

Abstract
Several mechanisms can underlie short-term synaptic depression, including vesicle depletion, receptor desensitization, and changes in presynaptic release probability. To determine which mechanisms affect depression under physiological conditions, we studied the synapse formed by auditory nerve fibers onto bushy cells in the anteroventral cochlear nucleus (the “endbulb of Held”) using voltage-clamp recordings of brain slices from P15–21 mice near physiological temperatures. Depression of both AMPA and NMDA EPSCs showed two phases of recovery. The fast component of depression for the AMPA EPSC was eliminated by cyclothiazide and aniracetam, suggesting it results from desensitization. The fast component of depression for the NMDA EPSC was reduced by the low-affinity antagonist L-AP5, suggesting it results from saturation. The remaining depression in AMPA and NMDA components is identical and therefore presynaptic in origin. It is likely to result from presynaptic vesicle depletion. Recovery from depression after trains of activity was slowed by the application of EGTA-AM, suggesting that the endbulb has a residual-calcium-dependent form of recovery. We developed a model that incorporates depletion, desensitization, and calcium-dependent recovery. This model replicated experimental findings over a range of experimental conditions. The model further indicated that desensitization plays only a minor role during prolonged activity, in large part because presynaptic release is so depleted. Thus, depletion appears to be the dominant mechanism of depression at the endbulb during normal activity. Furthermore, calcium-dependent recovery at the endbulb is critical to prevent complete run-down during high activity and to preserve the reliability of information transmission.
Keywords: Synapse, Depression, Saturation, Desensitization, Endbulb, Modeling
Introduction
Synaptic strength is subject to a number of mechanisms, both pre- and postsynaptic. These include facilitation, vesicle depletion, receptor desensitization, and receptor saturation (Regehr and Stevens 2001; Trussell et al. 1998; von Gersdorff and Borst 2002; Xu-Friedman and Regehr 2004; Zucker and Regehr 2002). Different mechanisms appear to predominate at different synapses (Regehr and Stevens 2001). However, it is still unclear which mechanisms are physiologically important and how they affect neuronal computation.
To study this issue, we examined the synapse formed by auditory nerve fibers onto bushy cells in the mouse anteroventral cochlear nucleus, termed the “endbulb of Held” (Lorente de Nó 1981). This synapse is particularly useful because in vitro experiments have implicated several mechanisms of depression, including desensitization (Isaacson and Walmsley 1996; Oleskevich et al. 2000), depletion (Bellingham and Walmsley 1999), and a decrease in presynaptic release probability (Bellingham and Walmsley 1999; Oleskevich et al. 2000). Desensitization and depletion have also been implicated at the homologous synapse in birds (Brenowitz et al. 1998; Otis et al. 1996; Trussell et al. 1993). Recovery from depletion is much slower than from desensitization, which could cause very different effects on synaptic strength during normal activity.
To evaluate the physiological significance of different mechanisms of depression, the endbulb is particularly useful because its activity in vivo has been well-characterized. Auditory nerve fibers can fire at rates of up to 300 Hz during sound stimulation (Johnson 1980; Joris et al. 1994a; Kiang 1965; Sachs and Abbas 1974; Taberner and Liberman 2005). Furthermore, bushy cells play a role in sound localization, where precise timing is important. Endbulb synapses appear to reduce jitter in the timing of bushy cell spikes (Joris et al. 1994a; Joris et al. 1994b) by mechanisms that depend on synaptic strength (Xu-Friedman and Regehr 2005a; b). Thus, it is important to understand how activity can affect synaptic strength and thereby the temporal precision of bushy cell spiking.
The amount of depression that results from a decrease in presynaptic release probability is somewhat controversial. Bellingham and Walmsley (1999) first described this mechanism at the endbulb, and suggested that it was responsible for all depression of both AMPA and NMDA components of the EPSC. One characteristic of the mechanism was that it was abolished by cyclothiazide application. Cyclothiazide (CTZ) is used to prevent AMPA receptor desensitization (Yamada and Tang 1993), and acts specifically in many (Chen et al. 2002; Dittman and Regehr 1998; Trussell et al. 1993; Xu-Friedman and Regehr 2003), but not all (Diamond and Jahr 1995; Ishikawa and Takahashi 2001), preparations. The cyclothiazide-sensitive form of depression described by Bellingham and Walmsley (1999) has not been described at any synapse besides the endbulb. It is therefore important to determine under what circumstances it is revealed.
To evaluate the relative importance of various mechanisms of depression, we conducted studies in mouse brain slices. We tested for the importance of AMPA receptor desensitization using cyclothiazide and aniracetam, which both prevent desensitization (Boxall and Garthwaite 1995; Yamada and Tang 1993) but through different mechanisms (Partin et al. 1996). By comparing AMPA and NMDA EPSCs, we could study presynaptic forms of depression. We saw no evidence of a cyclothiazide-sensitive change in presynaptic release probability. The presynaptic mechanism was most likely to be vesicle depletion, based on its recovery characteristics. In addition, recovery from depletion was activity-dependent. Based on our results, we developed a simple model of synaptic transmission that duplicates plasticity at the endbulb. This model indicated that, although desensitization is prominent at this synapse, it plays only a minor role during periods of high activity. Depletion dominates under these circumstances, and activity-dependent forms of recovery are critical to maintaining information transmission.
Materials and Methods
Slices were cut from the auditory brain stem of P15-P21 CBA/CaJ mice as described previously (Xu-Friedman and Regehr 2005a). Briefly, animals were anesthetized and decapitated, and the slices were removed and placed into low-sodium, ice-cold cutting solution containing (in mM): 76 NaCl, 26 NaHCO3, 75 sucrose, 1.25 NaH2PO4, 2.5 KCl, 25 glucose, 7 MgCl2, 0.5 CaCl2, bubbled with 95% O2 and 5% CO2 (pH 7.8, 305 mOsm). Slices were cut at a slight angle from sagittal, in order to best preserve straight projections of the auditory nerve, using an Integraslice 7500 MM (Campden Instruments, Loughborough, England) at 200 µm thickness and incubated at 30°C for 20 min in low-sodium solution and then for 40 min in standard recording solution containing (in mM): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 20 glucose, 1 MgCl2, 1.5 CaCl2, 4 Na L-lactate, 2 Na-pyruvate and 0.4 Na L-ascorbate bubbled with 95% O2 and 5% CO2 (pH 7.4, 310 mOsm). This Ca concentration is close to physiological conditions (Manthei et al. 1973). Slices were then maintained at room temperature until recording. Recordings were made in the standard recording solution, or in a solution with 3 mM CaCl2 and 0 mM MgCl2. These recording solutions are referred to by their calcium concentration, i.e. 1.5 Cae and 3 Cae. Recordings were preferentially made in the dorsal and anterior parts of the nucleus, where bushy cells are enriched. Bushy cells were identified by their characteristically large and fast EPSCs (Isaacson and Walmsley 1995). It is not possible in these conditions to discriminate between spherical and globular bushy cells.
All recordings were carried out at 34°C in the presence of 10 µM strychnine to block glycine receptors. We tested whether it was necessary to block group II/III mGluR autoreceptors or GABAB activation by applying 2 µM CGP55845 (GABAB antagonist), 30 µM CPPG, and 20 µM LY341495. These drugs had no effect on paired-pulse ratio (for Δt = 10 ms, PPR was 0.52 ± 0.10 in control, and 0.49 ± 0.08 in blockers, P > 0.2, paired t-test, N = 3) or steady-state EPSC amplitude (for 100 Hz, relative amplitude = 0.26 ± 0.07 in control and 0.22 ± 0.06 in blockers, P > 0.1; for 200 Hz, 0.12 ± 0.04 in control and 0.11 ± 0.03 in blockers, P > 0.2; for 333 Hz, 0.05 ± 0.01 in control and 0.05 ± 0.01 in blockers, P > 0.2, paired t-test, N = 3). Therefore, these blockers did not appear necessary and were not used in most experiments. The bath was perfused at 3–4 ml/min using a pump (403U/VM2, Watson-Marlow Inc., Wilmington, MA), with saline running through an in-line heater (SH-27B with TC-324B controller, Warner Instruments, Hamden, CT). Recordings of AMPA EPSCs were made in the presence of 5 µM 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphoric acid (CPP). Cyclothiazide was used at 50 µM, and aniracetam was used at 5 mM from a stock of 0.5 M in DMSO. Application of identical concentrations of DMSO alone had no significant effect on EPSCs or paired-pulse depression (for Δt = 10 ms, PPR changed from 0.35 ± 0.04 to 0.38 ± 0.03 in 0.14 M DMSO, P > 0.2 for this and all other intervals tested, paired t-test, N = 3). EGTA-AM was diluted from a stock solution of 100 mM in DMSO to a final concentration of 100 µM immediately before usage. Slices were viewed using an Olympus (Melville, NY) BX51WI microscope with a 60× objective. Whole-cell recordings were made from bushy cells using an Axopatch 700B amplifier (Molecular Devices, Sunnyvale, CA) using an internal solution containing (in mM): 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, and 1 QX-314 (pH 7.2, 302 mOsm). Pipettes were pulled from 1.5 mm OD, 0.86 ID borosilicate glass (Sutter Instruments, Novato, CA) to a resistance of 1–2 MΩ. Cells were voltage clamped at −70 mV, with access resistance 3–12 MΩ compensated to 70%. AN fibers were stimulated with an electrode placed in the AVCN with a 0.2 ms pulse of 6–14 µA (A360 stimulus isolator; World Precision Instruments, Sarasota, FL). For NMDA EPSCs, recordings were made in 3 Cae, and in the presence of 10 µM NBQX. Single or paired stimuli were applied every 10 sec. For paired NMDA EPSCs at short intervals, the amplitude of the second pulse was determined after subtracting the average single-pulse EPSC. Train stimuli were applied every 30 s. Stimulation and data collection were done using an ITC-18 (Instrutech Corp., Port Washington, NY) controlled using custom-written software running in Igor (Wavemetrics, Lake Oswego, OR). Most chemicals were purchased from Sigma (St. Louis, MO); CGP55845, CPP, CPPG, CTZ, LY341495, and NBQX were purchased from Tocris (Ellisville, MO); L-AP5 from Acros (Morris Plains, NJ); and EGTA-AM from Invitrogen (Carlsbad, CA).
