

Abstract
The origin of fibrogenic cells in liver fibrosis remains controversial. We assessed the emerging concept that hepatocytes contribute to production of extracellular matrix (ECM) in liver fibrosis through epithelial-mesenchymal transition (EMT). We bred triple transgenic mice expressing ROSA26 stop β-gal; Albumin Cre; Collagen α1(I) GFP, in which hepatocyte-derived cells are permanently labeled by β-galactosidase (β-gal) and type I collagen-expressing cells are labeled by GFP. We induced liver fibrosis by repetitive carbon tetrachloride (CCl4) injections. Liver sections and isolated cells were evaluated for GFP and β-gal as well as expression of α-smooth muscle actin (α-SMA) and fibroblast specific protein 1 (FSP-1). Upon stimulation with TGFβ-1, cultured hepatocytes isolated from untreated liver expressed both GFP and β-gal with a fibroblast-like morphological change but lacked expression of other mesenchymal markers. Cells from CCl4-treated livers never showed double-positivity for GFP and β-gal. All β-gal positive cells exhibited abundant cytoplasm, a typical morphology of hepatocytes and expressed none of mesenchymal markers including α-SMA, FSP-1, desmin, and vimentin. In liver sections of CCl4-treated mice, GFP-positive areas were coincident with fibrotic septa and never overlapped X-gal-positive areas. Conclusion: Type I collagen-producing cells do not originate from hepatocytes. Hepatocytes in vivo neither acquire mesenchymal marker expression, nor exhibit a morphological change clearly distinguishable from normal hepatocytes. Our results strongly challenge the concept that hepatocytes in vivo acquire a mesenchymal phenotype through EMT to produce the ECM in liver fibrosis.
Keywords: fibroblast, collagen, extracellular matrix, cell fate mapping
Liver fibrosis is the consequence of a sustained wound healing response to chronic liver injury, including viral, alcoholic, and autoimmune hepatitis(1). Progressive liver fibrosis leads to cirrhosis and its associated complications including portal hypertension, hepatic encephalopathy, and hepatocellular carcinoma. Currently several antifibrotic drugs are in development for the treatment of liver fibrosis but their efficacy has not been proven in patients(2). Further understanding of the cellular and molecular mechanism of liver fibrosis may lead to the development of more effective treatments.
A key issue in liver fibrosis is the origin of fibrogenic cells or activated myofibroblasts that produce extracellular matrix (ECM) such as type I collagen. Quiescent hepatic stellate cells (HSCs) are believed to be the main source of fibrogenic cells(3). However, there is accumulating evidence suggesting that HSCs are not the only origin. Portal myofibroblasts(4) or bone marrow-derived fibrocytes(5) could be other cellular sources of myofibroblasts in liver fibrosis. In addition, a new concept has been proposed that hepatocytes undergo a phenotypical change called epithelial-mesenchymal transition (EMT) to acquire a fibroblastic phenotype in liver fibrosis(6). Using a cell fate tracing technique it was demonstrated that hepatocyte-derived cells exhibited a fibroblast-like morphology accompanied by expression of fibroblast specific protein 1 (FSP-1). However, it is still unknown whether those hepatocyte-derived cells contribute to production of ECM in liver fibrosis and if FSP-1 is a marker for fibroblasts in fibrotic liver.
We have developed a reporter mouse in which green fluorescent protein (GFP) is expressed under collagen α1(I) promoter, enabling tracking of collagen-producing cells in vitro and in vivo(7, 8). The aim of the present study was to directly examine whether hepatocyte-derived cells express type I collagen in vitro and in vivo.
Materials and Methods
Materials
Recombinant transforming growth factor β-1 (TGFβ-1) was purchased from R&D systems (Minneapolis, MN). An adenovirus expressing Cre recombinase (Ad Cre) was generated by AdEasy™ adenoviral system (Stratagene, La Jolla, CA). Gliotoxin was purchased from Sigma-Aldrich (St. Louis, MO).
