. MCD mRNA expression was calculated with the comparative CT (threshold cycle) method and reported as the ratio of MCD transcripts per cyclophilin transcripts.
Statistical analysis
Data are presented as means±SEM. Statistically significant differences between groups were calculated by Tukey’s Multiple Comparison Test of One-Way ANOVA. A value of p<0.05 is considered significant.
Results
Effect on body weight, and fat mass
To examine the effects of PPARγ and α agonists on pre-diabetic OLETF rats, we fed 14-week-old OLETF rats with normal or high-fat diets and measured their body weight and fat mass after 2 weeks of treatment with rosiglitazone or fenofibrate (Table 1). We found that the body weights of all groups of OLETF rats were significantly higher than those of LETO controls. Rosiglitazone and fenofibrate treatment did not significantly affect the phenotype of rats fed a normal diet. However, fenofibrate, but not rosiglitazone, significantly reduced body weight and abdominal subcutaneous and epididymal fat mass, in rats fed a high-fat diet.
Table 1.
Body weight and fat mass after 2-week treatment.
| LETO | Normal diet |
High fat diet |
|||||
|---|---|---|---|---|---|---|---|
| Ctrl | RSG | FNF | Ctrl | RSG | FNF | ||
| Food intake (g/d/rat) | 26.7±0.3 | 30.9±0.7 | 28.7±1.8 | 27.9±2.3 | 28.9±2.3 | 27.6±3.4 | 26.4±3.4 |
| BW (g) | 362±15 | 438±29† | 430±18 | 400±25 | 479±14‡ | 473±22 | 394±13★ |
| SQ fat (g) | 4.8±0.3 | 8.6±0.6† | 7.6±0.5 | 7.3±0.7 | 15.9±0.6‡ | 17.5±1.4 | 8.4±0.8★ |
| Epid fat (g) | 4.1±0.1 | 5.7±0.5 | 5.4±0.3 | 5.3±0.3 | 10.0±0.3‡ | 9.6±0.9 | 5.4±0.4★ |
| Total fat (%) | 2.5±0.4 | 3.2±0.7† | 3.2±0.3 | 2.9±0.5 | 5.4±0.4‡ | 5.7±0.8 | 3.5±0.5★ |
p<0.05, compared with normal control (LETO),
P<0.05, compared with normal diet fed OLETF control, and
P<0.05, compared with high fat diet fed OLETF control.
Values are expressed as means±SEM of 10 rats in each group.
Effect on blood lipids, cholesterol, insulin levels, and pancreatic insulin content
We then measured the levels of blood lipids, total cholesterol, and insulin in serum and in the pancreas in pre-diabetic rats after treatment with rosiglitazone and fenofibrate (Table 2). Serum triglycerides, free fatty acids, total cholesterol, insulin and pancreatic insulin content were elevated in all high fad diet groups of OLETF rats, indicating insulin resistance and abnormalities in lipid metabolism. In normal-diet groups, the levels of triglycerides and free fatty acids were significantly reduced by treatment with either rosiglitazone or fenofibrate. However, serum triglyceride, free fatty acid, and total cholesterol levels, and insulin were significantly reduced in the presence of either rosiglitazone or fenofibrate in high-fat diet groups, suggesting a significant improvement in lipid metabolism and insulin sensitivity.
Table 2.
Blood chemistry and pancreatic insulin content after a 2-week treatment.
| LETO | Normal diet |
High fat diet |
|||||
|---|---|---|---|---|---|---|---|
| Ctrl | RSG | FNF | Ctrl | RSG | FNF | ||
| TG (mg/dL) | 43.7±3.4 | 97.4±7.7† | 59.4±3.73 | 53.7±2.93 | 133.3±23.5‡ | 59.0±10.8★ | 75.8±8.8★ |
| FFA μEq/L) | 1023±135 | 1898±235 | 1357±1863 | 880±993 | 2710±318‡ | 1224±160★ | 1638±27★ |
| TC (mg/dL) | 79.3±4.4 | 83.7±7.3 | 98.7±7.5 | 70.5±6.5 | 124.3±5.6‡ | 91.3±2.7★ | 76.8±5.4★ |
| S. Ins(ng/mL) | .73±0.08 | 1.17±0.06† | 1.32±0.07 | 1.19±0.05 | 2.16±0.06‡ | 1.89±0.09★ | 1.87±0.04★ |
| P. Ins (ng/mg) | 4.8±0.7 | 5.7±0.5 | 6.8±1.3 | 6.7±2.0 | 7.5±1.3‡ | 7.9±0.5 | 8.0±0.6 |
Blood were taken from rat’s hearts when the rats were sacrificed and serums were separated by centrifugation. Pancreatic islet insulin was extracted using a buffer composed of 1.5% hydrochloride and 70% alcohol.
p<0.05, compared with normal control (LETO);
p<0.05, compared with OLETF control fed a normal diet;
p<0.01 vs. normal diet fed OLETF control; and
p<0.05 vs. high fat diet fed OLETF control.
