


Abstract
Background:
Previous cross-sectional fMRI studies in subjects with prodromal Alzheimer disease (AD) have reported variable results, ranging from hypoactivation, similar to patients with AD, to paradoxically increased activation or hyperactivation compared to cognitively normal older individuals. We have hypothesized that subjects in early phases of prodromal AD may experience a period of hippocampal hyperactivation, followed by loss of hippocampal activation as the disease progresses.
Methods:
We studied 51 older individuals without dementia (Clinical Dementia Rating [CDR] at baseline of 0, n = 21, and 0.5, n = 30) with longitudinal clinical and neuropsychological assessments, as well as fMRI during a face-name associative memory paradigm. Whole brain and region-of-interest analyses were applied to the longitudinal fMRI data.
Results:
Subjects classified as CDR 0 at baseline showed no difference in fMRI activity over 2 years, whereas those who were CDR 0.5 at baseline demonstrated a decrease in fMRI activity in the right hippocampus (p < 0.001). Dividing the subjects on the basis of their clinical and neuropsychological change over the 2 years, we found that subjects with more rapid decline demonstrated both the highest hippocampal activation at baseline, and the greatest loss of hippocampal activation. These findings remained significant after accounting for age, hippocampal volume, and APOE ε4 carrier status.
Conclusions:
Clinical decline is associated with loss of hippocampal activation in older subjects. Longitudinal fMRI provides a reliable indicator of brain activation over time, and may prove useful in identifying functional brain changes associated with cognitive decline on the trajectory toward clinical Alzheimer disease.
GLOSSARY
- AD
= Alzheimer disease;
- ANOVA
= analysis of variance;
- BOLD
= blood oxygen level dependent;
- CDR
= Clinical Dementia Rating;
- CDR-SB
= Clinical Dementia Rating Sum of Boxes;
- CVLT
= California Verbal Learning Test;
- EPI
= echoplanar imaging;
- MCI
= mild cognitive impairment;
- MTL
= medial temporal lobe;
- NvR
= novel vs repeated;
- RAVLT
= Rey Auditory Verbal Learning Test;
- ROI
= region of interest;
- SPM
= statistical parametric mapping.
The pathophysiologic process of Alzheimer disease (AD) begins many years prior to the clinical diagnosis of dementia. Tools such as fMRI have the potential to detect early alterations in brain function that may precede cognitive decline. While several studies have suggested that baseline patterns of fMRI activity may predict clinical decline,1-3 there have been no longitudinal fMRI studies to document that clinical decline is accompanied by a measurable loss of functional activation.
There has been much debate surrounding functional activation patterns in the hippocampus and related medial temporal lobe (MTL) regions among subjects in early preclinical phases of AD. While some studies have reported decreased activation in subjects with mild cognitive impairment (MCI) compared to normal elderly controls,4-7 other studies have noted that subjects in the earliest stages of MCI demonstrate hyperactivation relative to elderly controls.3,8,9 We have postulated that the discrepant findings may relate to the heterogeneity of the subjects under the broad rubric of MCI and prodromal AD.10 Increased MTL activation has also been reported in asymptomatic individuals at genetic risk.11-13 We hypothesize that hyperactivation might represent an early response to AD pathology, which may predict impending hippocampal failure and memory decline. Longitudinal fMRI research is needed to determine the evolution of fMRI activity over the course of clinical decline. In this study, we acquired fMRI and clinical assessments in older individuals without dementia at baseline and 2-year follow-up to investigate the relationship of brain function activity to clinical change over time.
METHODS
Fifty-one elderly subjects without dementia (74.8 ± 5.4 years) were recruited from ongoing longitudinal studies on cognitive aging and prodromal AD.
At the baseline visit, subjects were evaluated clinically with the Clinical Dementia Rating (CDR) scale and assigned a global CDR rating and CDR Sum of Boxes (CDR-SB) score. Subjects also underwent neuropsychological assessment, including the California Verbal Learning Test (CVLT) and/or Rey Auditory Verbal Learning Test (RAVLT). Subjects were eligible for the longitudinal fMRI study if they were clinically normal (CDR = 0) or had very mild impairment (CDR = 0.5). The CDR 0.5 subjects selected for this study had subjective memory complaints corroborated by their study partner, but did not yet meet research diagnostic criteria for MCI with significant objective memory impairment (performing at least 1.5 standard deviations below age- and education-adjusted norms at baseline).14,15 Subjects returned 2 years after the baseline visit for follow-up scanning and repeat clinical assessment. As the tests of episodic memory evolved in the longitudinal cohorts over the course of the follow-up period (i.e., some subjects received the CVLT at baseline and RAVLT at follow-up), we calculated Z scores to allow for a standardized comparison of neuropsychological test scores.