Results
Depression at the endbulb of Held
We examined short-term synaptic plasticity at the endbulb synapse by recording from bushy cells in whole-cell voltage clamp in brain slices. We stimulated the presynaptic inputs using pairs of pulses with a range of interpulse intervals (Δt). We first examined the synaptic current carried by AMPA receptors by blocking all other synaptic conductances with strychnine and CPP, under conditions as close to physiological as possible (1.5 Cae, 34°C). At all intervals, the second EPSC recorded in the bushy cell was depressed with respect to the first (Fig. 1A, B). At Δt = 10 ms, the ratio of the second EPSC to the first (paired-pulse ratio, PPR) was 0.63 ± 0.03 (mean ± SEM, N = 17 cells). This depression appeared to recover in two phases, fast and slow. To enhance depression for easier study, we increased the external calcium to 3 mM (3 Cae). At this concentration, both fast and slow components of depression were greater (Fig. 1B). On average, the PPR at Δt = 10 ms was 0.38 ± 0.03 (N = 15). For endbulbs measured in both conditions, the first EPSC increased in amplitude as depression increased (Fig 1C). This increase in EPSC1 and depression with higher Cae is consistent with all mechanisms of depression that have been proposed to date.
Figure 1.
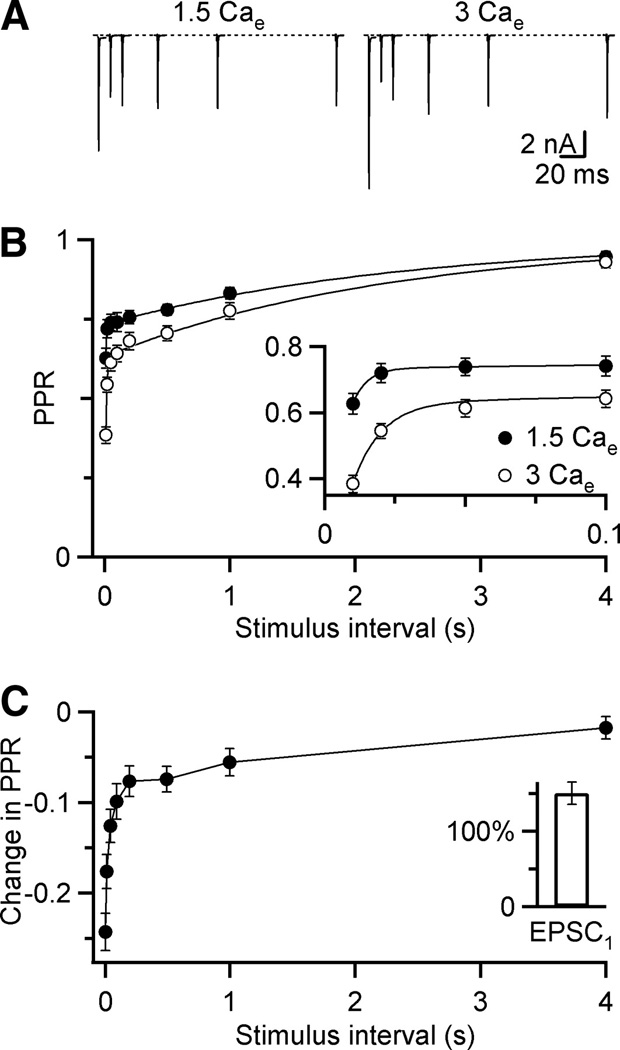
Kinetics of depression at the mouse endbulb of Held. A, AMPA EPSCs in response to paired-pulse stimuli for a range of interpulse intervals (Δt) in 1.5 Cae (left) and 3 Cae (right) for a single endbulb. Traces are averages of 4 or 5 trials. B, Average paired-pulse recovery in 1.5 Cae (black circles) or 3 Cae (open circles). Points are averages of 15 or 17 experiments. Error bars represent the standard error of the mean here and throughout. Inset, closeup of initial recovery. Curves are double-exponential fits to the experimental data (PPR = 1 – A1exp(Δt/τ1) 𢈒 A2 exp(Δt/τ2). In 1.5 Cae. A1 = 0.11 ± 0.02, τ1 = 5 ± 6 ms, A2 = 0.27 ± 0.01, τ2 = 2.4 ± 0.4 s. In 3 Cae, A1 = 0.25 ± 0.03, τ1 = 10 ± 3 ms, A2 = 0.37 ± 0.02, τ2 = 2.3 ± 0.3 s. C, Difference in PPR for cells measured in both 1.5 Cae and 3 Cae (N = 5 experiments). Inset, Amplitude of first EPSC in 3 Cae relative to 1.5 Cae.
We fit the time courses of recovery in the two different calcium concentrations using a double exponential curve (Fig. 1B), which yielded two components of depression that recovered with time constants of ~10 ms and ~2 s. This suggests that two distinct processes with different time courses contribute to depression.
Desensitization and depression
The depression of Fig. 1 could be attributable to presynaptic processes such as vesicle depletion or to postsynaptic processes such as receptor desensitization. The fast component seemed to match the time course of desensitization that has been seen at other synapses (Chen et al. 2002; Rozov et al. 2001; Trussell et al. 1993; Xu-Friedman and Regehr 2003). Desensitization has also been implicated at the endbulb synapse in rats (Isaacson and Walmsley 1996).
We tested for the importance of desensitization using 50 µM cyclothiazide (CTZ). Application of CTZ affected the timecourse and amplitude of AMPA EPSCs (Fig. 2A). Single-exponential fits of the decay phase of the EPSC were 0.34 ± 0.04 ms in control and 1.31 ± 0.40 in CTZ, which was a 3.29 ± 0.54-fold increase (N = 12). The amplitude of the first EPSC did not increase significantly (101 ± 3%). However, for very short intervals, the amplitude of the second pulse increased more than the first, leading to an increase in PPR (Fig. 2A, right). This effect was greatest at intervals < 50 ms, essentially eliminating the fast component of depression, but having no noticeable effect at longer intervals (Fig. 2B, C). For Δt = 10 ms, PPR was 0.42 ± 0.03 in control conditions, and 0.67 ± 0.05 in CTZ (N = 5). We quantified the effectiveness of CTZ by computing the percent change in PPR, which was greatest at Δt = 10 ms (67 ± 3%), and decayed to near 0 by 50 ms (Fig. 2D).
Figure 2.
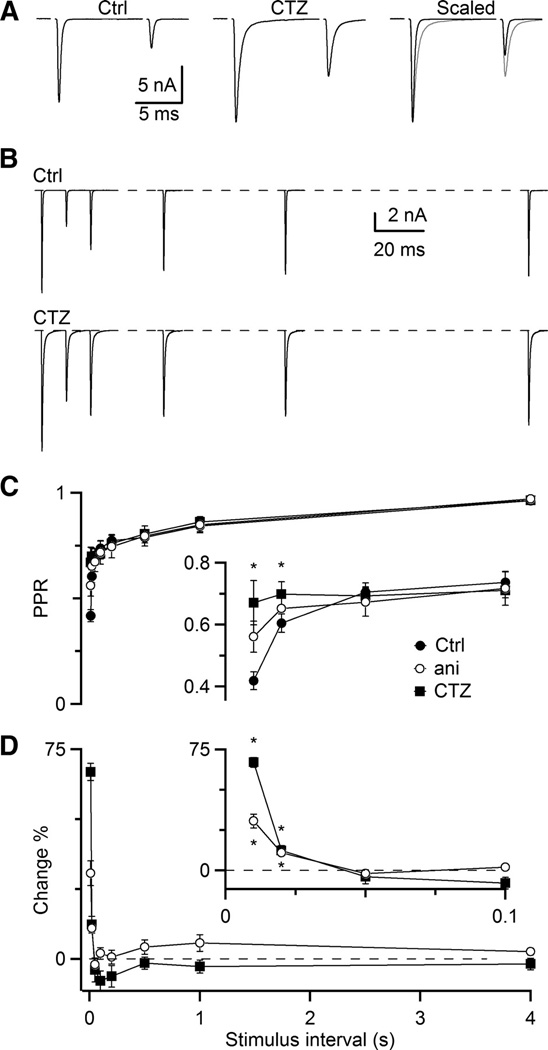
Cyclothiazide and aniracetam specifically reduce the fast component of AMPA EPSC depression. A, Paired-pulse depression at Δt = 10 ms in control conditions (left), in the presence of CTZ (middle), and scaled and overlaid (right, CTZ in red). B, Example paired-pulse recovery in 3 Cae in control and 50 µM CTZ. Traces are averages of 3 to 4 trials. C, Average PPR for 5 experiments in CTZ and 7 experiments in aniracetam (ani). Asterisks mark significant differences for both CTZ and ani (P < 0.01, one-sided, paired t-test). D, Relative increase in PPR in CTZ and ani with respect to control (PPRdrug/PPRctrl − 100%). Asterisks mark significant differences (P < 0.01, one-sided, paired t-test).
