

Abstract
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disorder. Familial AD (FAD) mutations in presenilins have been linked to calcium (Ca2+) signaling abnormalities. To explain these results, we previously proposed that presenilins function as endoplasmic reticulum (ER) passive Ca2+ leak channels. To directly investigate the role of presenilins in neuronal ER Ca2+ homeostasis, we here performed a series of Ca2+ imaging experiments with primary neuronal cultures from conditional presenilin double-knock-out mice (PS1dTAG/dTAG, PS2 −/−) and from triple-transgenic AD mice (KI–PS1M146V, Thy1–APPKM670/671NL, Thy1–tauP301L). Obtained results provided additional support to the hypothesis that presenilins function as ER Ca2+ leak channels in neurons. Interestingly, we discovered that presenilins play a major role in ER Ca2+ leak function in hippocampal but not in striatal neurons. We further discovered that, in hippocampal neurons, loss of presenilin-mediated ER Ca2+ leak function was compensated by an increase in expression and function of ryanodine receptors (RyanRs). Long-term feeding of the RyanR inhibitor dantrolene to amyloid precursor protein–presenilin-1 mice (Thy1–APPKM670/671NL, Thy1–PS1L166P) resulted in an increased amyloid load, loss of synaptic markers, and neuronal atrophy in hippocampal and cortical regions. These results indicate that disruption of ER Ca2+ leak function of presenilins may play an important role in AD pathogenesis.
Introduction
Alzheimer's disease (AD) is the most common form of age-related dementia in human beings over the age of 60 years. Most cases of AD are idiopathic, but the small fractions of AD cases are familial and characterized by an earlier onset and genetic inheritance. Mutations in presenilin-1 (PS1) and presenilin-2 (PS2) account for ∼40% of all known familial AD (FAD) cases in which a genetic cause has been identified (Tandon and Fraser, 2002). Presenilins are 50 kDa proteins that contain nine transmembrane domains (Laudon et al., 2005; Spasic et al., 2006) and reside in the endoplasmic reticulum (ER) membrane (Annaert et al., 1999). Presenilins undergo endoproteolytic cleavage to N-terminal (NTF) and C-terminal (CTF) fragments. The complex of cleaved presenilins with nicastrin, aph-1, and pen-2 subunits moves from ER to plasma membrane, in which it functions as a γ-secretase, which cleaves the amyloid precursor protein (APP) and releases the amyloid β-peptide (Aβ), the principal constituent of the amyloid plaques in the brains of AD patients. Consistent with the role of presenilins as catalytic subunits of a γ-secretase (De Strooper et al., 1998; Wolfe et al., 1999), FAD mutations in presenilins affect APP processing.
In addition to changes in APP processing, many FAD mutations in presenilins result in deranged Ca2+ signaling, and growing evidence indicate that neuronal Ca2+ signaling disruptions may play an early and important role in AD pathogenesis (Bezprozvanny and Mattson, 2008). The connection between presenilins and Ca2+ signaling was initially uncovered when it was reported that fibroblasts from FAD patients release supranormal amounts of Ca2+ in response to inositol-1,4,5-trisphosphate (InsP3) (Ito et al., 1994). Similar results were obtained in experiments with cells expressing FAD mutant presenilins (Leissring et al., 1999a,b) and in cortical and hippocampal neurons from presenilin FAD mutant knock-in mice (Guo et al., 1999; Chan et al., 2000; Schneider et al., 2001; Stutzmann et al., 2004, 2006; Smith et al., 2005; Chakroborty et al., 2009). To explain these results, it has been suggested that mutant presenilins affect store-operated Ca2+ influx (Leissring et al., 2000; Yoo et al., 2000; Herms et al., 2003; Akbari et al., 2004), increase activity and/or expression of intracellular Ca2+ release channels, such as ryanodine receptors (RyanRs) (Chan et al., 2000; Smith et al., 2005; Stutzmann et al., 2006; Hayrapetyan et al., 2008; Rybalchenko et al., 2008) and InsP3 receptors (InsP3Rs) (Cai et al., 2006; Cheung et al., 2008), or modulate function of the smooth endoplasmic reticulum Ca2+-ATPase (SERCA) pump (Green et al., 2008). Based on results obtained in bilayer reconstitution electrophysiological experiments and Ca2+ imaging experiments with mouse embryonic fibroblasts (MEFs) from PS double-knock-out (DKO) mice, we previously suggested that presenilins function as passive ER Ca2+ leak channels that control steady-state ER Ca2+ levels (Tu et al., 2006). We also found that many FAD mutations in presenilins result in loss of ER Ca2+ leak function, leading to ER Ca2+ overload and supranormal Ca2+ release from the ER (Tu et al., 2006; Nelson et al., 2007, 2010). The ER Ca2+ leak function of presenilins was independent of their γ-secretase function because it could be rescued by PS1–D257A mutant, which lacks γ-secretase activity, and it was maintained in Aph-1 triple-knock-out MEF cells, which lack γ-secretase but express full-length presenilins (Tu et al., 2006).
To directly investigate the role of presenilins in neuronal ER Ca2+ homeostasis, we here performed a series of Ca2+ imaging experiments with primary neuronal cultures from conditional presenilin double-knock-out mice (PScDKO mice) (PS1dTAG/dTAG, PS2 −/−) and from triple-transgenic AD mice (3xTg mice) (KI–PS1M146V, Thy1–APPKM670/671NL, Thy1–tauP301L) (Oddo et al., 2003). Obtained results supported the hypothesis that presenilins function as ER Ca2+ leak channels in neurons. We further discovered that a loss of ER Ca2+ leak function of presenilins in hippocampal neurons was compensated by increased expression and activity of RyanRs. Furthermore, long-term feeding of a double-transgenic AD mice (APPPS1 mice) (Thy1–APPKM670/671NL, Thy1–PS1L166P) (Radde et al., 2006) with the RyanR inhibitor dantrolene resulted in increased amyloid load, loss of synaptic markers, and neuronal atrophy in hippocampal and cortical regions of these mice. Our results provide new evidence that disruption of ER Ca2+ leak function of presenilins and resulting neuronal Ca2+ dyshomeostasis play an important role in AD pathogenesis. Our results obtained with the three different presenilin genetic mouse models (knockin, conditional knock-out, and transgenic) also provide the beginning of a common foundation for the currently contradictory view on the role that presenilins play in neuronal Ca2+ homeostasis.
Materials and Methods
Transgenic mouse lines.
The PScDKO mice (PS1dTAG/dTAG, PS2 −/−) were generated by crossing PS1dTAG/dTAG mice with PS2 −/− mice (Herreman et al., 1999). PS1dTAG/dTAG mice were generated by knockin strategy. The first exon of PSEN1 gene was surrounded by loxP sites (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). In addition, a double tag encoding for calmodulin binding protein followed by 3xFlag epitope tag was inserted immediately after the ATG start codon of PSEN1 (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). The generation and characterization of 3xTg mice (KI–PS1M146V, Thy1–APPKM670/671NL, Thy1–tauP301L) has been described previously (Oddo et al., 2003). The wild-type (WT) mice of the same mixed background strain (C7BL/6;129X1/SvJ;129S1/Sv) was used as a control for experiments with 3xTg mice. 3xTg mice and control WT mice were kindly provided by Frank LaFerla (University of California, Irvine, Irvine, CA). The generation and characterization of APPPS1 mice (Thy1–APPKM670/671NL, Thy1–PS1L166P) has been described previously (Radde et al., 2006). The APPPS1 mice were kindly provided by Mathias Jucker (University of Tübingen, Tübingen, Germany). The three transgenic mouse lines and WT mice were housed in a pathogen-free facility in a temperature-controlled room at 22–24°C with a 12 h light/dark cycle and were fed a standard laboratory chow diet and double-distilled water ad libitum. All procedures involving mice were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center at Dallas, in accord with the National Institutes of Health Guidelines for the Care and Use of Experimental Animals.
Antibodies.
Anti-CTF–PS1 monoclonal antibody (mAb) (MAB5232) and anti-actin mAb (MAB1501) were from Millipore Bioscience Research Reagents, anti-FLAG mAb (F3165) was from Sigma, anti-RyanR mAb (MA3-925) was from Affinity BioReagents, polyclonal antibody for InsP3R1 (T443) was described previously (Kaznacheyeva et al., 1998), polyclonal antibody for SERCA2b was provided by Dr. Frank Wuytack (Katholieke Universiteit Leuven, Leuven, Belgium), monoclonal antibody for postsynaptic density 95 (PSD95) (MA1-045) was purchased from Thermo Fisher Scientific, neuronal-specific nuclear protein (NeuN) mAb (MAB377) was from Millipore Corporation, dopamine and cAMP-regulated phosphoproteins-32 (DARPP-32) mAb (catalog #2306) was from Cell Signaling Technology, anti-Aβ 6E10 mAb (SIG-39300) was from Covance, Alexa Fluor-488 or Alexa Fluor-594 anti-rabbit and anti-mouse secondary antibodies (A21121, A11012, and A11008) were from Invitrogen, HRP-conjugated anti-rabbit and anti-mouse secondary antibodies (115-035-146 and 111-035-144) were from Jackson ImmunoResearch, and biotinylated anti-mouse IgG reagent was from Vector Laboratories (M.O.M. kit, BMK-2202). For Western blotting, analysis proteins were resolved on 6% (for RyanR and InsP3R1), 10% (for actin and SERCA2B), or 12% (for PS1) SDS-PAGE gels, transferred to nitrocellulose paper, and probed with the corresponding primary and secondary antibodies.
Lentivirus generation and infection of primary neuronal cultures.
Shuttle plasmid constructs encoding nuclear localization signal (NLS)–green fluorescent protein (GFP)–Cre and NLS–GFP were provided by Thomas Südhof (Stanford University, Stanford, CA). Lenti-Cre and Lenti-GFP viruses were generated by cotransfection of shuttle vectors with human immunodeficiency virus-1 packaging vector 8.9 and vesicular stomatitis virus glycoprotein G envelope glycoprotein plasmids into the packaging cell line HEK293T as we described previously (Tang et al., 2009). The generated viral stocks were aliquoted into 1 ml tubes, immediately frozen in liquid nitrogen, and stored at −80°C. Before use, the aliquots of generated lentiviruses were thawed and warmed in the 37°C incubator. Each batch of generated Lenti-Cre and Lenti-GFP lentiviruses was tested in pilot cortical neuronal culture infection experiments, and the titer with minimal toxicity and maximum infection efficiency (>95%) was used in all experiments.
The hippocampal and striatal cultures of PScDKO, 3xTg, and WT mice were established from postnatal day 0–1 pups and maintained in culture as we described previously (Tang et al., 2005; Zhang et al., 2006). Lenti-Cre and Lenti-GFP viruses were added to PScDKO cultures at day in vitro 4 (DIV4). Wild-type PS1 and mutant PS1 constructs (L166P, ΔE9, M146V, D257A, and D385A) were amplified by PCR using constructs in pcDNA3 (Tu et al., 2006; Nelson et al., 2007) and cloned into lentiviral shuttle vector. An N-terminal hemagglutinin (HA) tag was added to all lentiviral PS1 constructs by PCR. Control–short-hairpin RNA interference (Ctrl–shRNAi) (SHC002) and RyR–shRNAi (SHCLNG_NM_009109, TRCN0000103010) lentivirus shuttle constructs were obtained from Sigma. Lenti-PS1 and Lenti-shRNAi viruses were generated by following the same procedures as for Lenti-Cre and Lenti-GFP viruses. The Lenti-PS1 rescue viruses were added to Lenti-Cre-infected PScDKO hippocampal neuronal cultures at DIV6. Lenti-shRNAi viruses were added to WT and 3xTg hippocampal neuronal cultures at DIV6.
Fura-2 Ca2+ imaging experiments.