Animals
ROSA26 stop β-gal mice (stock number 003504), in which the β-galactosidase reporter gene is expressed under the ROSA26 promoter after Cre recombinase-mediated excision of the stop codon, were purchased from The Jackson Laboratory (Bar Harbor, ME)(9). Albumin-Cre mice (stock number 003574), in which Cre recombinase is expressed under the albumin promoter, were purchased from Jackson Laboratory. Coll GFP mice, in which GFP is expressed under collagen α1(I) promoter, were described previously(8).
Generation of Triple Transgenic Mice
A ROSA26 stop β-gal+/+ mouse was mated with an Alb Cre+/+ mouse to generate double transgenic mice: ROSA26 stop β-gal+/−; Alb Cre+/−. These double transgenic mice were mated with Coll GFP+/− mice. The offsprings were genotyped for ROSA26 stop β-gal and Alb Cre transgenes according to the protocols provided by Jackson Laboratory.
Presence of Coll GFP transgene is evaluated by observation of green fluorescence of the tails. Triple transgenic mice ROSA26 stop β-gal+/−; Alb Cre+/−; Coll GFP+/− were obtained in accordance with the Mendel’s law of inheritance. In these triple transgenic mice, cells that derived from albumin-expressing cells (i.e. hepatocyte-derived cells) are permanently labeled with β-galactosidase and collagen-expressing cells are labeled with GFP.
Isolation of Hepatocytes and Induction of EMT in vitro with TGFβ-1
Hepatocytes were isolated from the triple transgenic mice, as previously described(10). They were plated on collagen-coated plastic plates and cultured in Waymouth’s medium supplemented with 10% fetal bovine serum (FBS) and antibiotics/antimycotics solution. Twenty hours after plating, the culture medium was replaced with Waymouth’s medium supplemented with 0.5% FBS and 2 μg/ml insulin. The cells were cultured for 48 hours in the presence or absence of 3 ng/ml recombinant TGFβ-1.
TaqMan Real Time RT-PCR
Total RNA was extracted from cells by RNeasy Mini Kit with on-column DNA digestion (QIAGEN, Valencia, CA). Total RNA was reverse-transcribed to complementary DNA. Quantitative real time RT-PCR was performed using commercially available primer-probe sets and the ABI Prism 7000 Sequence Detector and software. The relative abundance of the target genes was obtained by calculating against a standard curve and normalized to 18S ribosomal RNA as an internal control.
Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis and Isolation of Liver Cells from Fibrotic Livers
Mice were injected intraperitoneally with 0.5μl of CCl4 per gram mouse (Sigma-Aldrich, St. Louis, MO) diluted in corn oil every 3 days to induce liver fibrosis. The CCl4-treated liver was perfused via the inferior vena cava sequentially with 0.04% pronase (EMD Chemicals, Gibbstown, NJ) and 0.05% collagenase (Roche, Indianapolis, IN). The liver was excised and further digested in 0.05% pronase and 0.05% collagenase solution with gentle stirring. The cell suspension was filtered through a cell strainer to obtain a “whole liver cell fraction”. An aliquot of the “whole liver cells” were centrifuged at 50 g for 1 minute to obtain a “hepatocyte fraction”. The supernatant was collected and centrifuged at 800 g for 5 minutes to get a “non parenchymal cell fraction”. The cells were washed, plated, and cultured overnight to allow attachment on plates. All animal studies have been approved by the University Committee on Use and Care of Animals at University of California, San Diego (S07088).