Values are means±SEM of 10 rats in each group.
Effects on oral glucose tolerance test
We next performed the oral glucose tolerance test (OGTT) (2 g/kg body weight) on 16-week-old pre-diabetic OLETF rats treated with rosiglitazone and fenofibrate. As shown in Fig. 1, both groups developed impaired glucose tolerance. This impairment was more severe in the group fed the high-fat diet, despite the fact that these rats still exhibited normal fasting glucose levels. However, treatment of pre-diabetic OLETF rats with rosiglitazone or fenofibrate significantly improved insulin sensitivity in both the normal (Fig. 1A) and the high-fat (Fig. 1B) diet groups. Our results clearly show that PPAR-γ and -α agonists can improve insulin sensitivity during the pre-diabetic period even in the presence of a high-fat diet.
Fig. 1.
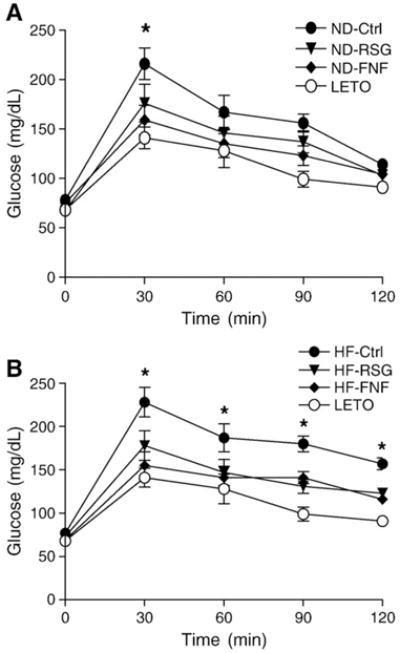
Results of the oral glucose tolerance test (OGTT). OGTT (2 g/kg body weight) was performed after pre-diabetic OLETF rats were treated for 2 weeks with rosiglitazone and fenofibrate. A. OGTT in 16-week-old OLETF rats fed a normal diet. *, p<0.05, treated groups were compared with normal diet OLETF (ND-Ctrl) rats of the same age. B. OGTT in 16-week-old OLETF rats fed a high-fat diet. *, p<0.05, treated groups were compared with high fat diet OLETF (HF-Ctrl) rats of the same age. Values are expressed as means± SEM (n=10). ND, normal diet; HF, high fat; Ctrl, control; RSG, rosiglitazone; FNF, fenofibrate; LETO, Long–Evans Tokushima Otsuka.
Effect on malonyl-CoA levels in skeletal muscle and liver
Since PPAR-γ and -α agonists significantly reduced serum levels of triglycerides and free fatty acids in pre-diabetic rats fed either a normal or a high-fat diet (Table 2), we investigated whether these agonists affect the levels of malonyl-CoA in skeletal muscle and liver. As shown in Fig. 2, malonyl-CoA levels were highly elevated in both the skeletal muscle and livers of pre-diabetic OLETF rats compared to control rats. This elevation was more significant in rats fed with a high-fat diet, suggesting an abnormal regulation of malonyl-CoA in the pre-diabetic OLETF rats. Malonyl-CoA levels in skeletal muscles of OLETF rats fed a normal diet were 25% lower in the rosiglitazone-treated animals and 33% lower in the fenofibrate-treated animals. In OLETF rats fed a high-fat diet, rosiglitazone treatment decreased malonyl-CoA levels in skeletal muscles by 35% and fenofibrate treatment by 43% (Fig. 2A). In OLETF rats fed a normal diet, rosiglitazone treatment decreased malonyl-CoA levels in the liver by 24% and fenofibrate treatment by 27% (Fig. 2B). In the high-fat diet group, rosiglitazone treatment reduced malonyl-CoA levels in the liver by 30% and fenofibrate treatment by 41% (Fig. 2B). Taken together, our results clearly indicate that both rosiglitazone and fenofibrate can reduce the levels of malonyl-CoA in skeletal muscle and liver in this pre-diabetic rat model.