Fura-2 Ca2+ imaging experiments with DIV12 or DIV13 hippocampal and striatal cultures were performed as described previously (Tang et al., 2005; Tu et al., 2006). Briefly, the cells were maintained in artificial CSF (aCSF) (in mm: 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.3). Fura-2 340 nm/380 nm ratio images were collected every 2 s for the duration of the experiment using a DeltaRAM-X illuminator, an IC-300 camera, and IMAGEMASTER PRO software (all from Photon Technology International, Inc.). The region of interest (ROI) used in the image analysis corresponded to the entire cell soma. For caffeine experiments, 25 mm caffeine in aCSF was applied. For ionomycin (IO) experiments, the cells were first washed with the Ca2+-free aCSF (omitted CaCl2 from aCSF and supplemented with 100 μm EGTA) for 30 s or 2 min as indicated, followed by addition of 5 μm IO. For KCl refill experiments, the neurons were first challenged with 15 mm KCl for 1 min in aCSF and then perfused with Ca2+-free aCSF for 30 s or 2 min as indicated, followed by addition of 5 μm IO. In dantrolene experiments, 50 nm dantrolene was added to the culture medium at DIV7, DIV9, and DIV11, and Ca2+ imaging experiments were performed at DIV13. All Ca2+ imaging experiments were done in room temperature. In caffeine experiments, maximal amplitude (peak) response was determined from fura-2 340 nm/380 nm ratios. The size of the ER Ca2+ pool was calculated by integrating an area under the IO-induced fura-2 Ca2+ response curve as we described previously (Tu et al., 2006). IO30 and IO120 correspond to the size of IO-releasable Ca2+ pool after 30 s and 120 s incubation in Ca2+-free aCSF, respectively. There was no significant difference in basal fura-2 ratio values in different groups of neurons before addition of ionomycin. The only exception were neurons infected with PS1–ΔE9 virus, which yielded higher basal Ca2+ levels and increased fura-2 ratios, presumably because this mutant acts as “superleaky” channel in the ER membrane (Tu et al., 2006).
D1ER Ca2+ imaging experiments.
D1ER expression plasmid was kindly provided by Dr Roger Tsien (University of California, San Diego, La Jolla, CA) (Palmer et al., 2004). D1ER is a fluorescence resonance energy transfer (FRET)-based cameleon Ca2+ indicator composed of enhanced cyan fluorescent protein (ECFP) and citrin fluorescent protein separated by a linker encoding calmodulin (CaM) and CaM-binding M13 peptide sequence (Palmer et al., 2004; McCombs and Palmer, 2008). Calreticulin (CRT) targeting sequence (MLLPVLLLGLLGAAAD) was added to N terminus and ER retention sequence (KDEL) was added to the C terminus of D1ER protein to facilitate ER targeting and retention.
WT and PS DKO MEF cells were described previously (Herreman et al., 2000; Tu et al., 2006). WT and DKO MEF cells were transfected with D1ER plasmid using Lipofectamine 2000. DIV7 WT and 3xTg primary hippocampal neuronal cultures were transfected with D1ER plasmid using the polyethylenimine method. Imaging experiments with transfected MEF cells and hippocampal neurons were performed 48 h after D1ER transfection using Deltavision RT wide-field epifluorescence deconvolution microscope (Applied Precision) equipped with a Photometrics CoolSNAPHQ monochromatic digital camera (Roper Scientific) controlled by the SoftWorx image acquisition software package (Applied Precision). The filter sets used for FRET imaging experiments were CFPx 436 nm/10 nm and yellow fluorescent protein (YFPx) 492 nm/18 nm for excitation (x) and CFPm 465 nm/30 nm and YFPm 535 nm/30 nm for emission (m). The series of three images CFPx/CFPm (CFP), YFPx/YFPm (YFP), and CFPx/YFPm (FRET) were acquired every 25 s. After collection of initial data points (100 s), the cells were washed with Ca2+-free aCSF and transferred to Ca2+-free aCSF containing 2 μm of the SERCA pump inhibitor thapsigargin for 600 s to deplete the ER Ca2+ stores. After thapsigargin treatment, the cells were exposed to 5 μm IO. At the conclusion of each experiment, the FRET/CFP ratio signals were calibrated in the presence 5 μm ionomycin using a series of calibration buffers (in mm: 125 KCl, 25 NaCl, 10 HEPES, and 0.8 EGTA, pH 7.3) with free Ca2+ clamped to defined concentrations (1 μm, 10 μm, 50 μm, 100 μm, 200 μm, 400 μm, 800 μm, and 5 mm). The free Ca2+ concentration in the calibration buffers was calculated by WEBMAXC STANDARD program. Calibration and analysis of the data were performed using NIH Image J software. The ER Ca2+ concentration was calculated from FRET/CFP ratio data for each cell using corresponding empirical calibration curves for the same type of cells.
Drug delivery and histological analysis.
Dantrolene was delivered to APPPS1 and WT mice by using an approach that we used previously for dantrolene evaluation in spinocerebellar ataxia type 3 (SCA3) and type 2 (SCA2) mice (Chen et al., 2008; Liu et al., 2009). Briefly, six APPPS1 hemizygous mice and six WT mice were fed with 100 μg of dantrolene suspended in 50 μl of PBS with 2% corn flour, resulting in the final dose of 5 mg/kg. The control group of six APPPS1 hemizygous mice and six WT mice were fed with 2% corn flour in PBS. All mice were fed orally twice a week from 2 to 8 months of age. At the conclusion of dantrolene feeding, all mice were terminally anesthetized and perfused transcardially with 30 ml of ice-cold PBS, followed by 50 ml of fixative (4% paraformaldehyde in 0.1 m PBS, pH 7.4) as we described previously (Chen et al., 2008; Liu et al., 2009; Tang et al., 2009). All brains were removed from the skull, postfixed overnight at 4°C in 4% paraformaldehyde, and equilibrated in 20–30% (w/v) sucrose in PBS. The brains were sliced to 30-μm-thick coronal sections using SM2000R sliding microtome (Leica).
For β-amyloid plaque staining, the 30 μm coronal sections from APPPS1 mice spaced throughout the forebrain were stained with 6E10 anti-Aβ mAb (1:1000 dilution), followed by staining with Alexa Flour-488 secondary anti-mouse IgG (1:2000 dilution). The quantitative analysis of amyloid load was performed blindly using the Isocyte laser scanner system (Molecular Devices). The average area of the amyloid plaques (AREA), the average intensity of fluorescent signal in the plaques (IINT-1), and the number of plaques was calculated automatically for each slice by using proprietary image analysis software (Molecular Devices). Twenty coronal sections from each of the 12 APPPS1 mice were quantified for the analysis, and the data were averaged within dantrolene-fed and control groups.
The immunostaining experiments were performed as we described previously (Chen et al., 2008; Liu et al., 2009; Tang et al., 2009). The hippocampal sections were stained with PSD95 mAb and Alexa Fluor-488 anti-mouse IgG. The hippocampal nuclei were stained with propidium iodide (PI). The striatal sections were stained with DARPP-32 mAb and Alexa Fluor-594 anti-mouse IgG. The images from hippocampal and striatal sections were collected using a Carl Zeiss Axoivert 100M confocal microscope. The striatal sections were also stained with anti-NeuN mAb and biotinylated anti-mouse IgG. Signal was amplified with an ABC Elite kit (Vector Laboratories) and detected with diaminobenzidine (Vector Laboratories). The Bielschowsky silver staining was performed on four sections for each APPPS1 mice and WT mice by following published procedures (Schwab et al., 2004). Briefly, the sections were closed and mounted to slides. The slides were placed in 0.1% silver nitrate solution, containing citric acid in a 65°C water bath for 7 min. The slides were rinsed in two changes of ddH2O, followed by rinse in 95% ethanol, and then rinsed with two changes of absolute ethanol. All slides were immersed in filtered 2.5% gum mastic ethanol solution for 5 min and then put into developing solution (10 ml of 2.5% gum mastic ethanol solution, 30 ml of 1.7% hydroquinone, plus 0.9 ml of 1% silver nitrate) in a 65°C water bath until a dark brown color developed (∼12 min). The slides were rinsed in water, dehydrated, and mounted. The pictures for NeuN and silver staining were taken using the Nikon Eclipse 80i microscope.
For quantification of PSD95 density, confocal data were collected using 40× objective with a Carl Zeiss 100M microscope. The collected data were exported in tiff file format using a Carl Zeiss confocal software. The tiff file containing PSD95 staining signal (green channel) was imported into NIH ImageJ. The ROI 200 × 200 pixels was randomly selected in NIH ImageJ, and the software automatically calculated the number of PSD95 puncta and area of PSD95-positive pixels in the selected ROI. For quantification of neurite density, the images of the hippocampal slides stained with Bielschowsky silver staining method were taken using a 40× objective using a Nikon Eclipse 80i microscope and exported as tiff files using Stereo Investigator software (MicroBrightField). The tiff files were imported into NIH ImageJ for analysis, and 200 × 200 pixel ROI was selected as described above. The smallest cell body in the ROI was manually chosen, and all objects with larger diameter than the selected cell body were excluded from the analysis to filter out signals from neuronal cell bodies and amyloid plaques. The remaining pixels correspond to the area covered by neurites. The area covered by neurites was automatically calculated for each ROI by MetaXpress. For PSD95 and silver staining quantification, four ROIs were randomly chosen for each analyzed section. Six CA1 hippocampal sections were analyzed from each mice, resulting in 24 ROIs for each mice. The data from all 24 ROIs were averaged for each mice. The obtained results from each of the six mice in vehicle- and dantrolene-treated groups were averaged and presented as mean ± SE (n = 6).
Statistical analysis.
Statistical comparisons of results obtained in experiments were performed by one-way ANOVA test or Student's t test. The p values are indicated in the text and figure legends as appropriate. The differences between control and experimental groups were determined to be nonsignificant in cases in which p > 0.05.
Results
Caffeine-induced Ca2+ release is enhanced in 3xTg and PS DKO hippocampal neurons
Previous Ca2+ imaging studies indicated that RyanR-mediated Ca2+ release is enhanced in cortical and hippocampal neurons from 3xTg mice (KI–PS1M146V, Thy1–APPKM670/671NL, Thy1–tauP301L) and KI–PS1M146V mice (Guo et al., 1999; Chan et al., 2000; Smith et al., 2005; Stutzmann et al., 2006; Chakroborty et al., 2009). Consistent with these findings, we discovered that application of RyanR agonist caffeine resulted in much greater Ca2+ responses in postnatal hippocampal neuronal cultures from 3xTg mice than from the WT mice (Fig. 1 A). On average, the peak amplitude of caffeine-induced Ca2+ responses was fourfold higher in 3xTg hippocampal neurons than in wild-type neurons (Fig. 1 C). By reverse transcription-PCR analysis, we established that APPKM670/671NL and tauP301L transgenes (driven by Thy1 promoter) are not expressed in these cultures before DIV18 (data not shown), and therefore observed differences in Ca2+ signals in DIV13 hippocampal neurons (Fig. 1 A) are primarily attributable to PS1M146V mutation. This is consistent with the previous comparison of Ca2+ signals in cortical neurons from KI–PS1M146V and 3xTg mice (Stutzmann et al., 2004, 2006; Smith et al., 2005). An increase in the RyanR expression levels has been proposed to be responsible for the enhanced caffeine response in 3xTg and KI–PS1M146V neurons (Chan et al., 2000; Smith et al., 2005; Stutzmann et al., 2006; Chakroborty et al., 2009). Western blot analysis of lysates prepared from DIV13 hippocampal cultures revealed a 1.8-fold increase in RyanR expression levels in 3xTg cultures when compared with WT cultures (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The levels of InsP3R1 were also increased in 3xTg cultures, although the difference with WT has not reach statistically significant levels (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The SERCA2b Ca2+ pump was expressed at similar levels in 3xTg and WT neurons (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Selective increase in neuronal RyanR expression levels is in agreement with previous studies with 3xTg and KI–PS1M146V mice (Chan et al., 2000; Smith et al., 2005; Stutzmann et al., 2006; Chakroborty et al., 2009).
Figure 1.