Evaluation for GFP and β-galactosidase Expression at Single Cell Level
We evaluated individual cells for both GFP expression by fluorescent microscopy andβ-galactosidase expression by X-gal staining. As GFP fluorescence is substantially interfered by 5-bromo-4-chloro-3-indolyl, the reaction product generated for X-gal staining(11), we could not photograph simultaneously for GFP and X-gal staining. Thus we photographed GFP first, then fixed the cell and reacted with X-gal. Matching fields for GFP images and X-gal images was the key issue to evaluate an individual cell for both GFP and X-gal. In order to match fields for GFP and X-gal staining, we put cross-striped scratches with a knife on the bottom of the plates before photographing for GFP. We photographed exactly matched fields for GFP and X-gal staining using the cross stripes as guides. Individual cells were identified morphologically and scored for positivity of GFP and X-gal staining. For liver sections, the livers were fixed with 4% neutral-buffered formalin, embedded in OCT compound, and sectioned at 6 μm. The sections were thawed in PBS, photographed for GFP, reacted with X-gal, and photographed for X-gal staining. Fields were matched for GFP and X-gal based on the alignment of the sections. X-gal staining was performed using the β-Gal staining kit (K1465-01, Invitrogen, Carlsbad, CA) according to manufacturer’s protocol.
Immunofluorescence
The cells were fixed with 4% paraformaldehyde, permeabilized with 0.05% Triton X-100, and blocked with Cytomation Protein Block Serum-Free (DAKO, Glostrup, Denmark). The primary antibody against α-smooth muscle actin (α-SMA) was clone 1A4 (DAKO) and that against fibroblast-specific protein 1 (FSP-1)(12) was a kind gift from Dr. Eric G. Neilson (Vanderbilt University, Nashville, TN). The primary antibody for desmin (RB9014) was purchased from LAB VISION (Fremont, CA). The primary antibodies were incubated overnight at 1:200 for α-SMA and desmin and 1:300 for FSP-1, respectively. The cells were then incubated with the respective secondary antibodies conjugated with Alexa Fluor 594 (red) (Invitrogen). In case we needed to combine GFP fluorescence images and immunostaining, we photographed for GFP before staining as GFP fluorescence is significantly attenuated after multiple washing steps of immunofluorescence. In order to match fields for GFP and immunofluorescence staining, we put scratches on the bottom of the plates before taking pictures for GFP.
Immunocytochemistry/Immunohistochemistry Combined with X-gal Staining
As β-galactosidase activity is attenuated after multiple washing and incubation steps of immunostaining, we performed X-gal staining first and immunostaining afterward. The cells were fixed with 4% paraformaldehyde and X-gal staining was performed. The cells were then permeabilized, blocked, incubated with a primary antibody. The primary antibodies for α-SMA, FSP-1, and desmin were described in the previous section. The primary antibody for vimentin (JM-3634-100) was purchased from MBL international (Woburn, MA) and used at 1:200. Biotin-conjugated secondary antibodies and streptavidin-biotin complex/horseradish peroxidase were bound. Diaminobenzidine was reacted to develop brown color. The blue precipitation of X-gal staining (5-bromo-4-chloro-3-indolyl) was stable during immunostaining procedures.
Fluorescence-Activated Cell Sorting (FACS)
The CCl4-treated liver was digested and whole liver cells were sorted by flow cytometry using a MoFlo (Beckman Coulter Miami, Florida) equipped with a Coherent Enterprise II Argon laser (488nm) and a processor module permitting doublet discrimination. GFP fluorescence was collected through a 530/40 nm bandpass filter. Cells were sorted at a rate of 30,000 events per second using a sort purify 1 drop window. GFP positive cells were sorted into Waymouth’s culture medium supplemented with 10% FBS and plated. X-gal staining was performed in the same manner as described above.
Sirius Red Staining
Liver tissues were fixed in 10% buffered formalin, embedded in paraffin and sectioned at 5μm thickness. Sections were stained with Sirius red solution (0.1% Direct Red 80 in saturated picric acid) to visualize collagen deposition.