CTZ can have non-specific effects on the presynaptic terminal (Bellingham and Walmsley 1999; Ishikawa and Takahashi 2001). We saw no evidence of this under our experimental conditions. If CTZ affected presynaptic release, it would have been expected to affect the entire recovery curve in Fig. 2C. However, CTZ only affected the fast component and left the slow component unchanged (Fig. 2D). Thus it seemed likely that under our experimental conditions, the effects of CTZ were specific to preventing postsynaptic receptor desensitization.
We confirmed these results further using aniracetam, which is not reported to have the non-specific effects on other channel types seen with CTZ. Aniracetam gave results similar to CTZ. The amplitude of the first EPSC did not increase significantly (105 ± 7%, N = 6). However, the decay phase of the EPSC was slower by a factor of 3.86 ± 0.47 (τ = 0.233 ± 0.022 ms in control, 0.917 ± 0.178 ms in aniracetam). Furthermore, PPR increased in aniracetam, but the fast component was not eliminated completely. The PPR at Δt = 10 ms was 0.56 ± 0.07 (N = 6), which was an increase of 31 ± 4% over control, again with no effect for Δt ≥ 50 ms. This is consistent with the lower efficacy of aniracetam, even at its limit of solubility (5 mM) (Lawrence et al. 2003). Thus, the effects of aniracetam were similar to CTZ, although it was less effective at reducing depression. These results provide additional support that the fast component of depression results from AMPA receptor desensitization, and that CTZ and aniracetam specifically prevent it.
CTZ could also change PPR by increasing the “affinity” of AMPA receptors for glutamate (Partin et al. 1994). Thus, PPR may appear to increase if CTZ caused EPSC1 and EPSC2 to both get larger, but EPSC1 saturates. This hypothesis predicts that EPSC2 should increase uniformly for different cells, because EPSC2 is not saturated. Thus, for a given cell, if CTZ causes a significant increase in PPR, it must result from a comparatively smaller increase in EPSC1; cells with a small increase in PPR should have a relatively larger increase in EPSC1. However, if CTZ relieves depression primarily through a change in desensitization, then EPSC1 should increase uniformly among all cells, while EPSC2 should increase more for cells where CTZ had a bigger effect (i.e. desensitization was greatest).
We tested these two alternatives by plotting the increases in EPSC1 and EPSC2 against the increase in PPR for individual cells (Fig. 3). The increase in EPSC1 was fairly uniform across all cells, regardless of the change in PPR (Fig. 3, open symbols). However, the change in EPSC2 was larger for cells in which CTZ had a larger effect (Fig. 3, closed symbols). These results are consistent with the target of CTZ being desensitization, and they are not consistent with PPR measurements being contaminated by saturation effects.
Figure 3.
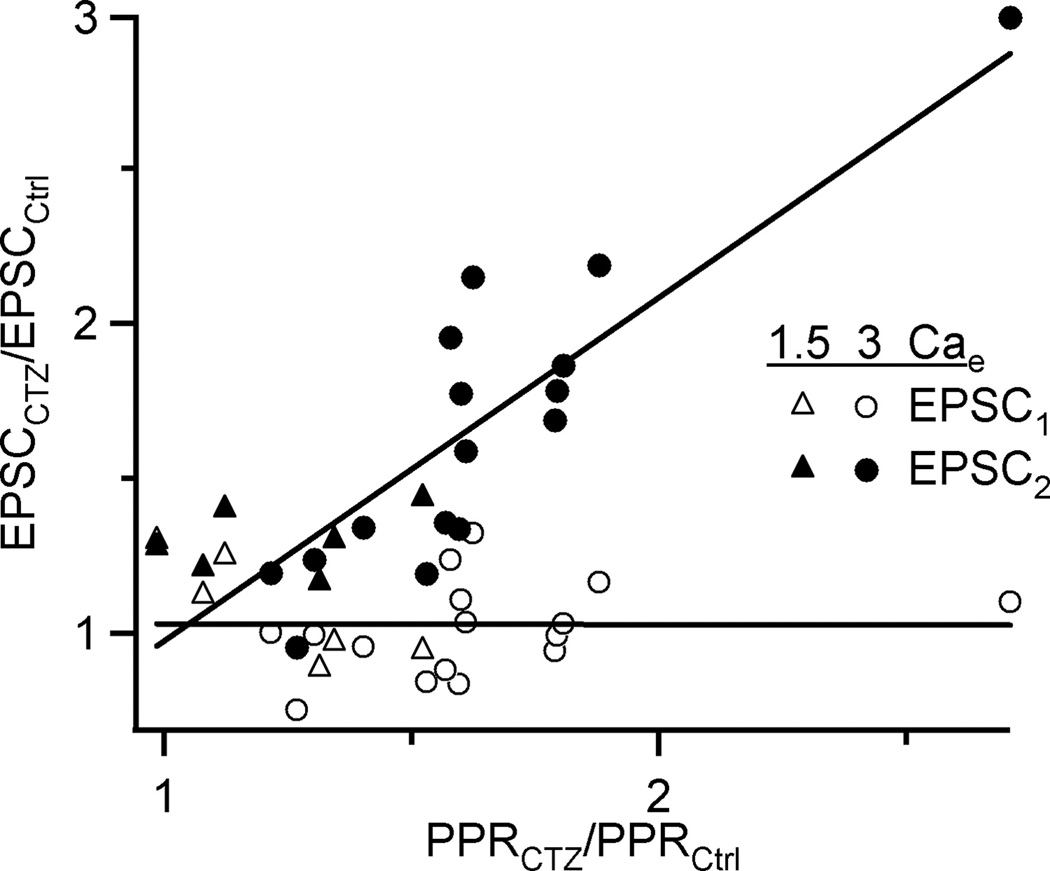
CTZ does not change PPR through saturation. The change in EPSC amplitude and PPR upon addition of CTZ was quantified for individual cells. Some cells showed large changes in PPR, whereas others showed less effect. The amplitude of EPSC1 (open symbols) changed uniformly regardless of the effect on PPR, while the amplitude of EPSC2 (closed symbols) correlated with effect on PPR. These results are not consistent with CTZ reducing depression by increasing saturation of the first pulse (see Results for explanation). These results are pooled from experiments conducted in 1.5 Cae (triangles) and 3 Cae (circles). Lines are fits to the data.
NMDA component of the EPSC
The NMDA component of the EPSC provides additional information about the mechanisms of depression. First, NMDA receptor kinetics are unaffected by CTZ, so the NMDA EPSC provides a useful control for the specificity of CTZ. Second, because both AMPA and NMDA receptors respond to the same glutamate signal, they can provide information about presynaptic forms of depression.
We studied the NMDA EPSC in 3 Cae in the presence of NBQX (Fig. 4). These experiments were conducted at a holding potential of −70 mV (3 Cae lacks magnesium). NMDA EPSCs at short intervals also show significant depression (Fig. 4A), though because of its slow kinetics, EPSC2 summates with EPSC1. Recovery from depression is best fit with a double exponential curve, with time constants of ~70 ms and 1.6 s. The slow component of recovery appears to have similar kinetics to the slow component observed for depression of the AMPA EPSC, and thus may share a common mechanism. However, the fast components have quite different kinetics, suggesting that they may result from different processes.
Figure 4.