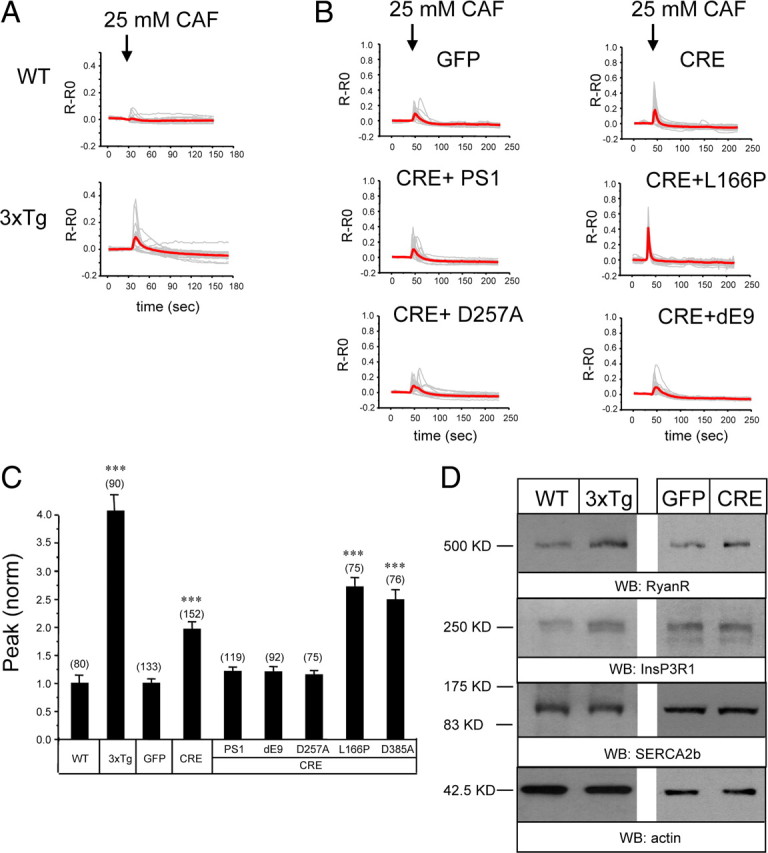
Caffeine-induced Ca2+ responses in 3xTg and PS DKO hippocampal neurons. A, B, Fura-2 340 nm/380 nm ratio traces are shown for WT and 3xTg hippocampal neurons (A) and for PScDKO hippocampal neurons infected with Lenti-GFP or Lenti-Cre viruses or lentiviral combinations (B). The neurons were challenged with 25 mm caffeine (CAF) as indicated. Individual cell traces (gray) and average traces (red) are shown for each group of neurons. C, The peak values of caffeine-induced Ca2+ transients are shown for WT, 3xTg, and PScDKO hippocampal neurons. In each experiment, the peak response was normalized to response in the WT group (for 3xTg experiments) or to the GFP-infected group (for PScDKO experiments). The normalized responses from different experiments were averaged and presented as mean ± SE (n indicates number of cells analyzed). The peak responses in 3xTg neurons were compared with peak responses in WT neurons, and the peak responses in PScDKO neurons were compared with the GFP-infected group. ***p < 0.001. D, Lysates from WT and 3xTg hippocampal cultures were analyzed by Western blotting (WB) with antibodies against RyanR, InsP3R1, and SERCA2b. Similar analysis was performed for PScDKO hippocampal neurons infected with Lenti-GFP and Lenti-Cre viruses. Actin was used as a loading control. Representative blot is shown. Quantification of multiple blots is shown in supplemental Figure 2 (available at www.jneurosci.org as supplemental material).
In the previous studies with MEFs, we discovered that presenilins function as ER Ca2+ leak channels and that disruption of ER Ca2+ leak function of presenilins results in increased ER Ca2+ levels (Tu et al., 2006; Nelson et al., 2007, 2010). To determine whether presenilins function as ER Ca2+ leak channels in neurons, we established primary neuronal cultures from PScDKO mice (PS1dTAG/dTAG, PS2 −/−). In control rescue experiments with DKO MEF cells, we confirmed that the mouse dTAG–PS1 construct retains ER Ca2+ leak channel activity (data not shown). To generate PS DKO neurons, the PScDKO neurons were infected with lentiviruses encoding nuclear-targeted GFP–Cre fusion protein (NLS–Cre). In parallel control experiments, PScDKO cultures were infected with lentiviruses encoding nuclear-targeted GFP protein (NLS–GFP). The absence of PS2 in these cultures was confirmed by Western blotting of lysates with anti-PS2 mAb (data not shown). To determine the time course of PS1 disappearance in cortical PScDKO cultures infected with Lenti-Cre, the lysates for Western blotting experiments were prepared at variable time points after infection. From this analysis, we determined that the levels of full-length dTAG–PS1 are reduced below detection limit by DIV12 in these cultures (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). In contrast, levels of dTAG–PS1 remained unchanged in PScDKO cultures infected with control Lenti-GFP virus (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Thus, we concluded that infection of PScDKO neurons with Lenti-Cre virus yielded PS DKO neurons. In Ca2+ imaging experiments, we discovered that application of caffeine induced significantly greater Ca2+ responses in PScDKO hippocampal neurons infected with Lenti-Cre viruses than in neurons infected with Lenti-GFP viruses (Fig. 1 B,C). On average, the peak amplitude of caffeine-induced Ca2+ response was twofold larger in Cre-infected PScDKO hippocampal neurons than in GFP-infected PScDKO neurons (Fig. 1 C). Western blot analysis revealed a trend toward increase in RyanR expression in Cre-infected PScDKO cultures, although the difference with GFP-infected cultures has not reached the level of statistical significance (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The levels of InsP3R1 and SERCA2b were similar in both Cre-infected and GFP-infected PScDKO cultures (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
We reasoned that enhanced caffeine responses in PS DKO cultures may be partially attributable to increased levels of RyanR expression (Fig. 1 D) but may also be attributable to loss of ER Ca2+ leak function of presenilins and overloaded ER Ca2+ stores. To determine an importance of ER Ca2+ leak function of presenilins, we performed a series of rescue experiments. In these experiments, PScDKO neurons were coinfected with Lenti-Cre viruses and lentiviruses encoding HA-tagged human PS1 rescue constructs. As a result, endogenous mouse dTAG–PS1 protein in these neurons was deleted and replaced with overexpressed human HA–PS1 protein. We found that infection with wild-type Lenti-PS1 viruses reduced caffeine responses in PS DKO neurons to wild-type levels (Fig. 1 B,C). In contrast, infection with PS1–L166P FAD construct, which lost Ca2+ leak channel function (Nelson et al., 2007), did not reduce caffeine responses in PS DKO neurons (Fig. 1 B,C). Expression of PS1–L166P instead resulted in an additional increase in the size of caffeine-induced Ca2+ response (Fig. 1 B,C), most likely caused by dominant-negative effects of this mutant on ER Ca2+ leak activity of remaining endogenous PS1. Infection with PS1–D257A construct, which lost γ-secretase function but not Ca2+ leak channel function (Tu et al., 2006), rescued caffeine responses in PS DKO neurons (Fig. 1 B,C). Infection with PS1–D385A construct, which lost both γ-secretase and Ca2+ leak channel functions (O. Nelson and I. Bezprozvanny, unpublished observations), did not reduce the size of the caffeine response and actually increased it, similar to PS1–L166P FAD mutant (Fig. 1 C). Infection with PS1–ΔE9 FAD construct, which encodes a gain-of-function ER Ca2+ leak mutant (Tu et al., 2006), rescued the size of the caffeine response in PS DKO neurons (Fig. 1 B,C). Based on these results, we concluded that loss of ER Ca2+ leak function and not γ-secretase function of presenilins resulted in increased caffeine responses in PS DKO neurons.
ER Ca2+ pool is increased and store-operated Ca2+ entry is reduced in 3xTg and PS DKO hippocampal neurons
The increased responses to caffeine observed in 3xTg and PS DKO hippocampal neurons (Fig. 1 A–C) could be due to increased levels of RyanR expression (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) or due to increased levels of ER Ca2+. To evaluate the ER Ca2+ content more directly, we measured the size of IO-sensitive Ca2+ pool in these neurons. Ionomycin is a Ca2+ ionophore that facilitates transport of Ca2+ across membranes. Addition of ionomycin induces exit of all ER-accumulated Ca2+ into the cytosol. The size of the IO-sensitive Ca2+ pool can be estimated by integrating a signal reported by cytosolic fura-2 after exposure to ionomycin. In the previous studies, we used a similar approach to evaluate ER Ca2+ content in wild-type and PS DKO MEF cells (Tu et al., 2006; Nelson et al., 2007). In experiments with hippocampal neurons, the cultures were transferred from aCSF (containing 2 mm Ca2+) to Ca2+-free media for 30 s and then challenged with 5 μm ionomycin. An increase in the cytosolic fura-2 signal observed immediately after ionomycin addition was integrated and designated as IO30. We found that application of ionomycin resulted in significantly greater responses in 3xTg hippocampal cultures than in WT cultures (Fig. 2 A). On average, the size of IO30 Ca2+ pool was 2.7-fold greater in 3xTg neurons than in WT neurons (Fig. 2 C).
Figure 2.
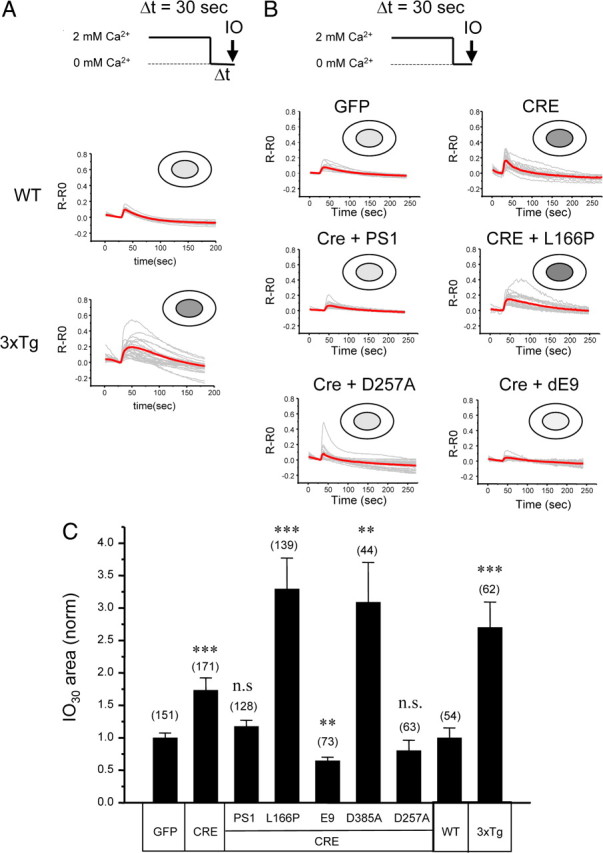
Ionomycin-sensitive Ca2+ pool in 3xTg and PS DKO hippocampal neurons. A, B, Fura-2 340 nm/380 nm ratio traces are shown for WT and 3xTg hippocampal neurons (A) and for PScDKO hippocampal neurons infected with Lenti-GFP or Lenti-Cre viruses or lentiviral combinations (B). The neurons were moved to Ca2+-free media for 30 s and challenged with 5 μm IO. Individual cell traces (gray) and average traces (red) are shown for each group of neurons. C, The average size of ionomycin-sensitive Ca2+ pool after 30 s incubation in Ca2+ free media (IO30). The area under IO-induced fura-2 signal was integrated for each neuron. In each experiment, IO30 values were normalized to IO30 values measured for the WT group (for 3xTg experiments) or to the GFP-infected group (for PScDKO experiments). The normalized responses from different experiments were averaged and presented as mean ± SE (n indicates number of cells analyzed). The IO30 values in 3xTg neurons were compared with IO30 values in WT neurons, and the IO30 values in PScDKO neurons were compared with IO30 values in the GFP-infected group. **p < 0.01; ***p < 0.001. n.s., Not significant.