Results
Primary Cultured Hepatocytes Exhibit Fibroblast-Like Morphological Change and Activate Collagen α1(I) Promoter upon Stimulation with TGFβ-1
Hepatocytes were isolated and cultured from triple transgenic mice ROSA26 stop β-gal; Alb Cre; and Coll GFP. Hepatocytes were GFP-negative before TGFβ-1 treatment or after 48 hours culture without TGFβ-1 (Figure 1, A, left and middle). Treatment of hepatocytes with 3 ng/ml TGFβ-1 for 48 hours induced a fibroblast-like morphological change (Figure 1, A, upper right), consistent with previous findings(6, 13). TGFβ-1 treatment not only induced a fibroblast-like morphological change, but also activated the collagen α1(I) promoter, as demonstrated by GFP expression in TGFβ-1-treated hepatocytes (Figure 1, A, middle right). Messenger RNA level of collagen α1(I) in TGFβ-1-treated hepatocytes was increased to a level comparable to culture-activated HSCs (Supplementary Figure S1, A). The increase was not blocked by gliotoxin, which eliminates potentially contaminating HSCs, as demonstrated by inhibition of the level of desmin, a marker for HSCs (Supplementary Figure S1, B). β-galactosidase expression of GFP-positive cells indicated that these cells were not contaminating non-parenchymal cells, but instead originally derived from hepatocytes (Figure 1, A, bottom, right). This result was confirmed by another experiment in which X-gal staining was followed by immunocytochemistry for GFP. Consistently, hepatocytes doubly positive for GFP and β-galactosidase appeared upon stimulation with TGFβ-1 (Figure 1, B, right). A few GFP-positive cells were seen even in the absence of TGFβ-1 (Figure 1, B, middle). However, they were never positive for β-galactosidase, indicating they were contaminating non-parenchymal cells.
Figure 1.

Collagen-Producing Cells in CCl4-Induced Liver Fibrosis Do Not Originate from Hepatocytes
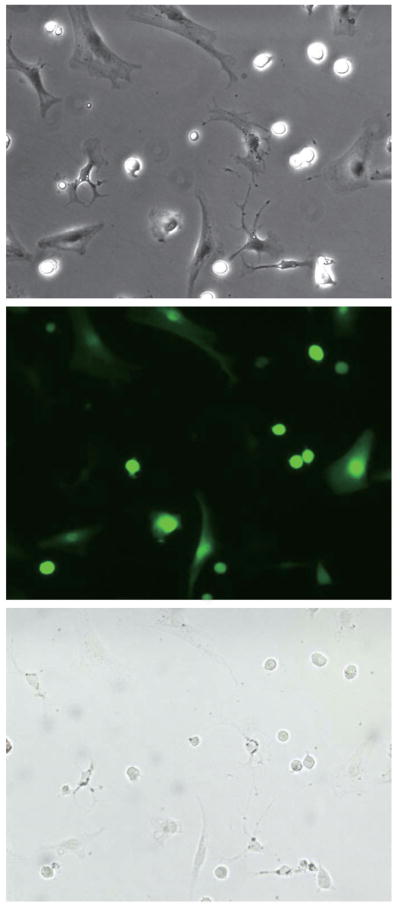
Liver fibrosis was induced in triple transgenic mice ROSA26 stop β-gal; Alb Cre; and Coll GFP by 8 injections with CCl4 (Supplementary Figure S2). GFP-positive cells (collagen-expressing cells) were seen along fibrotic septa (Figure 2, upper). However, they were never positive for β-galactosidase, as demonstrated by the lack of double positive cells in merged images of GFP and X-gal staining (Figure 2, bottom). Higher magnification images clearly show that the GFP-positive cells are present exclusively in X-gal-negative area (Supplementary Figure S3). Absence of hepatocyte-derived collagen-expressing cells (i.e. cells double positive for GFP and β-galactosidase) was confirmed by cell isolation; double positive cells were not seen in the whole liver cell fraction (Figure 3, A, left), hepatocyte fraction (Figure 3, A, middle), or non-parenchymal cell fraction (Figure 3, A, right). We photographed 224 different fields and evaluated 3,522 GFP-positive cells. None of them were positive for β-galactosidase. There were 1,737 β-galactosidase-positive cells in the evaluated fields and none of them were GFP-positve. Non-parenchymal cells expressed β-galactosidase upon Ad Cre-mediated excision of the stop codon (Figure 3, B), demonstrating that lack of X-gal staining in non parenchymal cells was due to persistence of the stop codon, not due to an insufficient level of β-galactosidase expression. This result eliminates the possibility that some GFP-positive cells were actually derived from hepatocytes but did not express sufficient β-galactosidase to be detected by X-gal staining. Absence of double positive cells was confirmed at different stages of liver injury; acute phase (single injection with CCl4) and chronic phase (16 times injection with CCl4), in both liver sections and in cells isolated from the injured liver (Supplementary Figure S4, S5, S6, and S7). Furthermore, GFP-positive cells were sorted by FACS and none of them (450,000 GFP-positive cells) were positive for β-galactosidase (Figure 4). These results clearly demonstrate that type I collagen expressing cells in CCl4-induced liver fibrosis do not originate from hepatocytes.