MRI procedures.
Subjects were scanned using a Siemens (Iselin, NJ) Trio 3.0-T scanner. Functional data were acquired using a T2*-weighted gradient echoplanar imaging (EPI) sequence (repetition time/echo time, 2,000/30 msec; flip angle, 90°). Twenty-eight slices were acquired in an oblique coronal orientation, perpendicular to the anterior-posterior commissural line (5 mm with 1 mm gap; voxel dimensions, 3.125 × 3.125 × 6 mm). Six functional runs (each 127 timepoints) were acquired for each subject.
fMRI memory paradigm.
fMRI data were collected serially at baseline and 2 years during an associative memory encoding paradigm as described in detail in previous studies.16-18 Subjects viewed a series of novel faces (faces unfamiliar to the participants) paired with fictional first names, and were explicitly instructed to try to remember the name associated with each face. In this study, we compared blocks of encoding of novel face-name pairs to repeated face-name pairs. After each scanning session, subjects underwent forced-choice recognition testing, in which faces were presented with the correct and an incorrect name printed underneath in a counterbalanced order. At baseline, only 14 face-name pairs were tested postscan, and at 2-year follow-up, all 84 novel face-name pairs were included in the postscan test, so longitudinal assessment of change in postscan test performance was not reliable.
fMRI data analysis.
fMRI data were preprocessed and analyzed using statistical parametric mapping (SPM2; http://www.fil.ion.ucl.ac.uk/spm). Data were realigned to the standard SPM2 EPI template, and smoothed with a Gaussian kernel of 8 mm. Data were modeled with the canonical hemodynamic response function, and a high pass filter of 260 s was used to filter out low-frequency variations. T1 structural data were processed through a semiautomated anatomic reconstruction and labeling procedure producing individual anatomic regions of interest (ROI) using FreeSurfer software (http://surfer.nmr.mgh.harvard.edu). Two subjects were excluded from analyses involving anatomic ROIs created in FreeSurfer because of poor quality volumetric data.
Statistical analysis.
We first examined longitudinal fMRI activity at the whole brain level, generating group-averaged activation maps for the novel vs repeated (NvR) contrast using SPM2 random effects models at baseline and follow-up. SPM2 whole brain repeated measures analyses were then used to investigate within-subject changes in function over the 2-year period. Statistical maps were thresholded at a significance level of p < 0.001 with a minimum extent threshold of 20 contiguous voxels.
We investigated activation changes within the hippocampus using 2 ROI approaches. First, we created an empirically defined functional ROI in the right hippocampus based on the CDR 0.5 group at baseline (see Results and figure 1C). Second, we utilized an unbiased, a priori defined anatomic ROI based on FreeSurfer volumetric analyses. We further constrained the anatomic ROI using a union mask based on the activated voxels at baseline and 2-year follow-up from the NvR contrast for all 51 subjects to examine hippocampal regions engaged in the task.

Figure 1 Group activation maps.
Group-averaged statistical parametric mapping activation maps showing activation at (A) baseline and (B) 2 years in groups defined by baseline Clinical Dementia Rating (CDR) status. (C) CDR 0 subjects showed no differences in hippocampal activation between baseline and follow-up scans, while CDR 0.5 subjects demonstrated a decrease in right hippocampal activation (p = 0.001).
We extracted percent signal change estimates within ROIs using the SPM ROI Toolbox (http://marsbar.sourceforge.net/). Analysis of variance (ANOVA) and paired t tests were used to assess between-group and within-group differences in percent signal change estimates. Pearson correlations were used to investigate the relationship between hippocampal signal and clinical status. A linear regression model with covariates was used to investigate predictors of clinical decline.
Standard procedures and subject consents.
All subjects provided informed consent in accordance with the Human Research Committee guidelines of the Brigham and Women's Hospital (Boston, MA).
RESULTS
Subjects.