We found that the IO-induced responses were also greater in PScDKO neurons infected with Lenti-Cre viruses than in neurons infected with Lenti-GFP viruses (Fig. 2 B). On average, the size of the IO30 Ca2+ pool was 1.7-fold greater in Cre-infected neurons (Fig. 2 C). Coinfection with wild-type PS1 virus or with the virus expressing PS1–D257A “γ-secretase mutant” rescued IO-induced responses back to the levels observed in GFP-infected cultures (Fig. 2 B,C). In contrast, coinfection with “loss of ER Ca2+ leak” function FAD mutant PS1–L166P did not rescue IO responses and instead resulted in enhanced responses (Fig. 2 B). On average, the size of the IO30 Ca2+ pool was threefold greater in PScDKO neurons coinfected with Cre and PS1–L166P viruses than in PScDKO neurons infected with GFP viruses (Fig. 2 C). Similar results were observed in experiments with PS1–D385A mutant, which lost both γ-secretase and ER Ca2+ leak functions (Fig. 2 C). As discussed above, it is likely that PS1–L166P and PS1–D385A mutants exert a dominant-negative effect on ER Ca2+ leak activity of endogenous PS1 still remaining in Cre-infected PScDKO neurons. Coinfection of Lenti-Cre with the “gain of Ca2+ leak function” PS1–ΔE9 FAD mutant reduced the size of IO responses to the levels even below levels observed in GFP-infected PScDKO neurons, consistent with superleaky ER membrane (Fig. 2 B,C). Based on these results, we concluded that loss of ER Ca2+ leak function and not γ-secretase function of presenilins resulted in the increased size of IO-sensitive Ca2+ pool in PS DKO neurons.
Ionomycin is a Ca2+ ionophore that, in our experiments, facilitates Ca2+ transport across all intracellular membranes and not only across ER membranes. Thus, some of the Ca2+ in the IO-sensitive Ca2+ pool may come from other intracellular compartments, such as Golgi apparatus or mitochondria. To confirm the results obtained with ionomycin, we took advantage of the recently developed ER-targeted genetic Ca2+ indicator D1ER (Palmer et al., 2004; McCombs and Palmer, 2008). D1ER is a FRET-based cameleon Ca2+ indicator composed of ECFP and citrin fluorescent proteins separated by a linker encoding CaM and CaM-binding M13 peptide sequence (Fig. 3 A). During Ca2+ binding to CaM, CaM binds to M13 target sequence and brings ECFP and citrin proteins in close proximity, which facilitates FRET between these proteins (Fig. 3 A). CRT targeting sequence was added to the N terminus, and ER retention sequence was added to the C terminus of D1ER protein to facilitate ER targeting and retention (Fig. 3 A) (Palmer et al., 2004). To validate utility of D1ER indicator for our experiments, we transiently transfected the D1ER plasmid to wild-type and PS DKO MEF cells. Consistent with previous results obtained using Mag-Fura-2 ER Ca2+ indicator (Tu et al., 2006), we discovered that ER Ca2+ levels reported by D1ER are several fold higher in PS DKO MEF cells when compared with wild-type cells (data not shown). In experiments with neurons, D1ER expression plasmid was transiently transfected to WT and 3xTg hippocampal neuronal cultures. Proper targeting of D1ER construct to neuronal ER compartment was confirmed in immunostaining experiments with polyclonal anti-CRT antibodies (Fig. 3 B). To minimize effects of cell-to-cell variability in D1ER expression levels, we normalized FRET signal from D1ER indicator to CFP signal observed for the same cell. When cultured in aCSF media containing 2 mm Ca2+, 3xTg neurons had a significantly higher FRET/CFP ratio than WT neurons, consistent with elevated ER Ca2+ concentration (Fig. 3 C). After transfer to Ca2+-free aCSF media and addition of 2 μm of the SERCA pump inhibitor thapsigargin, FRET/CFP ratios were reduced in both WT and 3xTg neurons, indicating store depletion caused by ER Ca2+ leak activity. In WT cells, FRET/CFP ratios were reduced to basal levels within 75 s after addition of thapsigargin (Fig. 3 C). In contrast, it took at least 250 s for FRET/CFP ratios to reduce to a new basal state in 3xTg neurons, consistent with impaired ER Ca2+ leak function (Fig. 3 C). Also, basal FRET/CFP ratios after thapsigargin application were significantly higher in 3xTg neurons than in WT neurons (Fig. 3 C). Addition of 5 μm ionomycin had no addition effect on FRET/CFP in WT neurons but caused addition reduction of FRET/CFP ratios in 3xTg neurons to the same levels as observed in WT neurons (Fig. 3 C). From these experiments, we concluded that, in 3xTg neurons, a significant portion of Ca2+ is accumulated in the thapsigargin-insensitive ER pool. This pool was not observed in our experiments with WT neurons, in which all ER Ca2+ could be released by thapsigargin (Fig. 3 C). Thus, appearance of this new pool is likely to be a result of compensation to impaired ER Ca2+ leak function in 3xTg neurons. Addition studies will be required to identify the nature of this ER subcompartment. Another possibility is that the rate of ER Ca2+ leak was so reduced in 3xTg neurons that 600 s incubation with thapsigargin was insufficient to deplete the stores completely. To convert FRET/CFP ratios to absolute ER Ca2+ values, we concluded each experiment with a calibration procedure using a series of extracellular solutions with defined Ca2+ concentrations. As a result of this procedure, calibration D1ER curves were constructed for WT and 3xTg neurons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Using these calibration curves, we estimated that, in the basal state, the ER Ca2+ levels were equal to 207 ± 47 μm (n = 100) in WT hippocampal neurons and 623 ± 108 μm (n = 51) in 3xTg neurons (p < 0.001). The threefold increase in ER Ca2+ levels in 3xTg neurons reported by D1ER indicator is in near perfect agreement with a 2.5-fold increase in the size of the IO-sensitive Ca2+ pool that we measured for the same neurons using fura-2 (Fig. 2 A,C). Thus, in future experiments, we focused on IO-sensitive Ca2+ pool measurements because of the relative simplicity of experimental procedures involved in these measurements.
Figure 3.
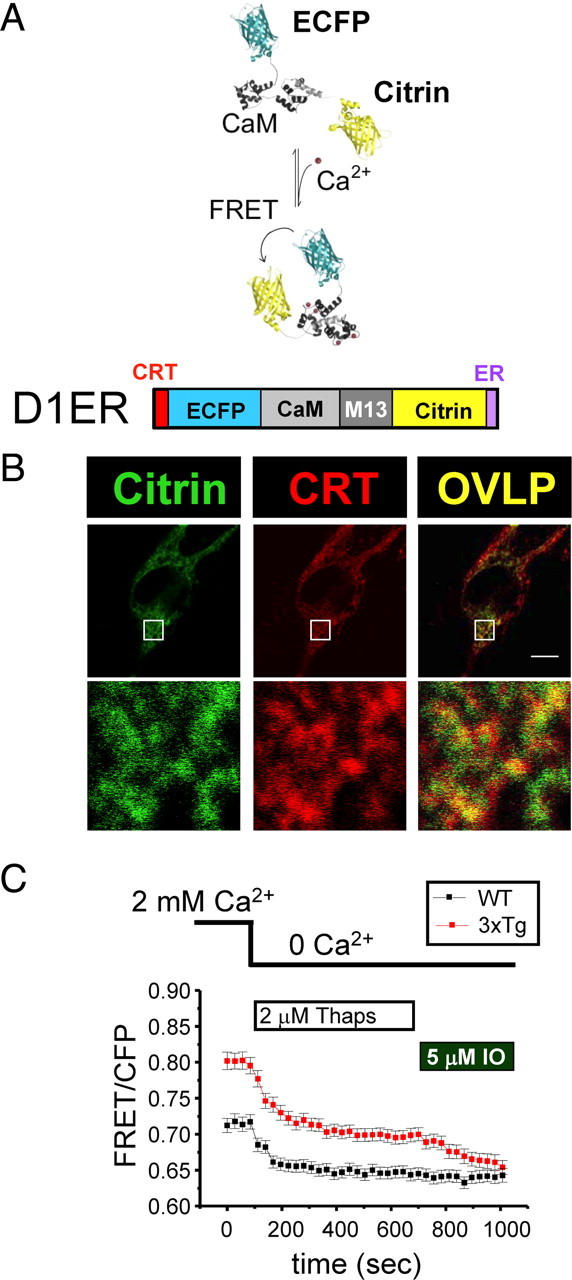
ER Ca2+ concentration is elevated in 3xTg hippocampal neurons. A, Principle of D1ER indicator (adapted from McCombs and Palmer, 2008). D1ER is a FRET-based cameleon Ca2+ indicator composed of ECFP (structure shown in cyan) and citrin fluorescent protein (structure shown in yellow) separated by a linker encoding CaM and CaM-binding M13 peptide sequence. CRT targeting sequence is added to the N terminus and the ER retension sequence is added to C terminus of D1ER protein to facilitate ER targeting and retension. B, D1ER is targeted to ER in hippocampal neurons. Hippocampal neurons were transiently transfected with D1ER plasmid, fixed, permeabilized, and stained with polyclonal antibodies against CRT. Subcellular localization of D1ER was determined by imaging on citrin channel (left, green). The location of CRT was visualized by rhodamine-conjugated secondary anti-rabbit antibodies (middle, red). The overlap between D1ER and CRT signals is shown on the right (OVLP) as yellow. Scale bar, 5 μm. The 10× magnified region is shown as indicated. C, D1ER signals from WT and 3xTg neurons. The FRET/CFP signal ratios for neurons transfected with D1ER plasmid are shown as a function of time as mean ± SE (n = 100 for WT cells and 51 for 3xTg cells) for WT neurons (black squares) and 3xTg neurons (red squares). The time of change to Ca2+-free aCSF and times of addition of 2 μm thapsigargin (Thaps) and 5 μm IO are as indicated.
The filling state of ER Ca2+ pool regulates the store-operated Ca2+ entry (SOC) pathway (Venkatachalam et al., 2002). To investigate effects of presenilin deletions and mutations on activation of the neuronal SOC pathway, we transferred fura-2-loaded hippocampal neurons from aCSF media to Ca2+-free media for 2 min and then returned neurons to aCSF media. The ER Ca2+ pool is depleted during incubation in Ca2+-free media, and “Ca2+ readdition” results in activation of SOC-mediated Ca2+ entry. We found that PScDKO neurons infected with Lenti-GFP respond to Ca2+ readdition more strongly than PScDKO neurons infected with Lenti-Cre (Fig. 4 A). We also discovered that Ca2+ readdition results in much stronger responses in WT hippocampal neurons than in 3xTg neurons (Fig. 4 B). On average, the size of the peak Ca2+ response was reduced twofold in Cre-infected PScDKO neurons and in 3xTg neurons when compared with control groups (Fig. 4 C). Our results (Fig. 4) are in agreement with previous studies of SOC in PS FAD mutant cells (Leissring et al., 2000; Yoo et al., 2000; Herms et al., 2003; Akbari et al., 2004), but our conclusion is that presenilins affect SOC in hippocampal neurons indirectly, by controlling the filling state of ER Ca2+ stores. A similar hypothesis has been proposed previously based on the analysis of SOC in KI–PS1M146V fibroblasts (Leissring et al., 2000).
Figure 4.