Fig. 2.

Malonyl-CoA levels in the skeletal muscle and livers of OLETF rats fed either normal or high-fat diets. A. Malonyl-CoA levels in skeletal muscle. B. Malonyl-CoA levels in liver. Values are expressed as means±SEM (n=10). †, p<0.05, vs. LETO; ‡, P<0.01, vs. OLETF controls in the normal-diet group; * and ★, p<0.01, vs. corresponding OLETF control. Ctrl, control; RSG, rosiglitazone; FNF, fenofibrate.
Effect on MCD and ACC activities
Since malonyl-CoA levels in tissues are regulated by MCD and ACC, we measured the activities of these two enzymes in skeletal muscle. As shown in Fig. 3A, rosiglitazone and fenofibrate significantly increased MCD activity in rats fed either a normal or a high-fat diet (Fig. 3A). Interestingly, MCD activity was more dramatically enhanced by fenofibrate than by rosiglitazone in rats fed a high-fat diet. However, ACC activity in skeletal muscle was not significantly changed after treatment (Fig. 3B). These results suggest that rosiglitazone and fenofibrate decrease malonyl-CoA levels at least in part by increasing MCD activity in skeletal muscle in OLETF rats fed either a normal or a high-fat diet.
Fig. 3.
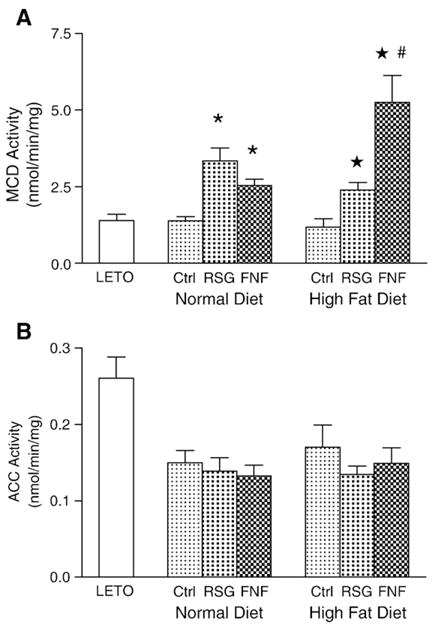
MCD and ACC activities in skeletal muscle. A. MCD activities. B. ACC activities. Values are means±SEM (n=10), * and ★, p<0.001 vs. corresponding OLETF control; #, p<0.001, vs. rosiglitazone treated group fed a high-fat diet. Ctrl, control; RSG, rosiglitazone; FNF, fenofibrate.
We next examined the effect of rosiglitazone and fenofibrate on MCD and ACC activities in the liver. We found that fenofibrate, but not rosiglitazone, significantly increased the activity of MCD in OLETF rats fed a normal diet. Both rosiglitazone and fenofibrate increased the activity of MCD in rats fed a high-fat diet (Fig. 4A). In contrast to ACC activity in rats fed a normal diet, ACC activity was elevated in rats fed a high-fat diet. Rosiglitazone suppressed this elevated ACC activity by 38% and fenofibrate by 87% (Fig. 4B). These results suggested that the decrease we observed in malonyl-CoA levels in the livers of pre-diabetic rats fed a high-fat diet is due to both an increase in degradation by MCD and a decrease in synthesis by ACC.
Fig. 4.
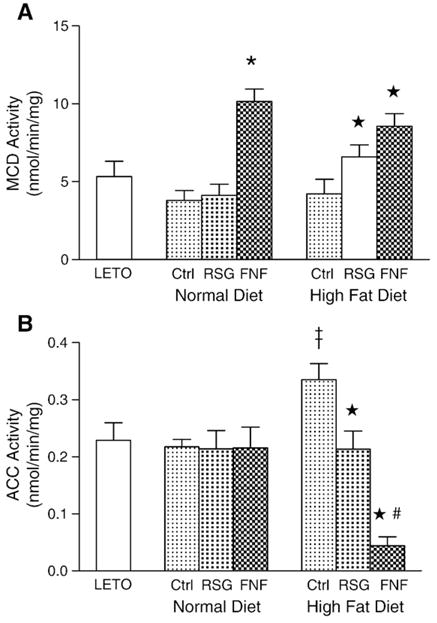
MCD and ACC activities in liver. Values are expressed as means±SEM (n=10). A. MCD activities.*, p<0.001, ★, p<0.01, vs. corresponding OLETF control. B. ACC activities. #, p<0.01, compared with OLETF controls in the normal-diet group. ‡, p<0.01 vs. normal diet fed OLETF control; ★, p<0.01 vs. high fat diet fed OLETF control, and #, p<0.01, vs. rosiglitazone-treated group fed a high-fat diet.