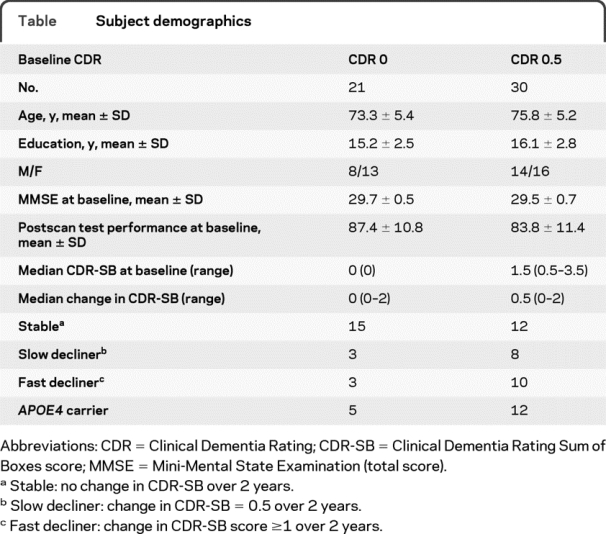
At baseline, 21 subjects were classified as CDR 0 and 30 as CDR 0.5. Subject demographics are shown in the table. CDR 0 subjects were similar to CDR 0.5 subjects in age, years of education, Mini-Mental State Examination, and postscan test performance at baseline (p > 0.05 for all comparisons). Of the 51 subjects, 17 were APOE ε4 carriers (CDR 0, n = 5/21; CDR 0.5, n = 12/30).
Table Subject demographics
Whole brain analyses based on baseline clinical status.
We first examined whole-brain fMRI activation patterns in the groups divided by baseline CDR rating (0 vs 0.5) at baseline and at 2-year follow-up (figure 1). Regions with greater activation for novel compared to repeated face-name pairs included the fusiform, prefrontal cortices, thalamic, and MTL regions, in a pattern very similar to our previous reports.16,18 In the CDR 0 group, repeated-measures analyses revealed no regions with significant differences in activation over 2 years. However, the CDR 0.5 subjects demonstrated a longitudinal decrease in activation that was specific to the right hippocampus (figure 1C). There were 2 clusters within the right hippocampus: anterior peak: 33, −9, −18; posterior peak: 39, −24, −15; p = 0.001. This anterior hippocampal region from the CDR 0.5 group was then used as the functional ROI mask to extract fMRI signal response for all subjects in subsequent analyses. No regions showed greater activation at follow-up compared to baseline in either group.
Relationship of change in fMRI activity to change in clinical status.
Subjects were then classified into 4 groups based on change in their clinical status over the 2-year follow-up: 1) stable CDR 0 subjects (CDR-SB 0 at baseline and follow-up, n = 15); 2) stable CDR 0.5 subjects (CDR 0.5, no change in SB, n = 12); 3) slow decliners (CDR 0/0.5 at baseline, decline in SB = 0.5, n = 11); and 4) fast decliners (CDR 0/0.5 at baseline, decline in SB ≥1, n = 13). We chose to define CDR-SB ≥1.0 as the cutoff for fast decline, as this degree of change was felt to be more clinically meaningful based on previous data predicting conversion to clinical dementia.19 Consonant with this classification, utilizing the National Alzheimer's Coordinating Center diagnostic criteria, we found that 5/11 slow decliners progressed to meet criteria for amnestic or multidomain MCI, and 1 progressed to probable AD, whereas 4/13 of the fast decliners progressed to MCI, and 4 progressed to dementia (3 to probable AD, 1 to probable vascular/mixed dementia).
We first focused on percent signal change estimates from the empirically defined functional ROI in the right hippocampus (figure 1C). The longitudinal change in hippocampal activation (NvR response at 2 years − NvR response at baseline) differed among the 4 groups (F3,47 = 5.29, p = 0.003; figure 2). Groups with a constant CDR-SB score had stable hippocampal activation over the 2 years, while the fast decliner group demonstrated a loss of hippocampal activation (p < 0.001). There was also a trend toward a difference in baseline activation among the 4 groups (p = 0.085), with a post hoc t test revealing higher baseline activation for the fast decliner group compared to the stable CDR 0 group (p = 0.02). No differences were observed between baseline and 2-year scans in a calcarine cortex ROI sampled as a control region, indicating that the observed effects were regionally specific.