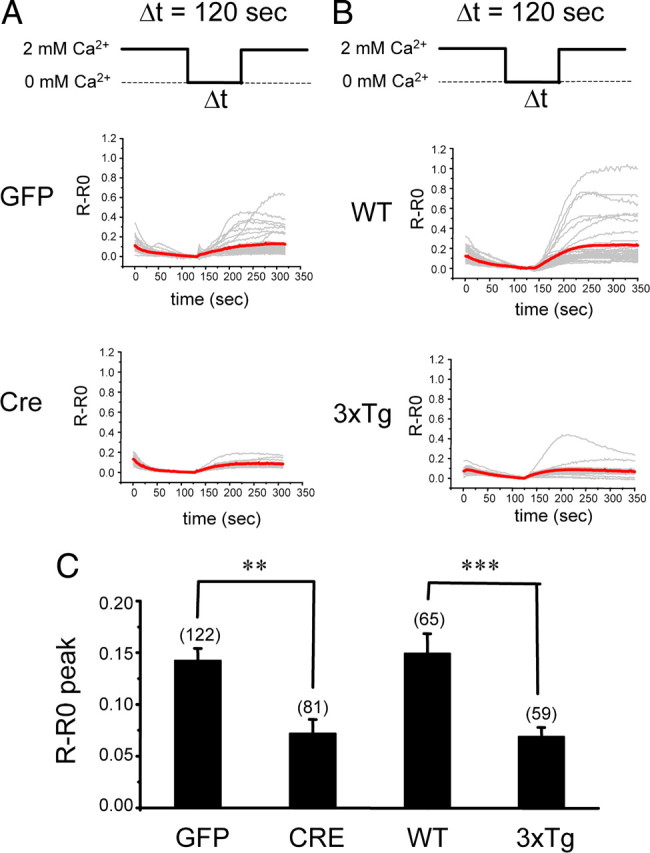
Store-operated Ca2+ entry in 3xTg and PS DKO hippocampal neurons. A, B, Fura-2 340 nm/380 nm ratio traces are shown for PScDKO hippocampal neurons infected with Lenti-GFP or Lenti-Cre viruses (A) and WT or 3xTg hippocampal neurons (B). The neurons were moved from aCSF to Ca2+-free media for 120 s and then returned to aCSF. Individual cell traces (gray) and average traces (red) are shown for each group of neurons. C, An average fura-2 peak responses to Ca2+ readdition are shown for PScDKO neurons infected with Lent-GFP or Lenti-Cre and for WT or 3xTg neurons. The average peak SOC responses are shown as mean ± SE (n indicates number of cells analyzed in each group). **p < 0.01; ***p < 0.001.
ER Ca2+ leak is impaired in 3xTg and PS DKO hippocampal neurons
The results obtained from IO30 and D1ER measurements (Figs. 2 and 3) are consistent with the hypothesis that the ER Ca2+ leak pathway is impaired in 3xTg and PS DKO hippocampal neurons. To measure the rate of passive ER Ca2+ leak in these neurons more directly, we compared the sizes of IO-induced Ca2+ responses after 30 and 120 s incubation in Ca2+-free media. We used the IO120/IO30 ratio as a quantitative measure of passive Ca2+ loss from the ER within 90 s time period attributable to endogenous ER Ca2+ leak pathway activity. Because the size of IO30 pool is very small in WT neurons and in PScDKO neurons infected with Lenti-GFP (Fig. 2), in these experiments, we refilled the stores with Ca2+ by 1 min exposure to 15 mm KCl in aCSF. Resulting depolarization induces opening of voltage-gated Ca2+ channels and rapid Ca2+ influx into the hippocampal neurons, which leads to filling of the ER Ca2+ stores. We did not see much difference in KCl-induced Ca2+ responses between different groups of cells (data not shown). Immediately after KCl depolarization, the neurons were transferred to Ca2+-free media for 30 s and challenged with ionomycin. Application of ionomycin in these conditions resulted in similar responses in PScDKO hippocampal neurons infected with Lenti-GFP and Lenti-Cre viruses (Fig. 5 A), indicating that the stores were completely full in both cultures. When length of incubation in Ca2+-free media was extended to 120 s, ionomycin induced significantly smaller responses in PScDKO neurons infected with Lenti-GFP than in neurons infected with Lenti-Cre (Fig. 5 A). On average, the IO120/IO30 ratio was equal to 0.35 for GFP-infected PScDKO neurons and 0.93 for Cre-infected PScDKO neurons (Fig. 5 B). We interpreted these results as an indication that PScDKO neurons infected with Lenti-GFP lost 65% of accumulated ER Ca2+ after 90 s incubation in Ca2+-free media. In contrast, PScDKO neurons infected with Lenti-Cre only lost 7% of accumulated ER Ca2+ during the same time period. Thus, the ER membrane in GFP-infected PScDKO neurons is much more leaky for Ca2+ than the ER membrane in Cre-infected PScDKO hippocampal neurons. This conclusion did not depend on using the KCl refill protocol, because the IO120/IO30 ratio was equal to 0.8 for PScDKO neurons infected with Lenti-Cre in the absence of KCl refill step (Fig. 5 B). The IO120/IO30 ratio could not be measured for PScDKO neurons infected with Lenti-GFP in the absence of KCl refill as IO30 pool was too small for these neurons (Fig. 2). Coinfection of Cre with the wild-type PS1 virus resulted in partial rescue of the leak function, yielding a IO120/IO30 ratio of 0.5 after KCl refill (Fig. 5 B). In contrast, coinfection of Cre virus with the mutant PS1–M146V virus did not rescue the leak function, yielding a IO120/IO30 ratio of 1.15 after KCl refill and 0.95 in the absence of KCl refill (Fig. 5 B). A similar paradigm was applied to compare ER Ca2+ leak in WT and 3xTg neurons. We found that the IO120/IO30 ratio was equal to 0.25 in WT neurons after the KCl refill protocol and 0.79 in 3xTg neurons (Fig. 5 B). Thus, within 90 s incubation in Ca2+-free media, WT neurons lost 75% of accumulated Ca2+ and 3xTg neurons only lost 21%. The IO120/IO30 ratio could not be measured in WT neurons without KCl refill because the size of the IO30 Ca2+ pool is too small in these neurons (Fig. 2). From these results, we concluded that ER Ca2+ leak function is impaired in 3xTg and PS DKO neurons. We further concluded that ER Ca2+ leak function in PS DKO neurons can be rescued by expression of wild-type PS1 but not PS1–M146V mutant.
Figure 5.
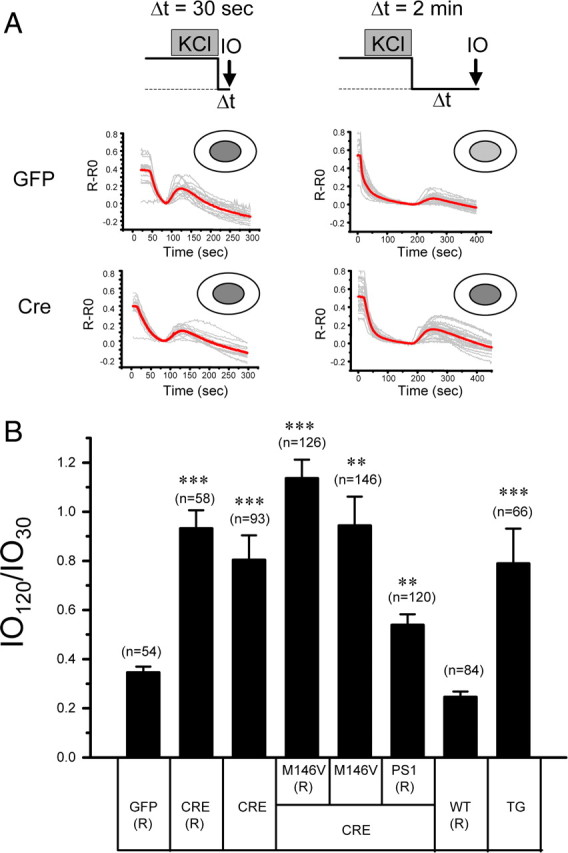
The ER Ca2+ leak measurements in 3xTg and PS DKO hippocampal neurons. A, Fura-2 340 nm/380 nm ratio traces are shown for PScDKO hippocampal neurons infected with Lenti-GFP or Lenti-Cre viruses. The neurons were stimulated with 15 mm KCl for 1 min in aCSF and then moved to Ca2+-free media for 30 or 120 s and challenged with 5 μm IO. Individual cell traces (gray) and average traces (red) are shown for each group of neurons. B, The average ER Ca2+ residual pool is shown as the IO120/IO30 ratio. In each experiment, IO30 and IO120 values were measured, and the IO120/IO30 ratio was determined for each batch of neurons. The IO120/IO30 ratios from different experiments were averaged and presented as mean ± SE (n indicates number of cells analyzed). The experiments that included the 15 mm KCl refill protocol are indicated with “(R).” The IO120/IO30 ratio in 3xTg neurons was compared with the IO120/IO30 ratio in the WT(R) group of neurons. The IO120/IO30 ratios in PScDKO neurons were compared with the IO120/IO30 ratio in the GFP(R) group. **p < 0.01; ***p < 0.001.
ER Ca2+ leak function in 3xTg and PS DKO medium spiny striatal neurons
AD preferentially affects hippocampal and cortical neurons, whereas other neuronal populations are not affected (Gomez-Isla et al., 2008). The reasons for the selective vulnerability of hippocampal and cortical neurons in AD are not well understood. Although the role of ER Ca2+ leak function of presenilins in AD pathogenesis is not yet clear, we reasoned that it may be of interest to evaluate ER Ca2+ leak function of presenilins in vulnerable and resistant neuronal populations. In experiments described above, we demonstrated that ER Ca2+ stores in hippocampal neurons are very leaky and that FAD mutations or genetic deletion of presenilins dramatically impair ER Ca2+ leak function in these neurons (Figs. 2, 5). For the next series of experiments, we focused on striatal medium spiny neurons (MSNs), which are selectively affected in Huntington's disease (Vonsattel and DiFiglia, 1998) but essentially spared in AD (Gomez-Isla et al., 2008). The MSN PScDKO neuronal cultures were established and infected with Lenti-GFP and Lenti-Cre viruses as described above for hippocampal neurons. Similar to procedures used for hippocampal neurons, MSNs were transferred from aCSF media to Ca2+-free media for 30 or 120 s and challenged with ionomycin. In contrast to hippocampal neurons, we found that PScDKO MSNs infected with Lenti-GFP respond strongly to ionomycin after 30 and 120 s incubation in Ca2+-free media (Fig. 6 A). On average, the IO120/IO30 ratio was equal to 0.75 for PScDKO MSNs infected with Lenti-GFP (Fig. 6 C). The PScDKO MSNs infected with Lenti-Cre virus also responded strongly to ionomycin after 30 and 120 s incubation in Ca2+-free media (Fig. 6 A). The average IO120/IO30 ratio for these neurons was equal to 1.2 (Fig. 6 C). Similar to PScDKO MSNs, we found that both WT and 3xTg MSNs responded strongly to ionomycin after 30 and 120 s in Ca2+-free media (Fig. 6 B). On average, the IO120/IO30 ratio was equal to 0.5 for WT neurons and 1.05 for 3xTg neurons (Fig. 6 C). From these results, we concluded that presenilins play a role as ER Ca2+ leak channels in MSNs, but the ER Ca2+ leak activity is much less potent in MSNs than in hippocampal neurons.
Figure 6.

The ER Ca2+ leak measurements in 3xTg and PS DKO striatal MSNs. A, B, Fura-2 340 nm/380 nm ratio traces are shown for PScDKO MSNs infected with Lenti-GFP or Lenti-Cre viruses (A) and for WT or 3xTg MSNs (B). The neurons were moved to Ca2+-free media for 30 or 120 s and challenged with 5 μm IO. Individual cell traces (gray) and average traces (red) are shown for each group of neurons. C, The average ER Ca2+ residual pool is shown as the IO120/IO30 ratio for MSNs. In each experiment, IO30 and IO120 values were measured, and the IO120/IO30 ratio was determined for each batch of neurons. The IO120/IO30 ratios from different experiments were averaged and presented as mean ± SE (n indicates number of cells analyzed). ***p < 0.001. D, PScDKO hippocampal neurons and MSNs were infected with Lenti-GFP or Lenti-Cre viruses. The lysates were prepared and analyzed by Western blotting with anti-FLAG antibodies and anti-CTF–PS1 antibodies. Actin was used as a loading control. Representative blot is shown.
To explain the differences between MSNs and hippocampal neurons, we evaluated expression of full-length PS1 in lysates prepared from hippocampal and MSN PScDKO neurons infected with Lenti-GFP and Lenti-Cre viruses. Actin was used as a loading control in these experiments (Fig. 6 D). The monoclonal anti-FLAG antibodies were used in these experiments for PS1 detection by taking an advantage of the dTAG epitope inserted into the PS1 N-terminal locus. Consistent with the previous studies (Annaert et al., 1999), we detected both full-length and cleaved forms of PS1 in lysates prepared from GFP-infected PScDKO hippocampal neurons and MSNs (Fig. 6 D). As expected, in both types of neurons, the cleaved PS1 form (PS1–NTF and PS1–CTF) was significantly more abundant than the holoprotein form (Fig. 5 D). Infection with Lenti-Cre resulted in complete loss of holoprotein and PS1–CTF forms and major reduction in the PS1–NTF form in both neuronal populations (Fig. 6 D). As we reported previously, only holoprotein presenilins support ER Ca2+ leak activity (Tu et al., 2006). It is of interest that the PS1 holoprotein was less abundant in MSN neuronal lysates than in hippocampal neuronal lysates (Fig. 6 D). Thus, it is possible that reduced ER Ca2+ leak activity in MSNs is related to comparatively lower levels of PS1 holoprotein expressed in these neurons.