Effect on AMPK activities in skeletal muscle and liver
It has been previously reported that AMPK regulates MCD and ACC activities under specific conditions, such as during exercise or the electric stimulation of skeletal muscles (Park et al. 2002). To examine whether AMPK regulates MCD and ACC activities in resting state in pre-diabetic OLETF rats, we examined its activity in skeletal muscle and liver. Surprisingly, the activity of AMPK in both tissues was dramatically lower in this pre-diabetic rat model than in control rats (Fig. 5). In addition, rosiglitazone and fenofibrate did not affect AMPK activity in the tissues of OLETF rats regardless of diet. These results suggest that the AMPK signaling network may be defective in the skeletal muscles and livers of OLETF rats.
Fig. 5.
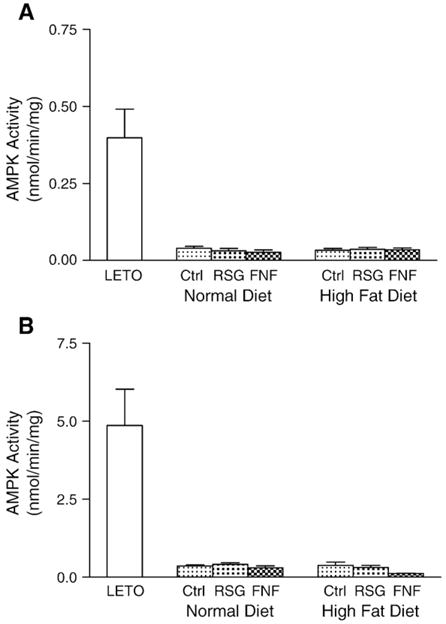
AMPK activities in skeletal muscle and liver. A. AMPK activities in skeletal muscle. B. AMPK activities in liver. Values are expressed as means±SEM (n=10).
MCD mRNA expression in skeletal muscle and liver
PPARs are transcriptional regulators. Since PPAR agonists increase MCD activity in both skeletal muscle and liver, we used real-time RT-PCR to examine whether they affect the transcription of MCD in OLETF rats fed a normal or high-fat diet. As shown in Fig. 6A, the levels of MCD mRNA in the skeletal muscles of OLETF rats were significantly lower than that LETO rats and restored by fenofibrate, but not rosiglitazone. Interestingly, we observed that MCD mRNA levels were significantly higher in rats fed a high-fat diet than in rats fed a normal diet. This finding suggests that, consistent with previous reports, increased availability of fatty acids may promote their utilization by increasing the expression of MCD in skeletal muscles (Young et al. 2001). As shown in Fig. 6B, the levels of MCD mRNA in the livers of OLETF rats are similar regardless of whether the rats are fed a normal or a high-fat diet. As in skeletal muscle, fenofibrate, but not rosiglitazone, significantly increased the expression of MCD in the livers of OLETF rats. Our data suggest that PPAR-α, but not PPAR-γ, agonists stimulate the transcription of MCD in skeletal muscle and liver, and that a high-fat diet alters this transcription in skeletal muscle.
Fig. 6.
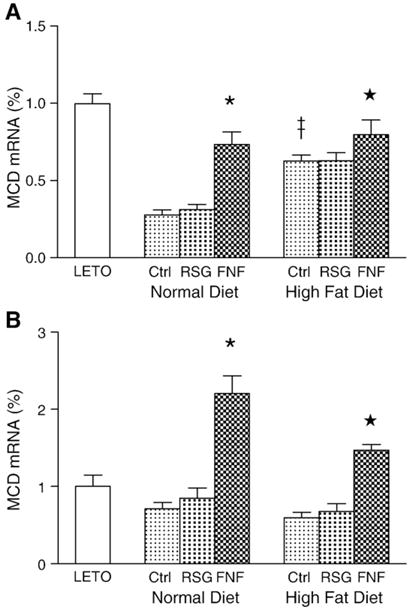
Expression levels of MCD mRNA in skeletal muscle and liver. The values are compared to control (LETO) rats. Values are expressed as means±SEM (n=10). A. Relative levels of MCD mRNA in skeletal muscle. ‡, p<0.01, vs. OLETF controls in the normal-diet;*, p<0.01, vs. corresponding OLETF control. B. Relative levels MCD mRNA in liver, *and ★, p<0.01 vs. corresponding OLETF control and rosiglitazone treated groups.