Figure 2 Hippocampal signal at baseline and follow-up
Percent signal change values extracted from functionally defined region of interest (ROI) in the right hippocampus. The ROI analyses confirmed that subjects who declined by ≥1 in Clinical Dementia Rating Sum of Boxes (CDR-SB) (fast decliners) showed the greatest decrease in right hippocampal activation (p < 0.001), and demonstrated increased activation in the region at baseline compared to those with no change in CDR-SB (p = 0.02).
We also compared the longitudinal changes in hippocampal signal dividing subjects into just 2 groups, comparing those with no change in CDR-SB score (n = 27) and those with any worsening (≥0.5) in CDR-SB score (n = 24). We again observed a greater loss of hippocampal signal among the subjects experiencing any clinical decline using both the functional ROI (p = 0.004) and anatomic ROI (p = 0.036) analyses.
We calculated the difference in fMRI response between the 2-year and baseline scans, and correlated this longitudinal difference in activation with the overall change in CDR-SB score. Across all 51 subjects, greater decline in CDR-SB score was associated with greater loss of hippocampal activation (R = −0.51; p < 0.001). Interestingly, greater baseline hippocampal activation was associated with greater decline on CDR-SB (R = 0.40; p = 0.003) and greater loss of hippocampal signal (R = 0.81; p < 0.001, figure 3), consistent with the hypothesis that hippocampal hyperactivation precedes clinical decline and loss of hippocampal activation.

Figure 3 Relationship between baseline hippocampal activity and change in hippocampal activity
Higher magnitude of activation in the right hippocampus at baseline correlates with a greater loss in percent signal change in this region over the 2-year period (r = 0.81; p < 0.001).
We then performed a series of analyses to determine if other demographic or volumetric factors might account for the observed results. Age was not correlated with baseline hippocampal signal (R = 0.13, p = 0.360), but there was a weak correlation between age and change in hippocampal signal (R = 0.28, p = 0.047). There were no significant differences in baseline fMRI or change in fMRI activation between APOE ε4 carriers and noncarriers, using either functionally or anatomically defined ROI.
To further investigate whether the observed longitudinal changes in activation were explained by changes in hippocampal volume, we examined fMRI activity within an anatomically defined hippocampal ROI determined by FreeSurfer. Subjects with stable CDR-SB scores showed no change in hippocampal activation within the anatomically defined ROI between baseline and 2-year scans, while the greatest loss of activation over the 2 years was observed in the fast decliner group (p = 0.011). The prespecified 2-group comparison between the fast decliners and stable CDR 0 subjects showed that baseline activation was higher for the fast decliners (p = 0.046). We also performed ANOVA on the hippocampal volumetric data and did not find any significant longitudinal changes in volume for any of the 4 groups, further suggesting that our observed decreases in hippocampal activation were not explained by volumetric loss.
Finally, the baseline hippocampal percent signal change for each subject was entered into a linear regression model, with change in CDR-SB score as the dependent variable. Controlling for other factors potentially relevant to decline (age, education, CDR-SB score at baseline, hippocampal volume, and APOE status), there was a main effect of hippocampal activation (functional ROI: β = 1.33, p = 0.005; anatomic ROI: β = 1.24, p = 0.029). No other covariates contributed to the model (p values > 0.121).
Relationship of hippocampal signal and neuropsychological memory testing.
We also related changes in hippocampal activation to behavioral performance on neuropsychological tests of verbal memory. The changes in neuropsychological test performance paralleled the changes in hippocampal function over time in the 4 groups (figure 4). The overall ANOVA for change in Z scores did not reach significance (F3,46 = 2.59, p = 0.065), but the prespecified comparison between the stable CDR 0 subjects and fast decliners indicated that fast decliners experienced greater decline in test performance (p = 0.01). This effect was due to both slight improvement in neuropsychological memory performance over the 2 years in the stable CDR 0 subjects (p = 0.057), suggestive of a practice effect commonly observed in cognitively normal adults, and a trend toward worsening neuropsychological performance among the fast decliners (p = 0.211).

Figure 4 Change in neuropsychological test scores and hippocampal signal
Mean change in standardized neuropsychological test score (A) and hippocampal signal (B). The fast decliners demonstrated more decline in objective memory performance compared to the Clinical Dementia Rating (CDR) 0 stable subjects (p = 0.01). This decrease in memory test performance corresponds to the observed decrease in hippocampal activation.