Figure 2.

Figure 3.

Figure 4.
Hepatocytes Do Not Express Mesenchymal Markers in CCl4-Induced Liver Fibrosis
To address if hepatocytes express mesenchymal markers during liver injury we performed immunostaining following X-gal staining. No cells were double positive for α-SMA and β-galactosidase in CCl4-treated liver of double transgenic mice (Figure 5, A). Similarly, no cells were double positive for FSP-1, desmin, or vimentin and β-galactosidase in CCl4-treated liver of double transgenic mice (Figure 5, B and Supplementary Figure S8). β-galactosidase positive cells in non-parenchymal cell fraction were never positive for α-SMA, FSP-1, desmin, or vimentin and must represent rare contaminating hepatocytes. Immunohistochemical staining and X-gal staining of liver sections supported absence of hepatocyte-derived α-SMA or FSP-1 positive cells in CCl4-treated liver (Supplementary Figure S9).
Figure 5.

Hepatocytes Do Not Express Mesenchymal Markers Even upon Stimulation with TGFβ-1 in vitro
Hepatocytes cultured in the presence of TGFβ-1 exhibited fibroblast-like morphological changes (Figure 6, A, upper) and expressed GFP driven by the collagen α1(I) promoter (Figure 6, A, second). Although some α-SMA positive cells were detected in the primary culture of hepatocytes (Figure 6, A, third), they appeared even in the absence of TGFβ-1 (data not shown) and were never positive for β-galactosidase (Figure 6, A, bottom), demonstrating that they were contaminating non-parenchymal cells and did not derive from hepatocytes. Similarly, hepatocytes treated with TGFβ-1 did not express FSP-1 (Figure 6, B, third) or desmin (Supplementary Figure S10, third). A few FSP-1 or desmin positive cells were present in the primary cultures of hepatocytes, but were never positive for β-galactosidase (Figure 6, B, bottom, and Supplementary Figure S10, bottom). Messenger RNA level of FSP-1 in hepatocyte culture was neither increased in a time dependent manner nor enhanced by addition of TGF β-1 (Supplementary Figure S11). This indicates that FSP-1 is not expressed by hepatocytes cultured on plastic dishes and FSP-1 is not induced by stimulation with TGFβ1.
Figure 6.

Discussion
Despite intense study, the origin of cells that produce the ECM in liver fibrosis is still a matter of debate. In the normal liver HSCs are characterized by vitamin A-containing lipid droplets and reside in a quiescent state in the space of Disse. Upon liver injury, HSCs acquire a myofibroblastic phenotype in response to inflammatory and profibrogenic stimuli, express activation markers including α-SMA, and synthesize ECM including type I collagens. Currently HSCs are considered to be the major source of myofibroblasts in the liver(3, 14). However, studies have demonstrated that HSCs are not the only cell type that contributes to the accumulation of ECM in liver fibrosis. Beaussier et al. suggested that portal mesenchymal cells contribute to liver fibrosis in ischemic or cholestatic liver injury(4). We recently identified fibrocytes as another potential source of ECM-producing population in the liver. These collagen-expressing cells derive from the bone marrow and migrate to the liver in response to injury(5).