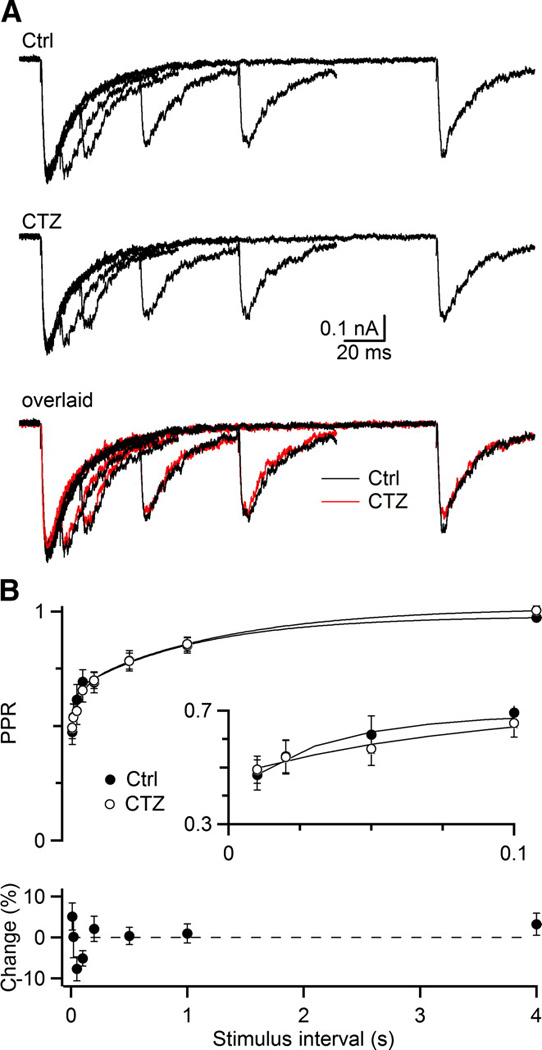
CTZ has no effect on the NMDA EPSC. A, Representative NMDA EPSCs measured in the presence of 10 µM NBQX and 3 Cae, shows the NMDA EPSC in control (upper), after addition of CTZ (middle), and these two sets of traces overlaid (bottom, CTZ in red). Traces are the average of 4 to 5 trials. B, Averaged PPR (upper) and relative change in PPR (bottom) from 6 experiments. Curves are double-exponential fits to the experimental data (see legend to Fig. 1). In control, A1 = 0.23 ± 0.06, τ1 = 71 ± 5 ms, A2 = 0.33 ± 0.05, τ2 = 1.6 ± 0.3 s. In CTZ, A1 = 0.25 ± 0.06, τ1 = 75 ± 5 ms, A2 = 0.30 ± 0.05, τ2 = 1.6 ± 0.3 s.
We first used the NMDA EPSC as a test for the specificity of CTZ. Application of 50 µM CTZ caused no significant change in NMDA EPSC depression in 3 Cae (Fig. 4A, B; P > 0.1 for all points, paired t-test, N = 7). To verify that NMDA receptor saturation did not mask changes in presynaptic release, we repeated these experiments in the standard recording solution containing 1.5 Cae. A holding potential of +35 mV was used in 1.5 Cae to relieve magnesium block of the receptor. The PPR in 1.5 Cae for Δt = 10 ms was 0.52 ± 0.04 in control, and 0.55 ± 0.03 in CTZ, with an average increase in PPR of 6.7 ± 3.9% (N = 6). Thus CTZ had no significant effect on depression for the NMDA component of the EPSC. This provides additional evidence that CTZ specifically affects postsynaptic AMPA receptor desensitization and that the fast component of depression for the AMPA EPSC results from desensitization.
NMDA Depression
We wanted to use the NMDA component of the EPSC to examine presynaptic contributions of depression. However, as noted above, recovery from depression of the NMDA EPSC appeared to follow a somewhat different time course compared to AMPA, especially in the fast components of recovery. We therefore tried to determine the mechanisms underlying the fast component of NMDA depression. One likely candidate is NMDA receptor saturation. Saturation arises when the number of receptors activated is sub-linearly related to the amount of neurotransmitter released. Glutamate has high affinity for the NMDA receptor and slow dissociation kinetics (Clements et al. 1992). Therefore, after an initial release of glutamate, fewer receptors may be available for the second pulse. This is consistent with the slow kinetics of the NMDA EPSC evident in Fig. 4.
To test this possibility, we used the low-affinity NMDA receptor antagonist L-AP5 (Olverman et al. 1988). L-AP5 prevents some NMDA receptors from binding glutamate and being activated during the first pulse. L-AP5 can dissociate rapidly from some of these receptors because it has a low affinity, thereby revealing NMDA receptors that are unbound to glutamate. These receptors can respond to a second pulse of glutamate. For this method to work, L-AP5 must block most, but not all, NMDA receptors. We determined the optimal concentration of L-AP5, by doing experiments similar to Fig. 5A – C. For single stimuli, the NMDA EPSC was blocked with a Kd of 112 ± 13 µM (Fig. 5D). We also looked at the effect of L-AP5 on paired stimuli with Δt = 10 ms. With increasing L-AP5, we saw a significant increase in PPR, indicating the presence of receptor saturation (Fig. 5C). Fits of the dose-response for the effect on paired pulses yielded an EC50 of 250 ± 190 µM (Fig. 5E).
Figure 5.
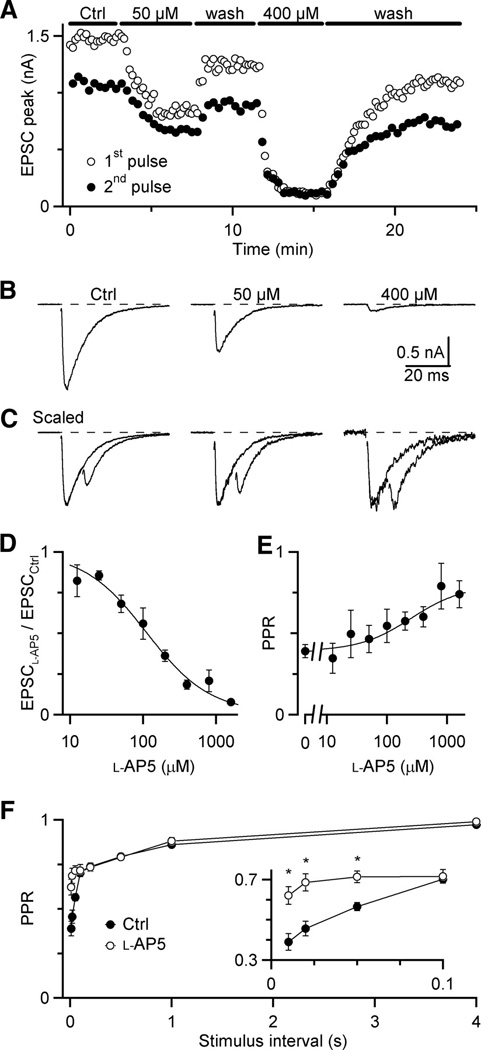
Fast component of NMDA EPSC depression is reduced in the presence of the low-affinity NMDA antagonist L-AP5. A, Representative experiment showing effects of L-AP5 on NMDA EPSC amplitudes. Trials alternated between single stimuli and paired pulse stimuli with Δt = 10 ms. L-AP5 is applied during periods indicated. B, Average NMDA EPSCs from the experiment shown in A, showing efficacy of L-AP5. C, Average paired-pulse EPSCs from the experiment shown in A, scaled to the same initial EPSC peak amplitude, to more clearly indicate changes in the second EPSC amplitude. D, Dose-response curve of L-AP5 for single EPSCs. Each point is the average of 2 to 8 experiments. Data are fitted with a Hill Equation of the form: relative block = 1 − (1 − min)/(1 + K/[L-AP5]), with K = 112 ± 13 µM and min = 0.0098 ± 0.018. E, Dose-response curve for NMDA PPR. Each point is the average of 2 to 8 experiments. Data are fitted with a Hill equation of the form PPR = 0.39 + (max − 0.39) / (1 + EC50/[L-AP5]), with max = 0.90 ± 0.12% and EC50 = 250 ± 190 µM. Number of experiments in D and E: 2 (12, 25, and 1600 µM), 3 (800 µM), 4 (100 and 200 µM), 6 (50 µM), 7 (0 µM in E), and 8 (400 µM). F, Paired-pulse recovery for NMDA EPSCs in control and 400 µM L-AP5 (average of 7 experiments). Asterisks mark significant differences (P < 0.01, paired t-test).
To determine whether saturation contributes to the fast component of NMDA EPSC depression, we recorded pairs of pulses of varying intervals in the presence of 400 µM L-AP5. We used this concentration because it reduced saturation significantly, but retained large enough EPSCs for reliable peak measurements. We found that L-AP5 significantly reduced the fast component of depression, but had little effect on the slow component. The greatest effect was for Δt =10 ms, where PPR increased from 0.39 ± 0.04 to 0.62 ± 0.04 (N = 7; Fig. 5F). This suggests that the fast component of NMDA EPSC depression results from saturation, while the slow component results from a separate process.
The recovery characteristics of NMDA and AMPA EPSCs are compared in Fig. 6. In control conditions, there is a significant difference in initial recovery rate, but not in the slow component of recovery (Fig. 6A). Our data suggested that the fast components of recovery from depression result from desensitization for the AMPA component of the EPSC, and saturation for the NMDA EPSC. If these two forms of postsynaptic depression could be blocked, the remaining depression is likely to be due to presynaptic forms of depression. We next compared the recovery of the AMPA EPSC in the presence of CTZ with the NMDA EPSC in the presence of L-AP5. We found that the difference was now very small (Fig. 6B), suggesting that the remaining depression is presynaptic in origin. Based on the timecourse of recovery, this mechanism is most likely to be vesicle depletion.
Figure 6.