Discussion
Clinically, rosiglitazone is used as an insulin-sensitizing drug in type 2 diabetes (Lehmann et al. 1995) due to its ability to regulate genes that control cellular energy homeostasis and insulin action in muscle and liver, whereas fenofibrate is used for the treatment of dyslipidemia mainly due to its ability to lower triglyceride levels (Berger and Moller 2002). The role of these agonists of PPARs in insulin resistance has been investigated extensively in diabetic animals (Guerre-Millo et al. 2000; Kramer et al. 2001; Wang et al. 2001) and in patients with type 2 diabetes and insulin resistance syndrome (Mudaliar and Henry 2001; Saltiel and Olefsky 1996). However, a few studies have emphasized the pre-diabetic phase, during which insulin resistance is already in place despite normal blood glucose levels due to the hyper-secretion of insulin by pancreatic β-cells.
In this study, we examined the effect of the specific agonists of PPAR-γ and PPAR-α, rosiglitazone and fenofibrate, respectively, on 14-week-old OLETF rats fed for 2 weeks with either a normal or a high-fat diet. OLETF rats spontaneously develop mild diabetes at 18 weeks of age and 14-week-old OLETF rats are pre-diabetic, but have normal fasting glucose levels (Kawano et al. 1992, 1994). Our results indicate that, in addition to their different effects on body weight and fat mass (Table 1), rosiglitazone and fenofibrate significantly improve the insulin resistance and reduce the levels of triglycerides, free fatty acids, and total cholesterol in the serum of these pre-diabetic animals (Table 2). Importantly, these agonists of PPAR-γ and PPAR-α also significantly reduce the levels of malonyl-CoA, a key regulator for fatty acid oxidation, in skeletal muscle and liver.
In the liver, malonyl-CoA is important both as an intermediate in the de novo synthesis of fatty acids and as an allosteric inhibitor of CPT1, the enzyme that controls the transfer of long-chain fatty acyl-CoAs into the mitochondria for oxidation. In contrast, malonyl-CoA functions mainly as a regulator of CPT1 in skeletal muscle, where de novo synthesis of fatty acids is minimal (Mcgarry 2002). Similar to what has been observed in obese humans or those who have type 2 diabetes (Bandyopadhyay et al. 2006), we found that the concentrations of malonyl-CoA in skeletal muscle and liver are highly elevated in pre-diabetic rats fed a normal diet, and that this effect is exacerbated by a high-fat diet. Increased malonyl-CoA levels may promote the de novo synthesis of fatty acids in liver and inhibit the oxidation of fatty acids in both liver and skeletal muscle, causing an accumulation of triglycerides and fatty acids that contribute to insulin resistance. Our data suggest that using PPAR-γ and PPAR-α agonists to lower malonyl-CoA levels may be one way to improve insulin resistance in pre-diabetic animals or humans.
Malonyl-CoA is synthesized by acetyl-CoA carboxylase (ACC) (Thampy 1989) and degraded to acetyl-CoA and carbon dioxide by malonyl-CoA decarboxylase (MCD) (Saha et al. 2000). In this study, we characterized the activities of these two enzymes in non-exercising, pre-diabetic OLETF rats. We found that MCD activities in both skeletal muscle and liver were significantly increased after 2 weeks of rosiglitazone and fenofibrate treatment even in rats fed a high-fat diet. In addition, we determined that the profound activation of MCD by fenofibrate that we observed in this study was due to an increase in MCD mRNA levels. This finding is consistent with our previous report that PPAR-α enhances MCD transcription (Lee et al. 2004). Our data suggest that PPAR-γ and PPAR-α agonists reduce malonyl-CoA levels in pre-diabetic OLETF by enhancing MCD transcription.
In contrast to MCD, the effects of PPAR-γ and PPAR-α agonists on ACC activity in liver differs from their effects in skeletal muscle. Although ACC activity was not affected by either rosiglitazone or fenofibrate in skeletal muscle, it was significantly elevated in the livers of rats fed a high-fat diet. This increased ACC activity was reduced to normal levels by rosiglitazone treatment and suppressed even further by fenofibrate.