Ryanodine receptors and ER Ca2+ leak in hippocampal neurons
Western blot analysis revealed a significant increase in RyanR expression levels in 3xTg hippocampal neurons (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). There was also a trend toward an increase in RyanR expression levels in PS DKO hippocampal neurons (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The effect on RyanR expression was specific, because expression of InsP3R1 in the same cultures was affected much less than expression of RyanR and expression of SERCA2b was not affected (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We hypothesized that an increase in RyanR expression may be a compensatory response to impaired ER Ca2+ leak function in these neurons. To test this hypothesis, we cultured WT and 3xTg hippocampal neurons for 6 d (DIV7–DIV13) in the presence of 50 nm dantrolene, a membrane-permeable RyanR inhibitor (Krause et al., 2004). After exposure to dantrolene, neurons were moved from aCSF to Ca2+-free media for 30 s and stimulated with ionomycin. We found that incubation with dantrolene had only minor effects on IO-induced responses in WT neurons (Fig. 7 A). Although some upward trend was observed, on average the size of the IO30 Ca2+ pool was not significantly different between control and dantrolene-exposed groups of WT hippocampal neurons (Fig. 7 B). In contrast, incubation with dantrolene dramatically enhanced IO-induced Ca2+ responses in 3xTg cells (Fig. 7 A). On average, the IO30 Ca2+ pool size was twofold larger in 3xTg hippocampal neurons exposed to dantrolene than in the control group of 3xTg neurons and eightfold larger than in the control group of WT neurons (Fig. 7 B). Consistent with the previous findings (Fig. 2 A,C), the IO30 Ca2+ pool size was fourfold larger in the control group of 3xTg neurons than in control group of WT neurons (Fig. 7 B).
Figure 7.

RyanRs compensate for impaired ER Ca2+ leak function in 3xTg hippocampal neurons. A, C, Fura-2 340 nm/380 nm ratio traces are shown for WT and 3xTg hippocampal neurons in control conditions (Ctrl) or after preincubation with 50 nm dantrolene (Dan) (A) and for WT and 3xTg hippocampal neurons infected with control shRNAi lentivirus (Ctrl) or RyanR-targeted shRNAi lentivirus (RyR) (C). The neurons were moved to Ca2+-free media for 30 s and challenged with 5 μm IO. Individual cell traces (gray) and average cell traces (red) are shown for each group of neurons. B, D, The average size of the ionomycin-sensitive Ca2+ pool after 30 s incubation in Ca2+-free media (IO30). The area under IO-induced fura-2 signal was integrated for each neuron. In each experiment, IO30 values were normalized to IO30 values measured for the WT Ctrl group. The normalized responses from different experiments were averaged and presented as mean ± SE (n indicates number of cells analyzed). ***p < 0.001. ns, Not significant.
Dantrolene is relatively specific for RyanR (Krause et al., 2004) but also inhibits other targets such as channels involved in store-operated Ca2+ entry (Zhao et al., 2006). To test the role of RyanR more specifically, we infected WT and 3xTg neuronal cultures with lentiviruses encoding shRNAi against RyanR (RyR–shRNAi) or control shRNAi (Ctrl–shRNAi). RyR–shRNAi is directed against mouse RyanR1 sequence, but, in control quantitative PCR experiments, we discovered that infection with RyR–shRNAi lentiviruses also reduced expression of RyanR2 and RyanR3 in hippocampal neurons (data not shown). An efficient knockdown of RyanRs in 3xTg hippocampal neurons infected with RyR–shRNAi was confirmed in control Western blotting experiments with pan-RyanR antibodies (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We found that IO-induced Ca2+ responses were increased in WT hippocampal neurons infected with RyR–shRNAi when compared with neurons infected with Ctrl–shRNAi (Fig. 7 C). On average, the size of the IO30 Ca2+ pool was fivefold higher in WT neurons infected with RyR–shRNAi, comparable in size with the IO30 Ca2+ pool in 3xTg neurons (Fig. 7 D). The 3xTg neurons infected with RyR–shRNAi responded to ionomycin more strongly than 3xTg neurons infected with Ctrl–shRNAi (Fig. 7 C). On average, the IO30 Ca2+ pool size was threefold larger in 3xTg neurons infected with RyR–shRNAi than in 3xTg neurons infected with Ctrl–shRNAi and >15-fold larger than in WT neurons infected with Ctrl–shRNAi (Fig. 7 D). From these results, we concluded that RyanRs play a role in the ER Ca2+ leak pathway in WT hippocampal neurons, but this role becomes more critical in 3xTg hippocampal neurons, which lost ER Ca2+ leak function of presenilins as a result of the PS1M146V mutation.
Evaluation of dantrolene in APPPS1 mice
The results obtained with 3xTg hippocampal neurons pointed to a close relationship between presenilin-mediated and RyanR-mediated Ca2+ leak pathways (Fig. 7). To understand the importance of this relationship in vivo, we set out to study the effect of dantrolene in an AD mouse model. For these studies, we focused on APPPS1 double-transgenic mice (Thy1–APPKM670/671NL and Thy1–PS1L166P) because of the early and robust amyloid accumulation in these mice (Radde et al., 2006; Serneels et al., 2009). Dantrolene was delivered to APPPS1 mice and WT control mice by using an approach that we used previously for dantrolene trials in SCA3 and SCA2 mice (Chen et al., 2008; Liu et al., 2009). In these experiments, six APPPS1 hemizygous female mice and six WT female mice were fed with dantrolene resuspended in PBS with 2% corn flour. The control group of six APPPS1 hemizygous female mice and six WT female mice were fed with 2% corn flour in PBS. Littermate mice were selected for four groups to reduce variability. All mice were fed orally twice a week from 2 to 8 months of age and then killed and processed for histological analysis.
To quantify amyloid load in these mice, the coronal slices from both groups of APPPS1 mice were stained with anti-Aβ monoclonal antibody, followed by staining with fluorescently labeled anti-mouse IgG. The stained slices were imaged by using a micrometer resolution laser scanning system. Visual inspection of obtained results indicated that amyloid plaques are more abundant and fluorescent in the dantrolene-fed group of APPPS1 mice than in the control group (Fig. 8 A). To quantify these results, we estimated the average number of plaques present in a slice and the average plaque fluorescent signal intensity. From this analysis, we concluded that there was a twofold increase in average plaque number in APPPS1 mice fed with dantrolene when compared with the control group of APPPS1 mice (Fig. 8 B). There was also a twofold increase in average size and fluorescent intensity of amyloid plaques in dantrolene-fed APPPS1 mice when compared with the control group (Fig. 8 B).
Figure 8.

Long-term dantrolene feeding increases amyloid accumulation in brains of APPPS1 mice. A, The coronal sections from dantrolene-fed (Dan) or vehicle-fed (Veh) APPPS1 mice were stained with anti-Aβ monoclonal antibody and fluorescently labeled secondary IgG. Representative fluorescent images of coronal slices from vehicle-fed and dantrolene-fed APPPS1 mice are shown. B, Quantification of average plaque size, pixel intensity, and plaque number in forebrain sections of dantrolene-fed and vehicle-fed APPPS1 mice. a.u., Arbitrary units. The data for each group are shown as mean ± SE (n = 6 mice). ***p < 0.001.
Amyloid load increase in dantrolene-fed APPPS1 mice (Fig. 8 A,B) suggested that neuropathological processes may also be accelerated in these mice. To test this hypothesis, we stained hippocampal slices from vehicle-fed and dantrolene-fed APPPS1 mice with antibodies against the excitatory synaptic marker PSD95. Loss of synaptic connections is one of the earliest events in human AD (Selkoe, 2002; Gomez-Isla et al., 2008), and we reasoned that earliest stages of the pathology may be detected by reduction in PSD95 staining. In control experiments, we also performed PSD95 staining of hippocampal slices from vehicle-fed and dantrolene-fed WT mice. We discovered that feeding dantrolene resulted in reduction in PSD95 staining density in both WT and APPPS1 mice but that reduction in APPPS1 mice was more severe (Fig. 9 A). To compare these results quantitatively, we normalized the PSD95-positive area in hippocampal slices from dantrolene-fed mice to the mean PSD95-positive area in hippocampal slices from vehicle-treated mice from the same group. We found that feeding of dantrolene reduced the PSD95-positive area to 73 ± 4% (n = 6) of vehicle control in WT mice and to 51 ± 4% (n = 6) of vehicle control in APPPS1 mice (p < 0.01). On average, a number of the PSD95-positive puncta per 100 μm2 was reduced from 55 ± 3 (n = 6) in vehicle-treated APPPs1 mice to 21 ± 2 (n = 6) in dantrolene-fed APPPS1 mice. The number of neuronal cell bodies visualized by PI staining was slightly reduced in the CA1 region of dantrolene-fed APPPS1 mice (Fig. 9 A). Thus, we concluded that feeding of dantrolene accelerated synaptic loss more significantly than neuronal loss at this early stage of the APPPS1 mice pathology in the hippocampus.
Figure 9.
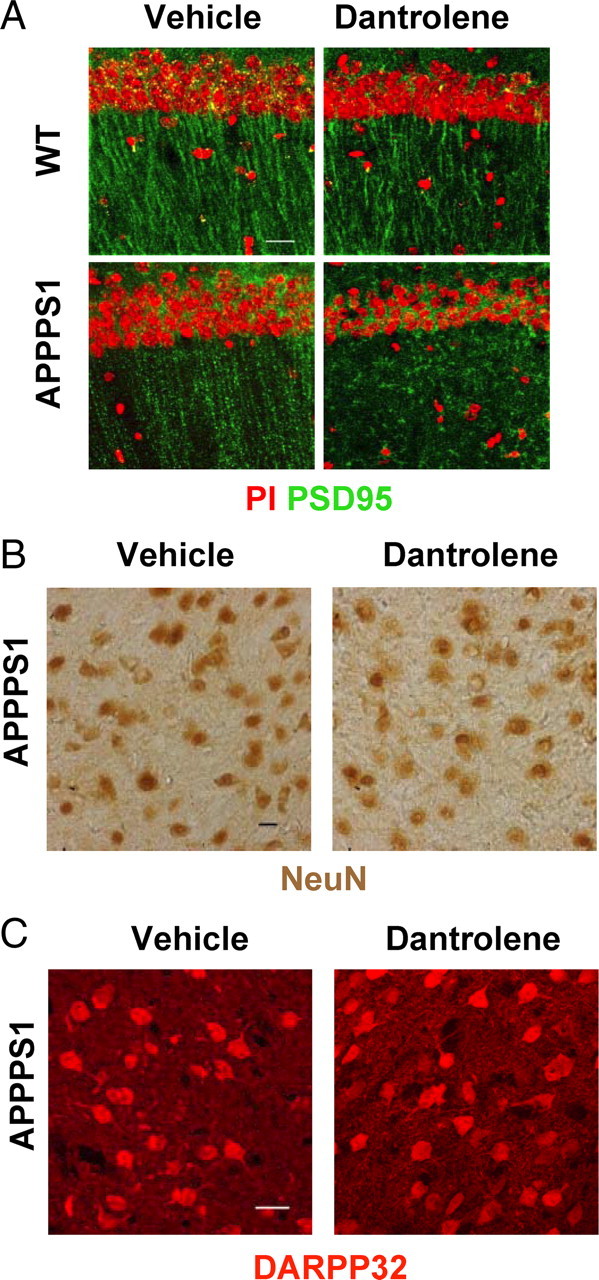
Neuropathological evaluation of long-term dantrolene feeding to APPPS1 mice: synaptic and neuronal markers. A, Hippocampal sections from vehicle-fed and dantrolene-fed WT and APPPS1 mice were stained with PI (red) and PSD95 (green) antibodies. Representative images are shown. Scale bar, 20 μm. B, C, Striatal sections from vehicle-fed and dantrolene-fed APPPS1 mice were stained with NeuN (C) and DARPP-32 (D) monoclonal antibodies. Scale bars, 20 μm.