An additional concept suggests that myofibroblasts can also originate from epithelial cells through phenotypical changes called EMT. Pioneering studies on EMT in the field of organ fibrosis was accomplished in the kidney(15), lens(16) and lung(17, 18). It was recently proposed that EMT also contributes to liver fibrosis(6, 13). Zeisberg et al. demonstrated that cells that formerly expressed albumin (that is, hepatocytes) acquire expression of FSP-1 in response to CCl4 in vivo or TGFβ-1 in vitro(6). Kaimori et al. reported that hepatocytes express collagen α1(I) in response to TGFβ-1 in vitro, and that Smad signaling mediates EMT (13) which was also reported by Dooley et al.(19). In addition, biliary epithelial cells were suggested to undergo EMT(20–22). Our current study did not investigate if cholangiocytes undergo EMT and contribute the pool of ECM-producing cells in the injured liver.
To the best of our knowledge, none of the above mentioned studies provide evidence that hepatocytes-derived cells express ECM genes and contribute to ECM production in liver fibrosis in vivo. For example, Zeisberg et al. relied solely on FSP-1 expression as a marker for fibrogenic cells(6). However, FSP-1 positive cells in the liver have not been characterized so far, and it is unknown if these cells synthesize ECM in vivo. Kaimori et al. showed that primary cultures of hepatocytes stimulated with TGFβ-1 express collagen α1(I) together with morphological changes consistent with EMT in vitro(13). However, as in all experiments with primary cultured cells, contamination with some other populations cannot be ruled out. Although this study also observed collagen α1(I) expression in a hepatocyte cell line as well, the increase in the expression level precedes and does not parallel the morphological changes of EMT. The transient increase in collagen α1(I) expression observed in the cell line might reflect a temporal activation of collagen α1(I) promoter irrelevant to EMT. Results from previous studies suggesting that cholangiocyte undergo EMT are inconclusive because they entirely rely on double immunofluorescence staining for cholangiocyte markers and surrogate markers for mesenchymal cells including FSP-1 (20–22). As pointed out above it remains unknown if FSP-1 positive cells express collagen and therefore also contribute to the ECM-producing cells in vivo. Cell fate tracking for cholangiocytes has not been performed so far and genetic evidence that cholangiocytes loose their epithelial characteristics and acquire a mesenchymal phenotype and start to synthesize ECM is therefore still missing. Our current study did not address the role of cholangiocytes in EMT.
To analyze if hepatocyte-derived cells indeed contribute to ECM production in liver fibrosis, we utilized a reporter mouse in which GFP is expressed under the murine collagen α1(I) promoter/enhancer in combination with a cell fate tracing technique used in the previous study by Zeisberg et al.(6). Our in vitro results using primary cultured hepatocytes initially appeared to support the concept of EMT. Hepatocytes not only exhibited fibroblast-like morphological changes, but also expressed collagen α1(I) in response to TGFβ-1. Cell fate tracing technique using ROSA26 stop β-gal and Alb Cre mice excluded the possibility that GFP-expressing cells were contaminating mesenchymal cells. However, the GFP-expressing hepatocytes did not express mesenchymal markers such as FSP-1 or α-SMA. A small number of the cells that became positive for FSP-1 or α-SMA in hepatocyte culture were not positive forβ-galactosidase and were rare contaminating cells. Thus, our observation that hepatocytes express collagen α1(I) with a “fibroblast-like” morphological change does not satisfy the definition of EMT as it is not associated with mesenchymal marker expression. A number of studies have demonstrated that cultured hepatocytes express mesenchymal markers in response to TGFβ-1(13, 23). However, these studies failed to show that cells positive for mesenchymal markers were truly hepatocyte-derived cells and do not represent contaminating cells. Only Zeisberg and colleagues employed the cell fate tracing technique to demonstrate that hepatocyte-derived cells express a mesenchymal marker (FSP-1)(6). The reliability of the immunostaining (FSP-1 andβ-galactosidase) is open to question (discussed later). The fact that we (Supplementary Figure S7) as well as Kaimori et al.(13) observed no increase in FSP-1 mRNA levels in hepatocytes treated with TGFβ-1 challenges the observation of Zeisberg et al. Taken together, our study in line with previous reports do not provide evidence that primary cultured hepatocytes undergo EMT and acquire expression of FSP-1.