DISCUSSION
This longitudinal fMRI study demonstrates that clinical decline in older individuals is accompanied by significant loss of hippocampal activation over time. Consistent with our previous findings, subjects with the highest activation at baseline demonstrated the greatest clinical decline, supporting the hypothesis that there is a period of hyperactivation in the earliest stages of prodromal AD. These subjects also demonstrated the greatest loss of hippocampal activation over 2 years, consistent with the hypothesis that hyperactivity may be an indicator of impending hippocampal failure. These longitudinal fMRI findings were not explained by age, APOE status, or hippocampal volume loss. Our results suggest that longitudinal fMRI is capable of detecting early brain dysfunction predictive of subsequent decline, and tracking clinically meaningful changes over the course of early cognitive impairment.
Previous work from our group using a scene-encoding task in a separate group of older subjects found that fMRI activation in the posterior MTL was predictive of subsequent cognitive decline,2,3 but these studies did not include longitudinal imaging. The current study replicates our finding that increased baseline MTL activation is associated with greater subsequent clinical decline, in a separate cohort of subjects and using a different paradigm. The face-name paradigm engages more anterior hippocampal regions at baseline and was associated with loss of anterior hippocampal activation over time, suggesting that the hyperactivity predictive of decline may be regionally specific to the demands of the cognitive task. It is possible that this finding represents a regression to the mean phenomenon; however, we did not observe any longitudinal differences in the cognitively stable normal older individuals, and the loss of hippocampal activation paralleled clinical decline on both the CDR and neuropsychological measures of episodic memory.
In addition to our own studies that have reported evidence of increased hippocampal activation in the earliest stages of MCI, relative to normal controls,2,3,20,21 several other groups have reported hyperactivation in MCI, in particular for successful memory formation.8,9,22 This paradoxical increase in activation has also been observed in carriers of the APOE ε4 allele,11,12 even among young carriers,13 and in asymptomatic offspring of individuals with confirmed familial late-onset AD.23 We did not observe a significant effect of APOE carrier status in this study, perhaps because of the more advanced age of our subjects. We have previously speculated that hyperactivity might reflect an increase in neuronal recruitment to maintain memory performance in the setting of early AD pathology,10 which is supported by the finding of regionally specific hyperactivity according to memory task demands. The finding that increased activity is a predictor of impending hippocampal failure, however, suggests that the hyperactivity may be an indicator of neuronal stress or be directly related to the pathophysiologic process of AD. Hyperactivity could be related to aberrant sprouting of cholinergic fibers,24 excitotoxicity from the excessive stimulation of NMDA receptors,25 or stress from Aβ-induced neuronal alterations (e.g., excitatory activity triggers compensatory inhibitory mechanisms, which jointly contribute to network dysfunction).26
It should be noted that blood oxygen level dependent (BOLD) is an indirect measure of neuronal activity, and may reflect other physiologic properties, including cerebrovascular changes.27 The BOLD signal may depend on baseline perfusion, cerebral blood volume, vascular compliance, and interactions between these variables.28 The increase in BOLD activation observed in our fast decliner group could represent a decreased resting oxygenation state, increased perfusion, or some other disruption in neurovascular coupling.
One other potential explanation for apparent decreases in functional activation over time could be loss of hippocampal volume. Controlling for volume both within ROI analyses and in a multivariate model, we still observed loss of hippocampal activation. We did not observe significant hippocampal atrophy over the 2 years using automated volumetric analyses, perhaps because many of our subjects were still at relatively early stages of prodromal AD, prior to significant neuronal loss. It has been postulated that hippocampal atrophy may occur later in the pathophysiologic sequence of AD.29,30 We hypothesize that those subjects who demonstrated the greatest decrease in function will subsequently manifest hippocampal atrophy, and are actively pursuing this hypothesis in ongoing longitudinal studies.
Over the course of the study, we modified our postscan memory test to include more test items at the follow-up scan. Unfortunately, this limited our ability to determine if loss of hippocampal signal accompanies worsening performance on postscan memory testing. Longitudinal fMRI studies now underway in our laboratory include more detailed postscan testing, and we will be able to analyze successful vs failed encoding using event-related analyses.