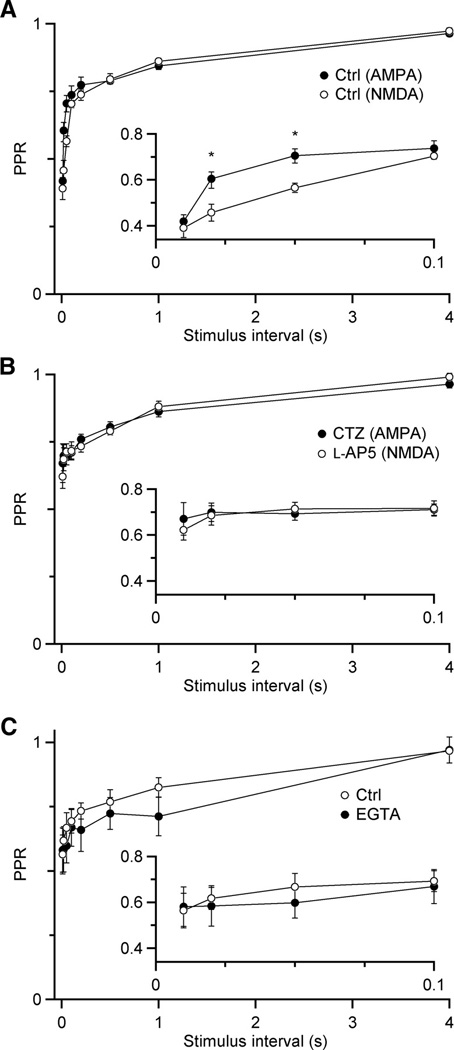
Comparison of AMPA and NMDA EPSC depression reveals presynaptic contribution. A, Paired-pulse recovery in control conditions for AMPA (N = 11 experiments) and NMDA (N = 7) EPSCs. The contribution of postsynaptic mechanisms of depression cause the curves to differ at short intervals. Asterisks mark significant differences (P < 0.01, unpaired t-test). B, Paired-pulse recovery for AMPA EPSCs in the presence of CTZ to prevent desensitization (N = 5) and NMDA EPSCs in the presence of 400 µM L-AP5 to prevent saturation (N = 7). With postsynaptic contributions to depression reduced, the remaining depression is likely of presynaptic origin. C, Paired-pulse recovery for AMPA EPSCs before and after treatment with EGTA-AM, all in the presence of CTZ (N = 5 experiments).
Activity-dependent recovery
The timecourse of presynaptic recovery may result from multiple factors, including the reuptake and recycling of released vesicles or the priming of reserve vesicles. Some of these pathways evidently take place in the absence of activity, but there are forms of recovery that are triggered by increased activity, through the accumulation of residual calcium (Dittman and Regehr 1998; Sakaba and Neher 2001; Stevens and Wesseling 1998; Wang and Kaczmarek 1998; see also Weis et al. 1999; Worden et al. 1997).
To test whether recovery at the endbulb depended on activity through a calcium-dependent process, we used EGTA-AM. EGTA is a slow calcium chelator, which has the effect of speeding the decay of residual calcium (Atluri and Regehr 1996). When EGTA-AM is applied to slices, it is taken up by all the cells, and the acetoxymethylester groups are cleaved, trapping EGTA within the cell. The postsynaptic cell is unaffected because the patch pipette sets its intracellular composition. Thus, EGTA application specifically causes residual calcium in the presynaptic terminal to decay more rapidly.
We tested for calcium-dependent forms of recovery using pairs of pulses, because single pulses are sufficient to induce accelerated recovery at some synapses (Dittman and Regehr 1998). These experiments were all conducted in 3 Cae the presence of 50 µM CTZ to prevent desensitization. We applied 100 µM EGTA-AM for 5 min, and then examined paired-pulse recovery (Fig. 6C). There was no significant change in recovery, notably at very early time points, where residual calcium is expected to have its largest effect. This lack of effect also suggests that facilitation plays a minor role at these synapses under these conditions.
While we saw no effect of EGTA-AM with pairs of pulses, some synapses require more extensive activity to trigger these activity-dependent processes (Wang and Kaczmarek 1998). Auditory nerve fibers normally fire at high rates, which would be likely to generate much higher levels of residual calcium, so it is possible that such a process would be triggered during trains of activity. To test this, we induced depression with a 5-pulse, 100 Hz conditioning train, and applied test pulses at different intervals (Fig. 7A). Depression at the end of the train was quite strong, but recovered rapidly (Fig. 7A). This strong depression, even in the presence of CTZ, suggests that forms of depression other than desensitization can play a large role during trains. On average the depression for Δt = 10 ms after the final pulse was 0.23 ± .07 relative to EPSC1 (N = 6 cells; Fig. 7D). The recovery followed a double exponential, with a large rapid component (τ = ~40 ms) and a smaller slow component (τ = ~2 s).
Figure 7.
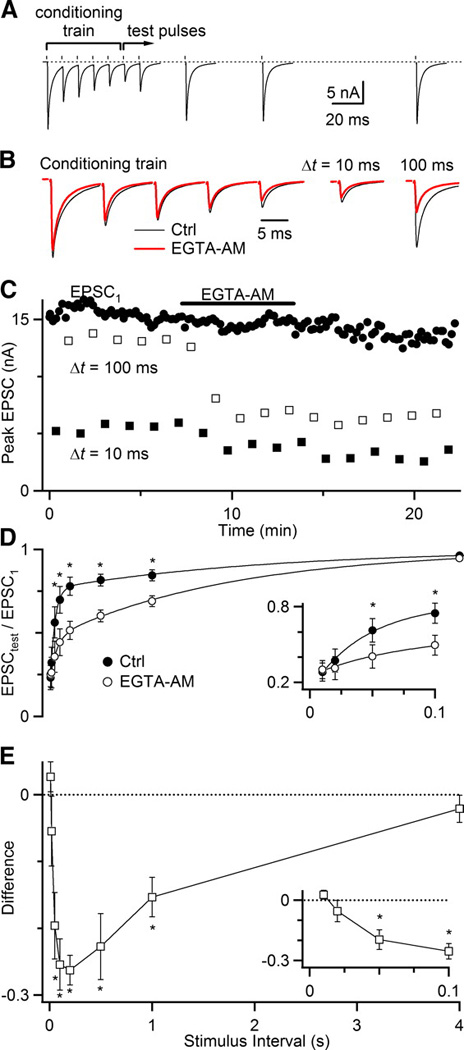
Endbulb synapses show calcium-dependent recovery from depression. A, Representative experiment showing rapid recovery after a conditioning train of 5 pulses at 100 Hz. Test pulses are conducted at a range of intervals after the conditioning train. All trials were conducted in the presence of CTZ. Test pulses are the average of 5–7 trials. B, Effects of 5 min application of 100 µM EGTA-AM on the conditioning train (left) and test pulses at Δt = 10 ms (center right) and Δt = 100 ms (far right). C, Experimental timecourse of EGTA-AM wash-in for the first EPSC in the conditioning train (EPSC1), as well as test pulses at Δt = 10 and 100 ms. D, Average recovery for 6 experiments before and after application of EGTA-AM. Recovery data are fit with a double-exponential curve (see legend to Fig. 1). In control, A1 = 0.54 ± 0.07, τ1 = 44 ± 18 ms, A2 = 0.24 ± 0.04, τ2 = 2.2 ± 0.3 s. After EGTA-AM, A1 = 0.23 ± 0.08, τ1 = 64 ± 6 ms, A2 = 0.53 ± 0.07, τ2 = 1.8 ± 0.3 s. Asterisks mark significant differences (P < 0.01, one-sided, paired t-test). E, The difference in recovery before and after EGTA-AM is applied from the same experiments as shown in D. Asterisks mark significant differences (P < 0.01, one-sided, paired t-test).
We applied EGTA-AM in the same way, and then tested recovery from train-induced depression. The effects of EGTA-AM reached steady-state after about 2 minutes of application. After EGTA-AM application, the EPSCs of the conditioning train did not change greatly (Fig. 7B, C). The initial amount of depression was also minimally affected (Fig. 7B, C). On average, the depression at Δt = 10 ms was 0.25 ± 0.07 relative to EPSC1. The major difference was that the rate of recovery was greatly slowed. For the sample experiment in Fig. 7A–C, there was significantly greater depression for Δt = 100 ms after application of EGTA-AM, even though there were only minimal effects on EPSC1, the conditioning train, and the depression at Δt = 10 ms. Recovery still followed a double exponential, but the rapid component (τ = ~60 ms) was greatly reduced, and the slow component (τ = ~2 s) dominated.
The greatest effect of EGTA-AM was at intermediate stimulus intervals. We quantified this by taking the absolute difference in depression at all time intervals (Fig. 7E). (We did not quantify using relative changes in depression because, at short intervals, they were highly subject to slight variations in EPSC amplitude because the absolute amplitudes involved were so small.) We found that the test pulses most affected by EGTA were at 0.05 to 1 s, peaking around 0.2 s. These results suggest that there are activity-dependent forms of recovery from depression that rely on residual calcium.