The density of amyloid plaques in dantrolene-fed APPPS1 mice was increased in cortical and hippocampal regions and in basal ganglia (Fig. 8 A). Thus, we investigated whether striatal neurons are also affected in these mice. By using the neuronal nuclear marker NeuN and the cytosolic MSN marker DARPP-32, we did not observe significant differences between the control and dantrolene-fed groups of APPPS1 mice (Fig. 9 B,C), suggesting that striatal neurons are not effected in these mice.
To further extend pathological characterization of these mice, we performed silver staining of hippocampal, cortical, and striatal regions from vehicle-fed and dantrolene-fed WT and APPPS1 mice. By using the Bielschowsky staining method, we observed significant reduction in neuronal fiber density and abundance of dystrophic neurites in hippocampal and cortical regions of dantrolene-fed APPPS1 mice (Fig. 10). Quantitative analysis of silver staining data revealed that the neurites occupied 42 ± 3% (n = 6) of surface area in hippocampal sections from the control group and only 31 ± 2% (n = 6) in the dantrolene-fed group. We also observed significant thinning of the CA1 cell body layer in the hippocampus and appearance of many spaces without cell bodies in the hippocampal and cortical neuropil of dantrolene-fed mice (Fig. 10). All of these changes were consistent with ongoing neuronal atrophy and neuronal loss in hippocampal and cortical regions of dantrolene-fed APPPS1 mice. Consistent with the immunostaining data (Fig. 9 C,D), silver staining showed that striatal MSNs were not affected in dantrolene-fed APPPS1 mice but that the cortical projection fibers were undergoing dystrophic changes (Fig. 10). The observed effects were specific for APPPS1 mice, because no significant abnormalities were observed in hippocampal, cortical, or striatal regions of dantrolene-fed WT mice (Fig. 10).
Figure 10.

Neuropathological evaluation of long-term dantrolene feeding to APPPS1 mice: silver staining. Hippocampal, cortical, and striatal sections from vehicle-fed and dantrolene-fed WT and APPPS1 mice were stained using Bielschowsky silver stain. Representative images are shown. Scale bars, 20 μm.
Discussion
Presenilins function as neuronal ER Ca2+ leak channels
In the previous studies with MEF cells from PS DKO mice, we discovered that presenilins control ER Ca2+ levels by acting as passive ER Ca2+ leak channels (Tu et al., 2006). We further demonstrated that many FAD mutations in presenilins result in loss of ER Ca2+ leak function, leading to ER Ca2+ overload and supranormal Ca2+ release from the ER (Tu et al., 2006; Nelson et al., 2007, 2010). Our hypothesis has been challenged based on experiments with PS-transfected Xenopus oocytes and the chicken B-cell DT40 cell line (Cheung et al., 2008). As an alternative explanation, Cheung et al. proposed that FAD mutant presenilins affect Ca2+ signaling by modulating channel openings of InsP3R1 and not by changing ER Ca2+ levels (Cheung et al., 2008). Results obtained in the present study strongly support our hypothesis that presenilins in fact function as ER Ca2+ leak channels. In experiments with hippocampal neuronal cultures from 3xTg and PScDKO mice, we demonstrated that PS1M146V mutation or genetic deletion of presenilins resulted in enhanced caffeine-induced Ca2+ release from the ER (Fig. 1), increase in the size of ionomycin-sensitive Ca2+ pool (Fig. 2), increased ER Ca2+ levels (Fig. 3), reduced store-operated Ca2+ entry (Fig. 4), and decreased passive ER Ca2+ leak rate (Fig. 5). The defects in Ca2+ signaling observed in PS DKO hippocampal neurons could be rescued by expression of wild-type PS1 or PS1–D257A mutant deficient in γ-secretase activity but not by PS1–L166P or PS1–M146V FAD mutants deficient in ER Ca2+ leak channel activity or PS1–D385A mutant deficient in both γ-secretase and ER Ca2+ leak activities (Figs. 1, 2, 5). From these results, we concluded that presenilins function as ER Ca2+ leak channels in hippocampal neurons, a function independent from their γ-secretase activity (Fig. 11 A). Our results provide a coherent explanation for numerous previous reports of abnormal Ca2+ signaling observed in hippocampal and cortical neurons expressing PS1 or PS2 FAD mutants (Begley et al., 1999; Guo et al., 1999; Chan et al., 2000; Yoo et al., 2000; Schneider et al., 2001; Herms et al., 2003; Stutzmann et al., 2004, 2006; Smith et al., 2005; Chakroborty et al., 2009). The main findings reported in these studies, such as enhanced ER Ca2+ release via InsP3R and RyanR and impaired SOC, can be accounted for by the loss of ER Ca2+ leak function of presenilins and overloaded ER Ca2+ stores. The same mechanism may also contribute to abnormal presynaptic ER Ca2+ signaling and impaired Ca2+-dependent synaptic plasticity in PS DKO hippocampal neurons (Zhang et al., 2009).
Figure 11.

Proposed role of presenilins and ryanodine receptors in neuronal ER Ca2+ homeostasis. A, In wild-type hippocampal neurons, steady-state ER Ca2+ levels were determined by a balance between SERCA pump activity and ER Ca2+ leak mediated by presenilins and RyanRs. B, In 3xTg or PS DKO hippocampal neurons, ER Ca2+ leak activity of presenilins is impaired and steady-state ER Ca2+ levels are increased twofold to threefold. The RyanR expression levels are increased, and the RyanR-mediated Ca2+ leak pathway partially compensates for impaired ER Ca2+ leak function of presenilins. C, Block of RyanR function by dantrolene (DAN) or knockdown of RyanR expression by RyR–shRNAi results in a 10- to 15-fold increase in the size of ER Ca2+ pool in 3xTg neurons. Feeding of dantrolene to APPPS1 mice results in an increase in amyloid accumulation, reduction in PSD95 density in hippocampus, and neuronal atrophy and loss in hippocampus and cortex.
Interestingly, the ER membrane of striatal MSNs is much less leaky for Ca2+ than ER membrane of hippocampal neurons, and presenilins appear to play a lesser role in controlling ER Ca2+ homeostasis in MSNs than in hippocampal neurons (Fig. 6). Full-length PS1 is present in relatively lower amounts in MSNs when compared with hippocampal neurons (Fig. 6 D), providing a possible biochemical explanation to these findings. In contrast to hippocampal and cortical neurons, striatal MSNs are not affected in AD (Gomez-Isla et al., 2008), and future studies will be needed to further investigate whether a correlation exists between the potency of the ER Ca2+ leak pathway and selective vulnerability of different neuronal populations in AD.
Presenilins, ryanodine receptors, and neuronal Ca2+ homeostasis
An increase in RyanR expression levels has been reported previously for 3xTg and KI–PS1M146V hippocampal and cortical neurons (Chan et al., 2000; Smith et al., 2005; Stutzmann et al., 2006; Chakroborty et al., 2009). We have also observed a significant increase in RyanR expression levels in 3xTg hippocampal neuronal cultures (Fig. 1 D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The increase in RyanR expression levels has been proposed previously to be responsible for enhanced caffeine-induced Ca2+ responses in 3xTg and KI–PS1M146V neurons (Chan et al., 2000; Smith et al., 2005; Stutzmann et al., 2006; Chakroborty et al., 2009). We would like to revise this view and suggest that the increase in RyanR levels plays a compensatory role for the loss of presenilin leak function. Our experiments with RyR–shRNAi indicated that, in wild-type hippocampal neurons, RyanRs contribute to the ER Ca2+ leak function (Fig. 11 A). In 3xTg hippocampal neurons, the PS1M146V mutation impairs presenilin-mediated ER Ca2+ leak and the role of RyanR becomes more critical. Most likely, an increase in RyanR expression levels is an attempt by 3xTg hippocampal neurons to boost the RyanR-mediated Ca2+ leak pathway, which would then conceivably compensate for the loss of presenilin-mediated ER Ca2+ leak more efficiently (Fig. 11 B). The sensitivity of RyanRs to cytosolic Ca2+ is known to be modulated by ER Ca2+ levels. When ER Ca2+ levels become too high, even basal cytosolic Ca2+ levels become sufficient to result in RyanR opening. This is the so-called “store-overload-induced Ca2+ release” (SOICR) mechanism, which has been well described in cardiac cells (Jiang et al., 2004; Jones et al., 2008). We postulate that a similar SOICR mechanism supported by RyanRs is also functional in neurons, in particular in the condition of impaired ER Ca2+ leak, such as that attributable to PS FAD mutations.
If RyanR-mediated Ca2+ leak is inhibited in this situation (by dantrolene or by RyR–shRNAi knockdown of RyanR expression), then ER Ca2+ stores become very full, with total ER Ca2+ content 10- to 15-fold higher than in wild-type cells (Fig. 11 C). Resulting Ca2+ dyshomeostasis likely affects other neuronal functions, such as synaptic transmission and plasticity (Chakroborty et al., 2009). Addition studies will be required to understand the mechanism involved in upregulation of RyanR resulting from PS1 FAD mutations. A threefold increase in RyanR2 mRNA has been discovered in samples extracted from adult hippocampus of 3xTg mice (Chakroborty et al., 2009), and a fourfold increase in RyanR3 mRNA was reported for samples from KI–PS1M146V adult mice hippocampus (Chan et al., 2000). We have also observed a twofold to threefold increase in RyanR2 and RyanR3 mRNA in quantitative PCR experiments with 3xTg hippocampal neuronal cultures (data not shown). These results suggest that impaired ER Ca2+ leak function and increased ER Ca2+ levels are linked with an increase in transcription of genes encoding RyanR2 and RyanR3 via an “ER Ca2+ homeostasis”-negative feedback mechanism. The molecular underpinnings of this feedback mechanism need to be clarified in addition studies.
Protective role of ryanodine receptors in Alzheimer's disease
Our results indicate that RyanRs may play a protective role in the context of AD. The Ca2+ homeostasis model that we propose (Fig. 11) predicts that blocking RyanRs in AD could have a disastrous effect on the pathology. We tested this hypothesis by long-term feeding of dantrolene to APPPS1 mice. We discovered that long-term feeding of dantrolene to APPPS1 mice results in threefold increase in amyloid load (Fig. 8 A,B), loss of excitatory markers of synaptic transmission in hippocampus (Fig. 9 A), and neuronal atrophy and loss in hippocampus and cortex (Fig. 10). The results with dantrolene are somewhat paradoxical, because high (1–10 μm) concentrations of dantrolene reduced glutamate-induced excitotoxicity in KI–PS1M146V mouse hippocampal neurons (Guo et al., 1999). We obtained similar results in studies with 3xTg hippocampal neuronal cultures (data not shown). Moreover, we reported previously that long-term feeding of dantrolene to SCA3 and SCA2 mouse models slowed progression of motor phenotype and alleviated age-dependent neuronal loss in these mice (Chen et al., 2008; Liu et al., 2009). These results suggest that, although in general dantrolene acts as an anti-excitotoxic agent, it specifically accelerated pathology in APPPS1 mice. These results agree with our hypothesis that the loss of ER Ca2+ leak function of presenilins in APPPS1 mice (attributable to the PS1L166P mutation) is compensated by increased RyanR-mediated Ca2+ flux from the ER. Blocking RyanRs by dantrolene in this situation leads to an addition increase in neuronal ER Ca2+ levels and accelerated pathology (Fig. 11 C).