If fMRI is to be useful as a tool for evaluating longitudinal change in brain function or the efficacy of pharmacologic interventions, it is essential to demonstrate the stability of fMRI measures over extended periods of time in the absence of clinical change. Two recent studies suggest that fMRI reproducibility is reasonable over short time frames in older populations.31,32 Our finding of reproducible patterns of activation over 2 years in clinically stable normal subjects is encouraging that fMRI can provide a reliable estimate of hippocampal activation over time, and thus may prove a useful tool for detecting clinically meaningful change in longitudinal pharmacologic studies in early prodromal AD.
Our results in subjects with very mild impairment demonstrate that clinical decline is related to the loss of hippocampal activation, and support previous findings that paradoxically increased activation may be predictive of impending clinical decline. These findings suggest that fMRI may prove valuable in tracking very early progression of brain dysfunction, prior to the point of irreversible neuronal loss, and at a time when disease-modifying therapies are likely to be most efficacious.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Jacqueline O'Brien and Reisa Sperling.
ACKNOWLEDGMENT
The authors thank Mary Foley and the Athinoula A. Martinos Center MRI Core for assistance with MRI; Jeanette Gunther, the Gerontology Research Unit staff, and the Massachusetts Alzheimer's Disease Research Center (ADRC) for assistance with subject recruitment and evaluation; and Akram Bakkour, Rebecca Betensky, Maija Pihlajamaki, Ali Atri, and Francine Grodstein for assistance with analyses and comments.
DISCLOSURE
Ms. O'Brien has received research support from the NIH (T32 MH017119 [trainee]). Ms. O'Keefe, Mr. LaViolette, and Ms. DeLuca report no disclosures. Dr. Blacker serves on the editorial boards of the American Journal of Geriatric Psychiatry and Experimental Gerontology; and receives research support from the NIH (4 R37 MH060009-09 [Co-PI], 5 P50 AG005134-25 [Co-PI of Core], 2 R01 AG007370-15 [PI of subcontract], 1R01AG029411-01 [Co-I], R01NS062028 [PI], and U01 AG032984 [PI of subcontract]), the Fidelity Foundation, and the Alzheimer's Association. Dr. Dickerson receives research support from the NIH (NIA R01 AG29411 [PI]). Dr. Sperling has served on a scientific advisory board for Archimedes Pharma Ltd (formerly Link Pharmaceuticals); serves on the editorial board of Alzheimer's Disease and Associated Disorders; has received speaker honoraria from Pfizer Inc; has served as a consultant for Elan Corporation, Wyeth, Bristol-Myers Squibb, and Bayer; her husband has served as a consultant for GE Healthcare. She receives research support from Elan Corporation, Wyeth, Bristol-Myers Squibb, the NIH (NIA R01AG027435 [PI]; NIA R01 AG29411 [Co-I]; P50 AG005134-25 [PI-Project 1]), and from the Alzheimer's Association.
Address correspondence and reprint requests to Dr. Reisa Sperling, Center for Alzheimer Research and Treatment, Brigham and Women's Hospital, Department of Neurology, 221 Longwood Ave., Boston, MA 02115 reisa@rics.bwh.harvard.edu
Editorial, page 1940
e-Pub ahead of print on May 12, 2010, at www.neurology.org.
Study funding: Supported by the National Institute on Aging AG-027435 and the Alzheimer's Association.
Disclosure: Author disclosures are provided at the end of the article.
Received August 4, 2009. Accepted in final form February 4, 2010.