We note that this process should not be confused with facilitation. They are similar, in that both depend on residual calcium, have similar time courses, and affect the size of EPSCs (Atluri and Regehr 1996; Felmy et al. 2003; Muller et al. 2007; Regehr and Stevens 2001; Zucker 1999). However, the effect of EGTA-AM application cannot be explained as eliminating facilitation. This is most easily thought about by considering the post-EGTA recovery curve (Fig. 7B), and adding a facilitation mechanism onto it. This should give larger EPSCs immediately after the conditioning train, which then decay back to the slower recovery curve. This is not what is observed in control conditions. Thus, facilitation and calcium-dependent recovery are distinct processes. The same reasoning applies to the mechanisms underlying post-tetanic potentiation. We also note that forms of depression that appear to be driven by calcium (Forsythe et al. 1998; Hsu et al. 1996; Xu and Wu 2005) can not explain the faster recovery rate after high levels of activity. If calcium-sensitive mechanisms of depression were related to the recovery rate seen here, then reduction of calcium should reduce depression. We find the opposite upon the addition of EGTA-AM, which is only consistent with calcium driving a rapid recovery process.
Modelling synaptic depression
In order to determine if the mechanisms studied above are sufficient to explain the depression of the AMPA EPSC, we aimed to develop a simple model incorporating presynaptic vesicle depletion, activity-dependent recovery, and postsynaptic desensitization. We calculated the relative size of the ith EPSC in a train of stimuli by:
| (1) |
where F is the probability of release, Di is the proportion of release sites that are ready to release on the i th pulse, Si is the proportion of receptors that are available (i.e. not desensitized). We did not model changes in F, as facilitation does not appear to be present under our conditions (Fig. 6C).
Recovery from depletion
Activity-dependent changes in Di are based on Dittman et al. (Dittman et al. 2000; Dittman and Regehr 1998), and are repeated here for completeness. The number of release-ready sites, D, is reduced by release, such that immediately after a release event, D decreases by FDi, and then recovers to resting levels at a rate depending on a calcium-dependent process:
| (2) |
where k0 is the resting recovery rate, and CaD(t) is the calcium-bound state of a sensor that drives a rapid recovery process of rate kmax. The interaction between the rapid recovery process and the calcium-bound sensor is modelled as a simple affinity given by KD. CaD increments after each EPSC, and decays to resting levels by a simple exponential, given by:
| (3) |
These equations yield an analytical expression for Di+1 based on the preceding pulse:
| (4) |
where Δt is the interval between pulses.
Desensitization
We supposed that postsynaptic AMPA receptor desensitization depended on the concentration of extracellular glutamate in the synaptic cleft. This departs from previous models that treated desensitization as a different receptor state with a single τ of recovery (Brenowitz and Trussell 2001; Hennig et al. 2007). Our model tries to capture desensitization as a result of extracellular glutamate concentration that is gradually cleared. Glutamate drives desensitization according to a simple binding reaction:
| (5) |
where S is the proportion of receptors that are available (not desensitized), and KS is the binding affinity of the receptor for extracellular glutamate. After the i th release event, the glutamate concentration increments by the amount just released, and then decays exponentially back to resting levels, so that the glutamate concentration at the (i + 1)th pulse is given by:
| (6) |
where Δt is the interval between pulses, and τS is the rate of glutamate clearance.
Model performance
We measured short-term changes in synaptic strength for several cells in Fig. 8, in response to paired-pulse stimuli in 1.5 or 3 Cae with and without CTZ (Fig. 8A, B), as well as in response to regular trains of activity in 1.5 Cae (Fig. 8C). The 1.5 Cae is a better match to physiological conditions (Manthei et al. 1973). EPSC amplitude showed considerable changes depending on the conditions.
Figure 8.
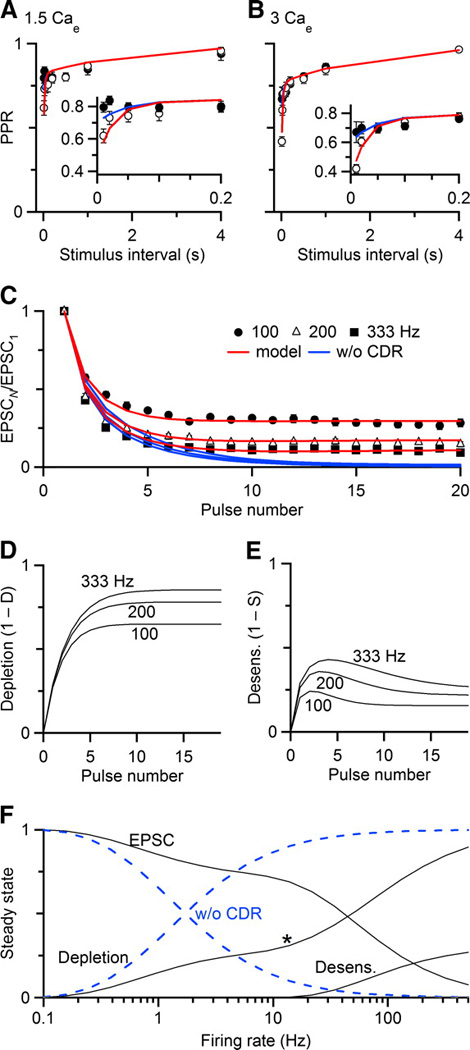
A simple model incorporating presynaptic vesicle depletion, calcium-dependent recovery, and postsynaptic receptor desensitization can replicate experimental data with few parameters. Data are collected in 1.5 Cae (A, C) and 3 Cae (B) to show paired-pulse recovery (A, B) and depression during regular trains of different frequencies (C). Details of the model are presented in the Results. Model parameters were fit by minimizing χ2 across all experimental conditions (see Table 1). Predictions of the model are plotted as lines. In A and B, blue lines indicate model responses without desensitization. In C, the blue line indicates responses of the model without calcium-dependent recovery (CDR). The model tracks the relative amounts of depletion (D) and desensitization (E) during regular trains. F, Black lines show the steady-state responses of the full model, tracking EPSC amplitude, depletion, and desensitization. Dashed blue lines show steady-state responses without CDR. The asterisk marks the frequency at which depletion increases more rapidly.
To test how well the model performed, we fit parameters to minimize χ2 across all experimental conditions simultaneously. We required that excluding the desensitization process should match the CTZ data, and that matching the data in 3 Cae should only require a change in the probability of release. Using the set of parameters in Table 1, the model was able to replicate the fast and slow components of paired-pulse depression (Fig. 8A, B). Eliminating desensitization from the model nearly eliminated the fast component of recovery, and aligned well with the CTZ data. By incorporating calcium-dependent recovery from depression, the model also captured the behavior during trains (red lines, Fig. 8C). When calcium-dependent recovery was excluded, the model could not match experimental data during trains (blue lines, Fig. 8C).
Table 1.
Parameters used for the model in Fig. 8.
| Parameter | Meaning | Value |
|---|---|---|
| F | probability of release | 0.3 in 1.5 Cae; 0.4 in 3 Cae |
| k0 | baseline recovery rate from depletion | 0.45/s |
| kmax | maximal recovery rate from depletion | 18/s |
| τD | decay time constant for calcium-dependent recovery | 35 ms |
| KD | affinity of fast recovery process for calcium sensor | 0.7 |
| τS | decay time constant of glutamate clearance | 15 ms |
| KS | affinity of receptor desensitization for glutamate | 0.6 |
The removal of calcium-dependent recovery results in much greater depression during brief trains. At first glance, this seems to conflict with the experiments of Fig. 7. In those experiments, application of EGTA-AM did not change EPSCs in the conditioning train. This apparent difference is likely to result from the fact that these manipulations are not equivalent. EGTA-AM accelerates the decay of, but does not completely eliminate, residual calcium. Brief increases in residual calcium that persist in EGTA-AM may be adequate to drive the fast recovery process during the conditioning train in Fig. 7, but not during the later test pulses (see Supplementary Fig. 1).
The model, despite its simplicity, may help give insights into the importance of different mechanisms of depression during activity. We considered the importance of depletion (1 – D) and desensitization (1 – S). As expected, depletion built up greatly during trains, especially at the highest frequencies (Fig. 8D). Desensitization had the greatest impact at the beginning of a train (Fig. 8E), presumably because glutamate release is greatest then, which causes the most desensitization. However, as depletion becomes significant later in a train, the amount of glutamate released decreases, so desensitization is less prominent.
We also estimated the steady-state amount of depression for regular trains, by taking the amplitude of the last pulse in a 100-pulse train. The steady-state EPSC was large at low frequencies, but showed increasing depression for frequencies above 30 Hz (Fig. 8F, black lines). However, responses were not abolished, even for activity at the very high firing rate of 500 Hz. We again considered how different mechanisms of depression contributed to this. Depletion made the greatest contribution at all frequencies considered. Depletion showed a gradual increase until ~20 Hz. The rate of this increase depends primarily on the value of k0, i.e. the activity-independent form of recovery. At about 20 Hz, the amount of depletion increases considerably (Fig, 8F, asterisk). The firing rate at which the amount of depletion begins this increase seems to depend the most on the value of τD, i.e. the time constant at which calcium-dependent recovery returns to baseline. Desensitization contributed very little to depression. Its largest contribution was at firing rates above 100 Hz, although at those rates, depletion was much greater.