Interestingly, the potentially protective role of RyanRs has been proposed previously based on studies of APP transgenic mice (TgCRND8 mice; PrP–APPKM670/671NL). These investigators discovered that the levels of RyanR3 mRNA and protein are significantly increased in the cortex of adult TgCRND8 mice and in TgCRND8 cortical neuronal cultures (Supnet et al., 2006). Moreover, direct exposure of wild-type cortical neuronal cultures to Aβ42 resulted in 3.5-fold increase in RyanR3 mRNA levels (Supnet et al., 2010). These effects were specific for RyanR3, because exposure to Aβ42 did not affect expression levels of RyanR2, InsP3R1, or SERCA2b (Supnet et al., 2006). Formation of amyloid plaques and accumulation of extracellular Aβ42 causes destabilization of neuronal Ca2+ signals and increase in neuronal excitability (Busche et al., 2008; Kuchibhotla et al., 2008), and Supnet et al. (2006) argued that upregulation of RyanR3 could be a compensatory response aimed to protect against increased cortical neurons excitability and abnormal Ca2+ signals induced by Aβ42 exposure. Consistent with this prediction, the same authors recently discovered that knockdown of RyanR3 by RNAi accelerated neuronal death of TgCRND8 cultured cortical neurons but not wild-type cultured cortical neurons (Supnet et al., 2010). Thus, upregulation of RyanRs may have protective effects in both mutant presenilin and mutant APP models of AD, as we discussed recently (Supnet and Bezprozvanny, 2010).
Neuronal Ca2+ signaling, amyloid accumulation, and AD pathology
Our main goal was to define the role played by presenilins in the neuronal ER Ca2+ leak pathway. In addition to this specific issue, some of our results have implications for the more general understanding of existing connections between neuronal Ca2+ signaling, amyloid accumulation, and neuronal pathology in AD. A few previous studies addressed potential connections between Ca2+ signaling and amyloid production (Green et al., 2007). Some of these studies concluded that increased cytosolic Ca2+ signals enhance production of Aβ42 and increase the Aβ42/Aβ40 ratio (Querfurth and Selkoe, 1994; Pierrot et al., 2004; Green et al., 2007). Other studies indicated that increased Ca2+ signals reduce production of Aβ42 and reduce the Aβ42/Aβ40 ratio (Dreses-Werringloer et al., 2008). Most of these studies have been performed in non-neuronal systems. In our experiments, we observed twofold to threefold increases in amyloid load in dantrolene-fed APPPS1 mice (Fig. 8 A,B). Although the exact mechanism responsible for these effects still needs to be clarified, these results suggest that abnormal ER Ca2+ signaling is able to drive amyloid accumulation in the brain.
Cortical and hippocampal neuronal atrophy is typical of human AD (Gomez-Isla et al., 1996; Price et al., 2001; Gómez-Isla et al., 2008) and correlates much better with cognitive decline than the amyloid load (Giannakopoulos et al., 2003; Gómez-Isla et al., 2008). However, neuronal atrophy has been very difficult to reproduce in APP and presenilin transgenic mouse modes (Takeuchi et al., 2000; Schwab et al., 2004; Gómez-Isla et al., 2008). Interestingly, we found that feeding of dantrolene to APPPS1 mice resulted in significant loss of hippocampal excitatory synaptic markers (Fig. 9 A) and in neuronal atrophy in hippocampal and cortical regions (Fig. 10). Despite comparable increases in amyloid load in the striatal region (Fig. 8 A), striatal MSNs were not significantly affected by dantrolene feeding to APPPS1 mice (Figs. 9 B,C, 10). As discussed above, the ER Ca2+ leak pathway is less potent in striatal MSNs than in hippocampal neurons, which may explain why striatal neurons are less affected when the ER Ca2+ leak pathway is impaired by combined effects of PS1L166P mutation and dantrolene feeding in APPPS1 mice.
In conclusion, here we report analysis of the role played by presenilins in neuronal Ca2+ signaling. We conclude that presenilins function as ER Ca2+ leak channels in hippocampal neurons and that the loss of ER Ca2+ leak function of presenilins was partially compensated by an increase in RyanR expression. We further found that, if both PS-mediated and RyanR-mediated ER Ca2+ leak pathways are impaired, then ER Ca2+ levels in hippocampal neurons increase dramatically. Simultaneous block of both PS-mediated and RyanR-mediated ER Ca2+ leak pathways in vivo resulted in an increase in amyloid accumulation, loss of synaptic markers in hippocampus, and neuronal atrophy in hippocampus and cortex. Our results further support a close connection between neuronal Ca2+ signaling, amyloid accumulation, and AD pathogenesis (Bezprozvanny and Mattson, 2008).
Footnotes
This work was supported by the Alzheimer's Association, the Alzheimer's Disease Drug Discovery Foundation, a McKnight Brain Disorders Award, and National Institutes of Health Grant R01AG030746 (I.B.), a Methusalem grant from the Flemish government and the Katholieke Universiteit Leuven, the Fund for Scientific Research (Flanders, Belgium), Federal Office for Scientific Affairs Grant IUAP P6/43 (Belgium), and MEMOSAD (for Memory Loss in Alzheimer Disease: Underlying Mechanisms and Therapeutic Targets) Grant F2-2007-200611 of the European Union (B.D.S.). I.B. is a holder of the Carla Cocke Francis Professorship in Alzheimer's Research. We are grateful to Frank LaFerla for providing 3xTg mice and Mathias Jucker for providing the APPPS1 mice, Frank Wuytack for providing the SERCA2b antibody, Marek Michalek for providing CRT antibodies, Thomas Sudhof for providing the PSD95 antibodies and Lenti-Cre constructs, Roger Tsien for providing D1ER plasmid, Omar Nelson and Huarui Liu for help with making reagents, and Leah Benson for administrative assistance.
References
- Akbari Y, Hitt BD, Murphy MP, Dagher NN, Tseng BP, Green KN, Golde TE, LaFerla FM. Presenilin regulates capacitative calcium entry dependently and independently of gamma-secretase activity. Biochem Biophys Res Commun. 2004;322:1145–1152. doi: 10.1016/j.bbrc.2004.07.136. [DOI] [PubMed] [Google Scholar]
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–294. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley JG, Duan W, Chan S, Duff K, Mattson MP. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J Neurochem. 1999;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Cai C, Lin P, Cheung KH, Li N, Levchook C, Pan Z, Ferrante C, Boulianne GL, Foskett JK, Danielpour D, Ma J. The presenilin-2 loop peptide perturbs intracellular Ca2+ homeostasis and accelerates apoptosis. J Biol Chem. 2006;281:16649–16655. doi: 10.1074/jbc.M512026200. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–18200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- Chen X, Tang TS, Tu H, Nelson O, Pook M, Hammer R, Nukina N, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. 2008;28:12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP(3) receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, Marambaud P. A polymorphism in CALHM1 influences Ca2+ homeostasis, A beta levels, and Alzheimer's disease risk. Cell. 2008;133:1149–1161. doi: 10.1016/j.cell.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, Spires T, De Calignon A, Hyman BT. Neuropathology of Alzheimer's disease. Handb Clin Neurol. 2008;89:233–243. doi: 10.1016/S0072-9752(07)01222-5. [DOI] [PubMed] [Google Scholar]
- Green KN, Smith IF, Laferla FM. Role of calcium in the pathogenesis of Alzheimer's disease and transgenic models. Subcell Biochem. 2007;45:507–521. doi: 10.1007/978-1-4020-6191-2_19. [DOI] [PubMed] [Google Scholar]
- Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- Hayrapetyan V, Rybalchenko V, Rybalchenko N, Koulen P. The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction. Cell Calcium. 2008;44:507–518. doi: 10.1016/j.ceca.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Herms J, Schneider I, Dewachter I, Caluwaerts N, Kretzschmar H, Van Leuven F. Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J Biol Chem. 2003;278:2484–2489. doi: 10.1074/jbc.M206769200. [DOI] [PubMed] [Google Scholar]
- Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci U S A. 1999;96:11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol. 2000;2:461–462. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PP, Jiang D, Bolstad J, Hunt DJ, Zhang L, Demaurex N, Chen SR. Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store-overload-induced Ca2+ release. Biochem J. 2008;412:171–178. doi: 10.1042/BJ20071287. [DOI] [PubMed] [Google Scholar]
- Kaznacheyeva E, Lupu VD, Bezprozvanny I. Single-channel properties of inositol (1,4,5)-trisphosphate receptor heterologously expressed in HEK-293 cells. J Gen Physiol. 1998;111:847–856. doi: 10.1085/jgp.111.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F. Dantrolene: a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373. doi: 10.1111/j.1365-2044.2004.03658.x. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudon H, Hansson EM, Melén K, Bergman A, Farmery MR, Winblad B, Lendahl U, von Heijne G, Näslund J. A nine-transmembrane domain topology for presenilin 1. J Biol Chem. 2005;280:35352–35360. doi: 10.1074/jbc.M507217200. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Parker I, LaFerla FM. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem. 1999a;274:32535–32538. doi: 10.1074/jbc.274.46.32535. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM. Alzheimer's presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999b;72:1061–1068. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombs JE, Palmer AE. Measuring calcium dynamics in living cells with genetically encodable calcium indicators. Methods. 2008;46:152–159. doi: 10.1016/j.ymeth.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230–1239. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson O, Supnet C, Liu H, Bezprozvanny I. Familial Alzheimer's disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J Alzheimers Dis. 2010 doi: 10.3233/JAD-2010-100159. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci U S A. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrot N, Ghisdal P, Caumont AS, Octave JN. Intraneuronal amyloid-beta1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004;88:1140–1150. doi: 10.1046/j.1471-4159.2003.02227.x. [DOI] [PubMed] [Google Scholar]
- Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58:1395–1402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, Selkoe DJ. Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry. 1994;33:4550–4561. doi: 10.1021/bi00181a016. [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalchenko V, Hwang SY, Rybalchenko N, Koulen P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Schneider I, Reverse D, Dewachter I, Ris L, Caluwaerts N, Kuiperi C, Gilis M, Geerts H, Kretzschmar H, Godaux E, Moechars D, Van Leuven F, Herms J. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem. 2001;276:11539–11544. doi: 10.1074/jbc.M010977200. [DOI] [PubMed] [Google Scholar]
- Schwab C, Hosokawa M, McGeer PL. Transgenic mice overexpressing amyloid beta protein are an incomplete model of Alzheimer disease. Exp Neurol. 2004;188:52–64. doi: 10.1016/j.expneurol.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horré K, Van Houtvin T, Esselmann H, Paul S, Schäfer MK, Berezovska O, Hyman BT, Sprangers B, Sciot R, Moons L, Jucker M, Yang Z, May PC, Karran E, Wiltfang J, D'Hooge R, De Strooper B. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. 2009;324:639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer's disease. J Neurochem. 2005;94:1711–1718. doi: 10.1111/j.1471-4159.2005.03332.x. [DOI] [PubMed] [Google Scholar]
- Spasic D, Tolia A, Dillen K, Baert V, De Strooper B, Vrijens S, Annaert W. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J Biol Chem. 2006;281:26569–26577. doi: 10.1074/jbc.M600592200. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010;47:183–189. doi: 10.1016/j.ceca.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supnet C, Grant J, Kong H, Westaway D, Mayne M. Amyloid-beta-(1–42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J Biol Chem. 2006;281:38440–38447. doi: 10.1074/jbc.M606736200. [DOI] [PubMed] [Google Scholar]
- Supnet C, Noonan C, Richard K, Bradley J, Mayne M. Up-regulation of the type 3 ryanodine receptor is neuroprotective in the TgCRND8 mouse model of Alzheimer's disease. J Neurochem. 2010;112:356–365. doi: 10.1111/j.1471-4159.2009.06487.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–339. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon A, Fraser P. The presenilins. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-11-reviews3014. reviews3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinás R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Guo C, Wang H, Chen X, Bezprozvanny I. Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model. J Neurosci. 2009;29:1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER calcium leak channels, a function disrupted by mutations linked to familial Alzheimer's disease. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. The cellular and molecular basis of store-operated calcium entry. Nat Cell Biol. 2002;4:E263–E272. doi: 10.1038/ncb1102-e263. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. CaV1.2 and CaV1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J Neurosci. 2006;23:2297–2310. doi: 10.1111/j.1460-9568.2006.04734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Weisleder N, Han X, Pan Z, Parness J, Brotto M, Ma J. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J Biol Chem. 2006;281:33477–33486. doi: 10.1074/jbc.M602306200. [DOI] [PubMed] [Google Scholar]