ACC is a cytosolic enzyme that catalyzes the carboxylation of cytosolic acetyl-CoA to form malonyl-CoA (Saha and Ruderman 2003). Two principal isoforms have been identified: a 265 kDa protein referred to as ACCα or ACC1, which is predominantly expressed in lipogenic tissues such as liver and adipose tissue; and a 275–280 kDa protein, ACCβ or ACC2, which, although it is present to some extent in liver and other tissues (Bianchi et al. 1990; Kim 1997), is the major isoform expressed in skeletal muscle. ACCα and ACCβ are the products of distinct genes and they have different affinities for their substrate, cytosolic acetyl-CoA (Kim 1997). ACCα plays a major role in fatty acid synthesis and ACCβ is important for fatty acid oxidation (Kim 1997; Ruderman et al. 1999).
The fact that a high-fat diet increases ACC activity in the liver but not the skeletal muscles of pre-diabetic OLETF rats, and that this increase is abrogated by treatment with PPAR-γ and PPAR-α agonists, suggests that these conditions specifically modulate ACCα activity and its ability to promote fatty acid synthesis. In particular, the dramatic decrease in ACCα activity we observed in response to fenofibrate may explain the significant reduction in body weight and fat mass in OLETF rats fed with a high-fat diet.
Expression of ACCα is regulated by multiple hormones and nutrient status mediated by three promoters and multiple transcriptional factors, such as sterol regulatory element binding proteins (SREBP1a and SREBP1c) and carbohydrate response element binding protein (ChREBP), as well as by mRNA splicing (Barber et al. 2005; Brownsey et al. 2006). In addition, its function is regulated through posttranslational modification, including protein maturation, allosteric regulation, multiple sites of phosphorylation, and protein–protein interaction (Brownsey et al. 2006; Tong 2005). Therefore, further studies are required to determine how this PPAR-α agonist inhibits ACCα activity in the liver under these conditions.
AMPK is a heterotrimeric protein kinase complex that acts as an energy sensor, responding to a rise in AMP levels by increasing ATP-generating pathways and reducing ATP-consuming pathways (Hardie et al. 2003; Kahn et al. 2005). AMPK activation inhibits fatty acid and cholesterol synthesis and gluconeogenesis in the liver and stimulates fatty acid uptake and oxidation, glucose uptake, and mitochondrial biogenesis in skeletal muscle (Kahn et al. 2005). It regulates malonyl-CoA levels by inactivating ACC and activating MCD during exercise or the electric stimulation of skeletal muscles (Hardie and Carling 1997; Saha et al. 2000). It has been hypothesized that dysregulation of the AMPK/malonyl-CoA fuel-sensing and signaling network is a key factor in the development of insulin resistance (Ruderman and Prentki 2004). Thiazolidinedione treatment reportedly stimulates AMPK activity in cultured cells, rat liver, and adipose tissues by increasing the ratio of AMP to ATP (Fryer et al. 2002; Saha et al. 2004).
In contrast, we found that AMPK activity levels are barely detectable in pre-diabetic OLETF rats, regardless of whether they were fed with a normal or a high-fat diet. Furthermore, these low basal levels were not affected by treatment with either rosiglitazone or fenofibrate. Consistent with this observation, the activity of ACC–a key target of AMPK activation–in skeletal muscle remained essentially unchanged in rats fed either a normal or a high-fat diet. Our results suggest that AMPK/malonyl-CoA signaling network is defective in this pre-diabetic rat model, which may contribute to the dysregulation of fatty acid metabolism in skeletal muscle and liver.
Conclusion
In this study, we demonstrate that rosiglitazone and fenofibrate improve insulin sensitivity and reduce plasma triglyceride and free fatty acid levels in pre-diabetic rats. This effect is likely due to a reduction of malonyl-CoA levels caused by an increase in MCD activity. In particular, fenofibrate stimulates the transcription of MCD by PPAR-α. The fact that ACC and AMPK activities in skeletal muscle were not affected by these agonists suggests a defect in the AMPK signaling network in this diabetic rat model. Further studies will be required to determine how MCD activity is stimulated by rosiglitazone and why fenofibrate inhibits ACCα activity in the livers of rats fed a high-fat diet. Our data strongly suggest that early intervention with PPAR agonists during the pre-diabetic period will significantly improve fatty acid metabolism and insulin sensitivity and, perhaps, prevent further development of diabetes.
Acknowledgments
This work was supported by the Korean government (MOST) (No. R13-2002-054-04002-0) and Engineering Foundation, the grant (DK074805) from the National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK), and the grant (7-06RA-87) from the American Diabetes Association.