This situation contrasts with when calcium-dependent recovery is not present (Fig. 8F, dashed blue lines). Under these conditions, the steady-state EPSC is much smaller than the full model, and depressed to near 0 by 100 Hz. This indicates that calcium-dependent recovery is important even at low frequencies, and is critical to synaptic function at physiological firing rates. Steady-state desensitization never reached significant levels in the absence of calcium-dependent recovery.
Discussion
We found that the mechanisms of depression at the mouse endbulb of Held were both pre- and postsynaptic. Depression of both AMPA and NMDA EPSCs recovered by a double-exponential process. For AMPA EPSCs, the faster process was likely to be desensitization, as it was eliminated by CTZ and aniracetam. For NMDA EPSCs, the faster process was likely to be saturation, as it was eliminated by the low-affinity antagonist L-AP5. With these two distinct postsynaptic mechanisms removed, the remaining depression reported by both AMPA and NMDA EPSCs was very similar, suggesting a mechanism that was presynaptic in origin. This mechanism is likely to be vesicle depletion. During trains of activity, recovery from presynaptic depression was accelerated, and recovery was slowed by the calcium chelator EGTA. We further showed that these mechanisms could be incorporated into a very simple model with few free parameters, and that this model could account for changes in synaptic strength during longer periods of activity. The model showed that depletion plays a greater role in depression than desensitization. In addition, the model indicated that calcium-dependent forms of recovery are critical to the function of the synapse. Without these processes, the amount of depression would be much greater, rendering the synapse nearly silent at physiological firing rates.
Our results are consistent with a number of studies that have identified postsynaptic processes as important in depression at the endbulb (Isaacson and Walmsley 1995; Oleskevich et al. 2000; Trussell et al. 1998). We found that desensitization contributes to depression particularly at short interpulse intervals. However, during prolonged activity, depression can be very strong even when desensitization is reduced by CTZ, as in the short conditioning trains of Fig. 7. This suggests that the presynaptic mechanism of depression dominates during extended activity, as the model also indicates.
A few presynaptic mechanisms of depression are possible, including extracellular calcium depletion, calcium-channel inactivation, and vesicle depletion. Calcium depletion seems unlikely as its induction appears to require more extended activity than we used here (Borst and Sakmann 1999). Furthermore calcium-channel inactivation at the calyx of Held follows a much slower time course than the depression we observed (Xu and Wu 2005). Our results are more consistent with vesicle depletion. The presynaptic component of depression recovers with a timecourse similar to vesicle reuptake and re-recruitment found at other synapses (Betz and Bewick 1992; Ryan et al. 1993). In addition, the presynaptic component of depression displays a calcium-dependent form of recovery, similar to that found at the cerebellar climbing fiber synapse (Dittman and Regehr 1998), where vesicle depletion has been identified using variance-mean analysis (Foster and Regehr 2004). The molecular basis for this rapid form of recovery is not known, but has been suggested to rely on a calmodulin-dependent pathway (Sakaba and Neher 2001), acting through Munc-13 (Junge et al. 2004).
Our results conflict with an earlier study of paired-pulse depression at the rat endbulb. Bellingham and Walmsley (1999) found that recovery from depression of both AMPA and NMDA EPSCs followed a single exponential time-course, and that CTZ abolished depression of both receptor components. By contrast, we found that depression follows two distinct time-courses, that CTZ was specific for eliminating the rapid component of AMPA depression, and that CTZ had no effect on NMDA EPSCs. Thus we saw no evidence for a CTZ-sensitive presynaptic form of depression. While we cannot be certain why our experiments differ, two factors seem important. First, Bellingham and Walmsley (1999) studied rats of somewhat younger ages using paired pulses of Δt ≤ 150 ms. In our results (Fig. 1, Fig. 2, Fig. 4–Fig. 6, Fig. 8), and theirs (see their Fig. 1, Fig. 2, and Fig. 4), recovery was not complete by 150 ms, possibly suggesting a second mechanism of depression, but it would have been less evident on that timescale. The difference between our observations of the effect of CTZ on NMDA depression is not clear. One possibility is that rats endbulbs are subject to non-specific effects of CTZ whereas mouse endbulbs are not. Alternatively, it is possible that additional auditory nerve fibers were recruited in the rat. Empirically, CTZ application can cause changes in fiber excitability, leading to the recruitment of additional axons following stimulation. In our experience, it is easier to stimulate multiple inputs in the rat than in the mouse. Furthermore, we took great care to isolate single fibers in our study. A clear answer to these questions would require side-by-side comparisons, which is beyond the scope of this study.
Modelling depression
The model we have developed differs from previous models in the implementation of desensitization. Experiments in the chick nucleus magnocellularis as well as in cerebellar mossy fibers have emphasized that accumulation of extracellular glutamate plays an important role in desensitization-induced depression (Trussell et al. 1993; Xu-Friedman and Regehr 2003). We used a very simple model of extracellular glutamate to drive desensitization of receptors. This approach was able to replicate the fast component of depression in AMPA EPSCs.
The model has proven useful in several ways. First, it indicated the critical importance of calcium-dependent recovery in the functioning of the endbulb. Leaving it out of the model, as we had in earlier iterations, made it impossible to fit all the experimental data. Without calcium-dependent recovery, the extent of depression during trains is much too great (Fig. 8), and indicates its importance duing the high rates of activity normally shown at this synapse.
Second, the model provides some confirmation that the processes we have identified experimentally are sufficient to account for synaptic plasticity under multiple circumstances. One of the strengths of this type of model is that its simplicity allows derivation of analytical solutions, while it evidently captures synaptic plasticity quite closely.
Third, the model allows us to “look under the hood” to evaluate the importance of different mechanisms of depression during complex activity. The model indicates that depletion plays a dominant role in depression, particularly at high firing rates, while desensitization makes a minor contribution only during high-frequency activity.
Fourth, because the model empirically duplicates synaptic behavior over a range of conditions, it is very useful for exploring how synaptic plasticity may influence postsynaptic behavior in response to realistic presynaptic activity. Arbitrary spike trains, such as ones based on recorded auditory nerve activity, can be translated into synaptic strength using the model, and then applied to postsynaptic cells using methods such as dynamic clamp. In combination with models of bushy cell activity (such as Xu-Friedman and Regehr 2005a), the model could convert auditory nerve activity into bushy cell activity, which might be useful in the development of prostheses that stimulate the cochlear nucleus directly.
Functional implications
It is somewhat surprising that desensitization plays such a minor role during trains of activity. Desensitization can be quite prominent in paired-pulse experiments, and it could have been the case that further glutamate accumulation during trains would result in desensitization being of primary importance. However, the experimental and modelling results suggest that the dominant mechanism of depression is depletion. The most likely reason for this is that rested synapses can release large amounts of glutamate. As the presynaptic terminal becomes increasingly depleted during bouts of activity, the amount of glutamate released becomes smaller, and desensitization declines. Thus, even though brainstem auditory synapses typically show significant desensitization, this probably has a minor effect during the prolonged activity common in the auditory system, where depletion effectively reduces desensitization. Desensitization may increase in its contribution during a brief burst of high frequency spikes, such as might be encountered during irregular activity.
These findings may also shed light on the functional importance of the particular glutamate receptor composition at synapses. The AMPA receptors at the endbulb likely contain the gluR4flop isoform (Petralia et al. 2000). This receptor type appears to confer to EPSCs both brief duration, as well as desensitization-induced paired-pulse depression. This raises the question of what the critical selective pressure is to drive expression of gluR4flop. It could be that the brief EPSC is critical to fine temporal precision, and desensitization-induced depression is a side-effect. Alternatively, it could be that the properties of desensitization-induced depression are critical to the information-processing properties of synapses, for example, because they have much faster recovery compared to depleting synapses. The results presented here suggest that the prominent fast component of depression plays a comparatively minor role during more realistic activity, in which case selective pressure likely favored rapidly-desensitizing receptors mainly for the sake of temporally precise EPSCs.
Finally, these experiments indicate that calcium-dependent recovery is absolutely critical to the function of the endbulb. It has been clear since the initial reports that calcium-dependent recovery increases synaptic strength (Dittman and Regehr 1998; Stevens and Wesseling 1998; Wang and Kaczmarek 1998). This is particularly dramatic at the endbulb where high rates of activity are common (Johnson 1980; Joris et al. 1994a; Sachs and Abbas 1974). Without calcium-dependent recovery, the synapse would be rapidly depleted at physiological rates of activity. This contrasts with synapses made by bipolar cells in the retina and hair cells in the cochlea. These synapses are notable for reliability even during constant, high rates of activity, and they also have morphological features that differ from most synapses, i.e. large electron-dense structures (the “ribbon” or “synaptic body”). One question is whether the ribbon is necessary for reliability or synchronization, although it does clearly affect vesicle fusion at hair cells (Khimich et al. 2005). Endbulb synapses contain no obvious ribbon (Nicol and Walmsley 2002), yet they are able to support fast, highly synchronous release, with rapid recovery. Endbulbs could be similar to cerebellar mossy fiber synapses, which appear to draw upon an extraordinarily large reserve of vesicles to maintain release during high rates of activity (Saviane and Silver 2006). The mobilization of such a pool probably requires the presence of calcium-dependent recovery, so perhaps ribbon synapses and non-ribbon central synapses solve a similar problem in different ways. Future work will be necessary to compare the recovery mechanisms at these two types of synapses.