More importantly, our in vivo experiments detected no hepatocyte-derived collagen type I expressing cells. Absence of double-positive (β-galactosidase and GFP) cells was verified at various stages of CCl4-induced liver injury (1, 8, and 16 injections). Thus, it is unlikely that hepatocyte-derived fibrogenic cells were actually present but their appearance was transient and escaped our notice. In addition, our experiments showed lack of hepatocyte-derived FSP-1-positive cells using the endogenous gene and not a transgene, which contradicts the previous study(6). The reason for this discrepancy is of importance. Zeisberg utilized double immunofluorescence staining to detect FSP-1 andβ-galactosidase. In contrast, we used X-gal staining in combination with immunocytochemistry for FSP-1 instead of double immunofluorescence. We tested two different antibodies that sufficiently detected adenovirally-expressed β-galactosidase, but neither was able to detect β-galactosidase in the ROSA26 stop β-gal mice that were used in the present as well as Zeisberg’s study. To obtain the blue signal in X-gal staining, the sections or cells required overnight incubation whereas just two hours were enough for hepatocytes or liver sections infected with adenovirus expressingβ-galactosidase. From these observations, we concluded that the expression level ofβ-galactosidase in the ROSA26 stop β-gal mice was not high enough to be detected with immunofluorescence and therefore X-gal staining was the method of choice. Thus, it can be concluded that the absence of hepatocyte-derived FSP-1 positive cells in our study was not a false negative caused by inappropriate methodology. Rather, we would suspect that immunofluorescence for β-galactosidase in Zeisberg’s study might be non-specific staining or bleed through from another fluorescent probe, since the staining patterns of the two different antigens are nearly identical (see Figure 5E in reference (6) and compare staining patterns of beta-gal and FSP-1). Furthermore, it is concerning that the staining pattern for β-galactosidase on liver sections does not overlap with that of X-gal staining (compare Figure 5C and D). Taken together, we suspect Zeisberg et al. drew incorrect conclusions by the limitations of their immunostaining.
We appreciate the limitation of our study as well. As we have already shown in a previous study employing Coll GFP and α-SMA double transgenic mice, GFP and RFP does not overlap entirely (7). However, we wish to emphasize that staining of α-SMA in this study was exclusively observed in GFP positive cells, suggesting difference between activity of the promoter used to generate α-SMA RFP mice and the expression of α-SMA protein. Thus exclusive reliance on a reporter mouse system might result in potentially missing collagen-producing mesenchymal cells. Another weakness of our study is that the cell fate tracing technique utilizing ROSA26 stop β-gal and Alb Cre mouse does not mark 100% of hepatocytes. It can therefore not be excluded that potentially hepatocyte-derived mesenchymal cells were overseen. However, we would like to reemphasize that Zeisberg et al. used the same methodology to trace hepatocytes and all mentioned shortcomings of this approach also apply for their study.
In conclusion, we believe that previous studies proposing EMT as a significant source for collagen producing myofibroblasts in the liver are limited by the approach, methodology and quality of data. These studies rely on double-immunofluorescence staining of epithelial markers and surrogate markers for EMT but lack functional evidence for ECM production by hepatocytes-derived cells. Our study clearly identifies collagen expressing cells, but none of which are derived from hepatocytes in vivo. Although we recognize the limitation of our study, our results strongly challenge the concept that hepatocytes in vivo acquire a mesenchymal phenotype through EMT to produce ECM in liver fibrosis.
Supplementary Material
Acknowledgments
This research was supported by US National Institutes of Health grant R01GM041804.
Abbreviations used
- α-SMA
α-smooth muscle actin
- CCl4
carbon tetrachloride
- ECM
extracellular matrix
- EMT
epithelial-mesenchymal transition
- FBS
fetal bovine serum
- FSP-1
fibroblast specific protein 1
- GFP
green fluorescent protein
- HSC
hepatic stellate cell
- TGFβ-1
transforming growth factor β-1
Footnotes
No conflicts of interest exist.