References
- Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. 2006;55 (8):2277–2285. doi: 10.2337/db06-0062. [DOI] [PubMed] [Google Scholar]
- Barber MC, Price NT, Travers MT. Structure and regulation of acetyl-CoA carboxylase genes of metazoa. Biochimica et Biophysica Acta (BBA)—Molecular and Cell Biology of Lipids. 2005;1733 (1):1–28. doi: 10.1016/j.bbalip.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanisms of action of PPARs. Annual Review of Medicine. 2002;53 (1):409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Bi S, Chen J, Behles RR, Hyun J, Kopin AS, Moran TH. Differential body weight and feeding responses to high-fat diets in rats and mice lacking cholecystokinin 1 receptors. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2007;293 (1):R55–63. doi: 10.1152/ajpregu.00002.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, Evans JL, Iverson AJ, Nordlund AC, Watts TD, Witters LA. Identification of an isozymic form of acetyl-CoA carboxylase. Journal of Biological Chemistry. 1990;265 (3):1502–1509. [PubMed] [Google Scholar]
- Blaschke F, Takata Y, Caglayan E, Law RE, Hsueh WA. Obesity, peroxisome proliferator-activated receptor, and atherosclerosis in type 2 diabetes. Arteriosclerosis. Thrombosis, and Vascular Biology. 2006;26 (1):28–40. doi: 10.1161/01.ATV.0000191663.12164.77. [DOI] [PubMed] [Google Scholar]
- Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM. Regulation of acetyl-CoA carboxylase. Biochemical Society Transactions. 2006;34 (Pt 2):223–227. doi: 10.1042/BST20060223. [DOI] [PubMed] [Google Scholar]
- Chaput E, Saladin R, Silvestre M, Edgar AD. Fenofibrate and rosiglitazone lower serum triglycerides with opposing effects on body weight. Biochemical and Biophysical Research Communications. 2000;271 (2):445–450. doi: 10.1006/bbrc.2000.2647. [DOI] [PubMed] [Google Scholar]
- Chien D, Dean D, Saha AK, Flatt JP, Ruderman NB. Malonyl-CoA content and fatty acid oxidation in rat muscle and liver in vivo. American Journal of Physiology. Endocrinology, Metabolism and Gastrointestinal. 2000;279 (2):E259–265. doi: 10.1152/ajpendo.2000.279.2.E259. [DOI] [PubMed] [Google Scholar]
- Fryer LGD, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. Journal of Biological Chemistry. 2002;277 (28):25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Goodwin GW, Taegtmeyer H. Regulation of fatty acid oxidation of the heart by MCD and ACC during contractile stimulation. American Journal of Physiology. 1999;277 (4 Pt 1):E772–777. doi: 10.1152/ajpendo.1999.277.4.E772. [DOI] [PubMed] [Google Scholar]
- Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, Herbert JM, Winegar DA, Willson TM, Fruchart JC, Berge RK, Staels B. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. Journal of Biological Chemistry. 2000;275 (22):16638–16642. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D. The AMP-activated protein kinase. Fuel gauge of the mammalian cell? European Journal of Biochemistry. 1997;246 (2):259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- Hardie DG, John WS, David AP, Emma RH. Management of cellular energy by the AMP-activated protein kinase system. FEBS Letters. 2003;546 (1):113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- Jepsen K, Rosenfeld MG. Biological roles and mechanistic actions of co-repressor complexes. Journal of Cell Science. 2002;115 (4):689–698. doi: 10.1242/jcs.115.4.689. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metabolism. 2005;1 (1):15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kawano K, Hirashima T, Mori S, Natori T. OLETF (Otsuka Long–Evans Tokushima Fatty) rat: A new NIDDM rat strain. Diabetes Research and Clinical Practice. 1994;24(Suppl):S317–S320. doi: 10.1016/0168-8227(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long–Evans Tokushima Fatty (OLETF) strain. Diabetes. 1992;41 (11):1422–1428. doi: 10.2337/diab.41.11.1422. [DOI] [PubMed] [Google Scholar]
- Kim KH. Regulation of mammalian acetyl-coenzyme A carboxylase. Annual Review of Nutrition. 1997;17:77–99. doi: 10.1146/annurev.nutr.17.1.77. [DOI] [PubMed] [Google Scholar]
- King MT, Reiss PD, Cornell NW. Determination of short-chain coenzyme A compounds by reversed-phase high-performance liquid chromatography. Methods in Enzymology. 1988;166:70–79. doi: 10.1016/s0076-6879(88)66012-5. [DOI] [PubMed] [Google Scholar]
- Kramer D, Shapiro R, Adler A, Bush E, Rondinone CM. Insulin-sensitizing effect of rosiglitazone (BRL-49653) by regulation of glucose transporters in muscle and fat of Zucker rats. Metabolism. 2001;50 (11):1294–1300. doi: 10.1053/meta.2001.27202. [DOI] [PubMed] [Google Scholar]