Supplementary Material
Acknowledgments
The authors thank S. Chanda, C. Hsu, and S.R. Oh, and L. Pliss for helpful comments.
Grants
This work was supported by National Institute of Health grants R03 DC 007991-01 and R01 DC 008125-01A1.
Footnotes
Disclosures
N/A
References
- Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol. 1999;521(Pt 1):123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxall AR, Garthwaite J. Synaptic excitation mediated by AMPA receptors in rat cerebellar slices is selectively enhanced by aniracetam and cyclothiazide. Neurochem Res. 1995;20:605–609. doi: 10.1007/BF01694543. [DOI] [PubMed] [Google Scholar]
- Brenowitz S, David J, Trussell L. Enhancement of synaptic efficacy by presynaptic GABA(B) receptors. Neuron. 1998;20:135–141. doi: 10.1016/s0896-6273(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Brenowitz S, Trussell LO. Minimizing synaptic depression by control of release probability. J Neurosci. 2001;21:1857–1867. doi: 10.1523/JNEUROSCI.21-06-01857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Blitz DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron. 2002;33:779–788. doi: 10.1016/s0896-6273(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RAJ, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC, Regehr WG. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci. 2000;20:1374–1385. doi: 10.1523/JNEUROSCI.20-04-01374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmy F, Neher E, Schneggenburger R. Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron. 2003;37:801–811. doi: 10.1016/s0896-6273(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fiber synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Hennig M, Postlethwaite M, Forsythe ID, Graham B. A biophysical model of short-term plasticity at the calyx of Held. Neurocomputing. 2007;70:1626–1629. [Google Scholar]
- Hsu SF, Augustine GJ, Jackson MB. Adaptation of Ca(2+)-triggered exocytosis in presynaptic terminals. Neuron. 1996;17:501–512. doi: 10.1016/s0896-6273(00)80182-8. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Amplitude and time course of spontaneous and evoked excitatory postsynaptic currents in bushy cells of the anteroventral cochlear nucleus. J Neurophysiol. 1996;76:1566–1571. doi: 10.1152/jn.1996.76.3.1566. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Receptors underlying excitatory synaptic transmission in slices of the rat anteroventral cochlear nucleus. J Neurophysiol. 1995;73:964–973. doi: 10.1152/jn.1995.73.3.964. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Takahashi T. Mechanisms underlying presynaptic facilitatory effect of cyclothiazide at the calyx of Held of juvenile rats. J Physiol. 2001;533:423–431. doi: 10.1111/j.1469-7793.2001.0423a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DH. The relationship between spike rate and synchrony in responses of auditory-nerve fibers to single tones. J Acoust Soc Am. 1980;68:1115–1122. doi: 10.1121/1.384982. [DOI] [PubMed] [Google Scholar]
- Joris PX, Carney LH, Smith PH, Yin TC. Enhancement of neural synchronization in the anteroventral cochlear nucleus. I. Responses to tones at the characteristic frequency. J Neurophysiol. 1994a;71:1022–1036. doi: 10.1152/jn.1994.71.3.1022. [DOI] [PubMed] [Google Scholar]
- Joris PX, Smith PH, Yin TC. Enhancement of neural synchronization in the anteroventral cochlear nucleus. II.Responses in the tuning curve tail. J Neurophysiol. 1994b;71:1037–1051. doi: 10.1152/jn.1994.71.3.1037. [DOI] [PubMed] [Google Scholar]
- Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell. 2004;118:389–401. doi: 10.1016/j.cell.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- Kiang NY-s. Discharge Patterns of Single Fibers in the Cat's Auditory Nerve. Cambridge, MA: MIT Press; 1965. p. 154. [Google Scholar]
- Lawrence JJ, Brenowitz S, Trussell LO. The mechanism of action of aniracetam at synaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors: indirect and direct effects on desensitization. Mol Pharmacol. 2003;64:269–278. doi: 10.1124/mol.64.2.269. [DOI] [PubMed] [Google Scholar]
- Lorente de Nó R. The Primary Acoustic Nuclei. New York: Raven Press; 1981. p. 176. [Google Scholar]
- Manthei RC, Wricht DC, Kenny AD. Altered CSF constituents and retrograde amnesia in rats: a biochemical approach. Physiol Behav. 1973;10:517–521. doi: 10.1016/0031-9384(73)90214-x. [DOI] [PubMed] [Google Scholar]
- Muller M, Felmy F, Schwaller B, Schneggenburger R. Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of held that accelerates the decay of Ca2+ and short-term facilitation. J Neurosci. 2007;27:2261–2271. doi: 10.1523/JNEUROSCI.5582-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol MJ, Walmsley B. Ultrastructural basis of synaptic transmission between endbulbs of Held and bushy cells in the rat cochlear nucleus. J Physiol. 2002;539:713–723. doi: 10.1113/jphysiol.2001.012972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Clements J, Walmsley B. Release probability modulates short-term plasticity at a rat giant terminal. J Physiol. 2000;524:513–523. doi: 10.1111/j.1469-7793.2000.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olverman HJ, Jones AW, Watkins JC. [3H]D-2-amino-5-phosphonopentanoate as a ligand for N-methyl-D-aspartate receptors in the mammalian central nervous system. Neuroscience. 1988;26:1–15. doi: 10.1016/0306-4522(88)90123-6. [DOI] [PubMed] [Google Scholar]
- Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of alpha-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol. 1994;46:129–138. [PubMed] [Google Scholar]
- Petralia RS, Rubio ME, Wang YX, Wenthold RJ. Differential distribution of glutamate receptors in the cochlear nuclei. Hear Res. 2000;147:59–69. doi: 10.1016/s0378-5955(00)00120-9. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Stevens CF. Physiology of synaptic transmission and short-term plasticity. In: Cowan WM, Sudhof TC, Stevens CF, editors. Synapses. Baltimore: Johns Hopkins University Press; 2001. pp. 135–175. [Google Scholar]
- Rozov A, Jerecic J, Sakmann B, Burnashev N. AMPA receptor channels with long-lasting desensitization in bipolar interneurons contribute to synaptic depression in a novel feedback circuit in layer 2/3 of rat neocortex. J Neurosci. 2001;21:8062–8071. doi: 10.1523/JNEUROSCI.21-20-08062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- Sachs MB, Abbas PJ. Rate versus level functions for auditory-nerve fibers in cats: tone-burst stimuli. J Acoust Soc Am. 1974;56:1835–1847. doi: 10.1121/1.1903521. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Saviane C, Silver RA. Fast vesicle reloading and a large pool sustain high bandwidth transmission at a central synapse. Nature. 2006;439:983–987. doi: 10.1038/nature04509. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Taberner AM, Liberman MC. Response properties of single auditory nerve fibers in the mouse. J Neurophysiol. 2005;93:557–569. doi: 10.1152/jn.00574.2004. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Brenowitz S, Otis T. Postsynaptic mechanisms underlying synaptic depression. In: Faber DS, Korn H, Redman SJ, Thompson SM, Altman JS, editors. Central Synapses: Quantal Mechanisms and Plasticity. Strasbourg: Human Frontier Science Program; 1998. pp. 149–158. [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Borst JG. Short-term plasticity at the calyx of Held. Nat Rev Neurosci. 2002;3:53–64. doi: 10.1038/nrn705. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Weis S, Schneggenburger R, Neher E. Properties of a model of Ca(++)-dependent vesicle pool dynamics and short term synaptic depression. Biophys J. 1999;77:2418–2429. doi: 10.1016/S0006-3495(99)77079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden MK, Bykhovskaia M, Hackett JT. Facilitation at the lobster neuromuscular junction: a stimulus-dependent mobilization model. J Neurophysiol. 1997;78:417–428. doi: 10.1152/jn.1997.78.1.417. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Dynamic-clamp analysis of the effects of convergence on spike timing. I. Many synaptic inputs. J Neurophysiol. 2005a;94:2512–2525. doi: 10.1152/jn.01307.2004. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Dynamic-clamp analysis of the effects of convergence on spike timing. II. Few synaptic inputs. J Neurophysiol. 2005b;94:2526–2534. doi: 10.1152/jn.01308.2004. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Structural contributions to short-term synaptic plasticity. Physiol Rev. 2004;84:69–85. doi: 10.1152/physrev.00016.2003. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Ultrastructural contributions to desensitization at cerebellar mossy fiber to granule cell synapses. J Neurosci. 2003;23:2182–2192. doi: 10.1523/JNEUROSCI.23-06-02182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu L-G. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Yamada KA, Tang CM. Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci. 1993;13:3904–3915. doi: 10.1523/JNEUROSCI.13-09-03904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.