The authors have no financial relationship to disclose.
Contributor Information
Kojiro Taura, Email: ktaura@kuhp.kyoto-u.ac.jp, ktaura@ucsd.edu.
Kouichi Miura, Email: kmiura@ucsd.edu.
Keiko Iwaisako, Email: kiwaisako@ucsd.edu.
Christoph H Österreicher, Email: coesterr@ucsd.edu.
Yuzo Kodama, Email: ykodama@ucsd.edu.
Melitta Penz-Österreicher, Email: mpenzoes@ucsd.edu.
David A Brenner, Email: dbrenner@ucsd.edu.
References
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. 1985;82:8681–8685. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beaussier M, Wendum D, Schiffer E, Dumont S, Rey C, Lienhart A, Housset C. Prominent contribution of portal mesenchymal cells to liver fibrosis in ischemic and obstructive cholestatic injuries. Lab Invest. 2007;87:292–303. doi: 10.1038/labinvest.3700513. [DOI] [PubMed] [Google Scholar]
- 5.Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, Brenner DA. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 7.Magness ST, Bataller R, Yang L, Brenner DA. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology. 2004;40:1151–1159. doi: 10.1002/hep.20427. [DOI] [PubMed] [Google Scholar]
- 8.Yata Y, Scanga A, Gillan A, Yang L, Reif S, Breindl M, Brenner DA, et al. DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells. Hepatology. 2003;37:267–276. doi: 10.1053/jhep.2003.50067. [DOI] [PubMed] [Google Scholar]
- 9.Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci U S A. 1999;96:5037–5042. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osawa Y, Uchinami H, Bielawski J, Schwabe RF, Hannun YA, Brenner DA. Roles for C16-ceramide and sphingosine 1-phosphate in regulating hepatocyte apoptosis in response to tumor necrosis factor-alpha. J Biol Chem. 2005;280:27879–27887. doi: 10.1074/jbc.M503002200. [DOI] [PubMed] [Google Scholar]
- 11.Matei VA, Feng F, Pauley S, Beisel KW, Nichols MG, Fritzsch B. Near-infrared laser illumination transforms the fluorescence absorbing X-Gal reaction product BCI into a transparent, yet brightly fluorescent substance. Brain Res Bull. 2006;70:33–43. doi: 10.1016/j.brainresbull.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089–22101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- 14.Friedman SL, Rockey DC, McGuire RF, Maher JJ, Boyles JK, Yamasaki G. Isolated hepatic lipocytes and Kupffer cells from normal human liver: morphological and functional characteristics in primary culture. Hepatology. 1992;15:234–243. doi: 10.1002/hep.1840150211. [DOI] [PubMed] [Google Scholar]
- 15.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW. Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs. 2005;179:43–55. doi: 10.1159/000084508. [DOI] [PubMed] [Google Scholar]
- 17.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, Ueberham E, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135:642–659. doi: 10.1053/j.gastro.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 20.Omenetti A, Porrello A, Jung Y, Yang L, Popov Y, Choi SS, Witek RP, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008 doi: 10.1172/JCI35875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rygiel KA, Robertson H, Marshall HL, Pekalski M, Zhao L, Booth TA, Jones DE, et al. Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest. 2008;88:112–123. doi: 10.1038/labinvest.3700704. [DOI] [PubMed] [Google Scholar]
- 22.Robertson H, Kirby JA, Yip WW, Jones DE, Burt AD. Biliary epithelial-mesenchymal transition in posttransplantation recurrence of primary biliary cirrhosis. Hepatology. 2007;45:977–981. doi: 10.1002/hep.21624. [DOI] [PubMed] [Google Scholar]
- 23.Nitta T, Kim JS, Mohuczy D, Behrns KE. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology. 2008;48:909–919. doi: 10.1002/hep.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.