- Lee GY, Kim NH, Zhao ZS, Cha BS, Kim YS. Peroxisomal-proliferator-activated receptor alpha activates transcription of the rat hepatic malonyl-CoA decarboxylase gene: A key regulation of malonyl-CoA level. Biochemical Journal. 2004;378 (Pt 3):983–990. doi: 10.1042/BJ20031565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Molecular and Cellular Biology. 1995;15 (6):3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) Journal of Biological Chemistry. 1995;270 (22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- McGarry JD. Banting lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51 (1):7–18. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- Mudaliar S, Henry RR. New oral therapies for type 2 diabetes mellitus: The glitazones or insulin sensitizers. Annual Review of Medicine. 2001;52:239–257. doi: 10.1146/annurev.med.52.1.239. [DOI] [PubMed] [Google Scholar]
- Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman NB, Saha AK. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. Journal of Biological Chemistry. 2002;277 (36):32571–32577. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- Rasmussen BB, Holmback UC, Volpi E, Morio-Liondore B, Paddon-Jones D, Wolfe RR. Malonyl coenzyme A and the regulation of functional carnitine palmitoyltransferase-1 activity and fat oxidation in human skeletal muscle. Journal of Clinical Investigation. 2002;110 (11):1687–1693. doi: 10.1172/JCI15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Molecular Cell. 1999;4 (4):611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- Ruderman N, Prentki M. AMP kinase and malonyl-CoA: Targets for therapy of the metabolic syndrome. Nature Reviews. Drug Discovery. 2004;3 (4):340–351. doi: 10.1038/nrd1344. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. American Journal of Physiology. 1999;276 (1 Pt 1):E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- Saddik M, Gamble J, Witters LA, Lopaschuk GD. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. Journal of Biological Chemistry. 1993;268 (34):25836–25845. [PubMed] [Google Scholar]
- Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB. Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochemical and Biophysical Eesearch Communications. 2004;314 (2):580–585. doi: 10.1016/j.bbrc.2003.12.120. [DOI] [PubMed] [Google Scholar]
- Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: An expanding partnership. Molecular and Cellular Biochemistry. 2003;253 (1–2):65–70. doi: 10.1023/a:1026053302036. [DOI] [PubMed] [Google Scholar]
- Saha AK, Schwarsin AJ, Roduit R, Masse F, Kaushik V, Tornheim K, Prentki M, Ruderman NB. Activation of malonyl-CoA decarboxylase in rat skeletal muscle by contraction and the AMP-activated protein kinase activator 5-aminoimidazole-4-carboxamide-1-beta -D-ribofuranoside. Journal of Biological Chemistry. 2000;275 (32):24279–24283. doi: 10.1074/jbc.C000291200. [DOI] [PubMed] [Google Scholar]
- Sakamoto J, Barr RL, Kavanagh KM, Lopaschuk GD. Contribution of malonyl-CoA decarboxylase to the high fatty acid oxidation rates seen in the diabetic heart. American Journal of Physiology. Heart and Circulatory Physiology. 2000;278 (4):H1196–1204. doi: 10.1152/ajpheart.2000.278.4.H1196. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45 (12):1661–1669. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- Thampy KG. Formation of malonyl coenzyme A in rat heart. Identification and purification of an isozyme of A carboxylase from rat heart. Journal of Biological Chemistry. 1989;264 (30):17631–17634. [PubMed] [Google Scholar]
- Tong L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell Mol Life Sci. 2005;62 (16):1784–1803. doi: 10.1007/s00018-005-5121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PR, Guo Q, Ippolito M, Wu M, Milot D, Ventre J, Doebber T, Wright SD, Chao YS. High fat fed hamster, a unique animal model for treatment of diabetic dyslipidemia with peroxisome proliferator activated receptor alpha selective agonists. European Journal of Pharmacology. 2001;427 (3):285–293. doi: 10.1016/s0014-2999(01)01249-3. [DOI] [PubMed] [Google Scholar]
- Young ME, Goodwin GW, Ying J, Guthrie P, Wilson CR, Laws FA, Taegtmeyer H. Regulation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fatty acids. American Journal of Physiology. Endocrinology, Metabolism and Gastrointestinal. 2001;280 (3):E471–479. doi: 10.1152/ajpendo.2001.280.3.E471. [DOI] [PubMed] [Google Scholar]