REFERENCES
- 1.Petrella JR, Prince SE, Wang L, Hellegers C, Doraiswamy PM. Prognostic value of posteromedial cortex deactivation in mild cognitive impairment. PLoS ONE 2007;2:e1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller SL, Fenstermacher E, Bates J, Blacker D, Sperling RA, Dickerson BC. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J Neurol Neurosurg Psychiatry 2008;79:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickerson BC, Salat DH, Bates JF, et al. Medial temporal lobe function and structure in mild cognitive impairment. Ann Neurol 2004;56:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Small SA, Perera GM, DeLaPaz R, Mayeux R, Stern Y. Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer's disease. Ann Neurol 1999;45:466–472. [DOI] [PubMed] [Google Scholar]
- 5.Machulda MM, Ward HA, Borowski B, et al. Comparison of memory fMRI response among normal, MCI, and Alzheimer's patients. Neurology 2003;61:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson SC, Schmitz TW, Trivedi MA, et al. The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J Neurosci 2006;26:6069–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrella JR, Krishnan S, Slavin MJ, Tran TT, Murty L, Doraiswamy PM. Mild cognitive impairment: evaluation with 4-T functional MR imaging. Radiology 2006;240:177–186. [DOI] [PubMed] [Google Scholar]
- 8.Hamalainen A, Pihlajamaki M, Tanila H, et al. Increased fMRI responses during encoding in mild cognitive impairment. Neurobiol Aging 2007;28:1889–1903. [DOI] [PubMed] [Google Scholar]
- 9.Kircher TT, Weis S, Freymann K, et al. Hippocampal activation in patients with mild cognitive impairment is necessary for successful memory encoding. J Neurol Neurosurg Psychiatry 2007;78:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickerson BC, Sperling RA. Functional abnormalities of the medial temporal lobe memory system in mild cognitive impairment and Alzheimer's disease: insights from functional MRI studies. Neuropsychologia 2008;46:1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bookheimer SY, Strojwas MH, Cohen MS, et al. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med 2000;343:450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondi MW, Houston WS, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology 2005;64:501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filippini N, Zarei M, Beckmann CF, et al. Regional atrophy of transcallosal prefrontal connections in cognitively normal APOE epsilon4 carriers. J Magn Reson Imaging 2009;29:1021–1026. [DOI] [PubMed] [Google Scholar]
- 14.Grundman M, Petersen RC, Ferris SH, et al. Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch Neurol 2004;61:59–66. [DOI] [PubMed] [Google Scholar]
- 15.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004;256:183–194. [DOI] [PubMed] [Google Scholar]
- 16.Sperling RA, Bates J, Cocchiarella A, Schacter D, Rosen B, Albert M. Encoding novel face-name associations: a functional MRI study. Hum Brain Mapp 2001;14:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sperling RA, Greve D, Dale A, et al. fMRI detection of pharmacologically induced memory impairment. Proc Natl Acad Sci USA 2002;99:455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sperling RA, Bates JF, Chua EF, et al. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer's disease. J Neurol Neurosurg Psychiatry 2003;74:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daly E, Zaitchik D, Copeland M, Schmahmann J, Gunther J, Albert M. Predicting conversion to Alzheimer disease using standardized clinical information. Arch Neurol 2000;57:675–680. [DOI] [PubMed] [Google Scholar]
- 20.Dickerson BC, Salat D, Greve D, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 2005;65:404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Celone KA, Calhoun VD, Dickerson BC, et al. Alterations in memory networks in mild cognitive impairment and Alzheimer's disease: an independent component analysis. J Neurosci 2006;26:10222–10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heun R, Freymann K, Erb M, et al. Mild cognitive impairment (MCI) and actual retrieval performance affect cerebral activation in the elderly. Neurobiol Aging 2007;28:404–413. [DOI] [PubMed] [Google Scholar]
- 23.Bassett SS, Yousem DM, Cristinzio C, et al. Familial risk for Alzheimer's disease alters fMRI activation patterns. Brain 2006;129:1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto M, Masliah E. Cycles of aberrant synaptic sprouting and neurodegeneration in Alzheimer's and dementia with Lewy bodies. Neurochem Res 2003;28:1743–1756. [DOI] [PubMed] [Google Scholar]
- 25.Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int 2004;45:583–595. [DOI] [PubMed] [Google Scholar]
- 26.Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 2007;55:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleisher AS, Podraza KM, Bangen KJ, et al. Cerebral perfusion and oxygenation differences in Alzheimer's disease risk. Neurobiol Aging 2009;30:1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci 2004;5:347–360. [DOI] [PubMed] [Google Scholar]
- 29.Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain 2009;132:1310–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jack CR, Jr., Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009;132:1355–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrella JR, Prince SE, Krishnan S, Husn H, Kelley L, Doraiswamy PM. Effects of donepezil on cortical activation in mild cognitive impairment: a pilot double-blind placebo-controlled trial using functional MR imaging. AJNR Am J Neuroradiol 2009;30:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clement F, Belleville S. Test-retest reliability of fMRI verbal episodic memory paradigms in healthy older adults and in persons with mild cognitive impairment. Hum Brain Mapp 2009;30:4033–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]