

Abstract

Enantioenriched allenylsilanes are used as carbon nucleophiles in three component reactions with in situ generated N-sulfonylimines to selectively form syn-homopropargylic sulfonamides. The reactions proceed with a variety of aldehyde and sulfonamide reaction partners. These novel reaction products are obtained with useful levels of diastereoselectivity, and the axial chirality of the allenylsilane is fully transferred to point chirality, forming products with >97% ee.
Recent developments in the synthesis and use of allenylmetal reagents have allowed their re-emergence as an important class of reagents for a variety of organic transformations, capable of producing highly functionalized alkynes and heterocycles.1 In particular, allenylsilanes have been used as carbon nucleophiles in additions to a variety of oxonium ions, producing homopropargylic alcohols,2 homopropargylic ethers,3 furans,4 and dihydrofurans.5 In cases where enantioenriched allenylsilane reagents were used, the axial chirality of the allene was typically fully transferred into point chirality in the products, leading to a variety of highly enantioenriched building blocks for organic synthesis.
The reaction of allenylmetal reagents with iminium ions has been underdeveloped. The addition of allenylsilanes to N-acyl iminium ions has been shown to favor either a [3 + 2] annulation pathway, providing dihydropyrroles, or a [3 + 3] pathway to provide dihydrooxazines.5,6 Achiral allenylsilanes and stannanes have also been used in a [3 + 2] annulation with a N-sulfonylimines,7 and in the case of the stannane7a the formation of the acyclic propargylation product was favored under certain reaction conditions. In these reported cases, a single imine reaction partner was used. Experiments with allenylboranes have been shown to specifically form homopropargylic amines,8 and in one case where enantioenriched allenylboranes were used, highly enantioenriched products were obtained.9
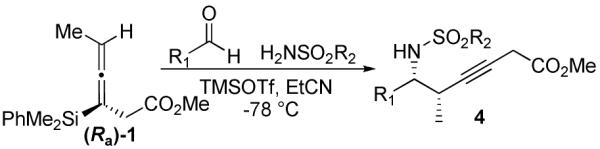
In our continued interest in developing chiral carbon nucleophiles as reagents to enhance the field of acyclic stereocontrol, we sought to acquire a method for the synthesis of homopropargylic amines from enantioenriched allenylsilanes. We have previously reported a multigram synthesis of enantioenriched allenylsilane 1.3 In exploring the [3 + 2] annulation of this reagent with N-acyl iminium ions we learned that the acyclic propargylation product was obtained as a byproduct under certain conditions. This led us to seek reaction conditions that would selectively lead to homopropargylic amines. Herein we report the achievement of that objective, forming the desired products in high yields with full transfer of chirality (axial to point), thereby complementing existing methods utilizing allyl and crotyl silanes that produce homoallylic amines.10
When allenylsilane (Sa)-1 was reacted with an iminium ion formed in situ from methyl carbamate and 2-bromobenzaldehyde in the presence of TMSOTf, the result was a mixture of homopropargylic amine 2a (Scheme 1) and dihydrooxazine 2b. When the solvent was changed from EtCN to DCM the oxazine is formed almost exclusively, however after considerable experimentation we were unable to find conditions that lead to selctive formation of the the homopropargylic amine. Accordingly, a number of amine sources were screened, with the hope of finding conditions that would result in an exclusive propargylation pathway. The most promising amine source for these reactions proved to be sulfonamides.11
Scheme 1.
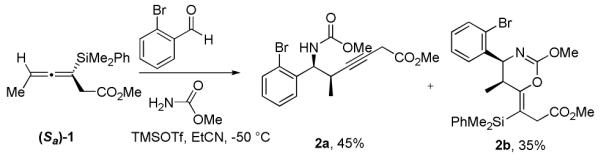
Reactions with N-Acyl Iminium Ions
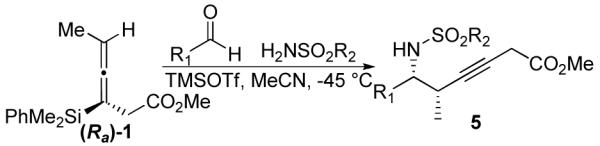
When the combination of aliphatic aldehydes and methanesulfonamide were used for the in situ formation of iminium ions mediated by TMSOTf, followed by reaction with allenylsilane (Ra)-1, the result was nearly exclusive formation of homopropargylic sulfonamides 3a-3h. This reaction was effective for a variety of aldehyde partners, including secondary aliphatic aldehydes (Table 1, entries 1-2) which gave high yields and form the products as a single diastereomer. Primary aliphatic aldehydes (entries 3-6) gave moderate to high yields, and lower but still useful levels of diastereoselectivity. Tertiary aliphatic aldehydes (entries 7-8) also gave high yields and a single diastereomer, although the reactions required slightly higher temperatures to reach completion.
Table 1.
Enantioselective Propargylations with Methanesulfonamide
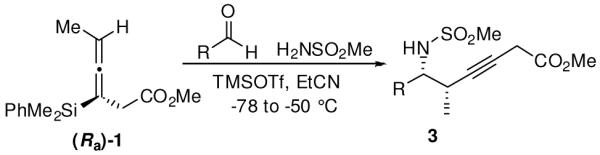 | ||||
|---|---|---|---|---|
| entry | aldehyde | yield (%)a | drb | product |
| 1 |
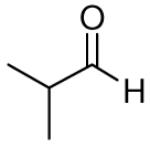
|
81 | >20:1 | 3a |
| 2 |
|
77 | >20:1 | 3b |
| 3 |
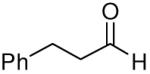
|
81 | 10:1 | 3c |
| 4 |
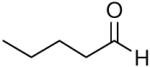
|
69 | 5:1 | 3d |
| 5 |
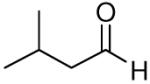
|
64 | 7:1 | 3e |
| 6 |
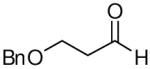
|
65 | 9:1 | 3f |
| 7 |
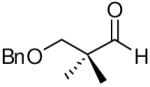
|
82c | >20:1 | 3g |
| 8 |
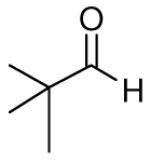
|
82c | >20:1 | 3h |
Isolated yields after purification over silica gel.
Diastereomeric ratios determined by 1H NMR analysis on crude material.
Reaction run at −45 °C in MeCN.
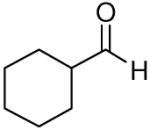
Propargylation reactions with aliphatic aldehydes were also tolerant to a number of different sulfonamides, in each case exclusively forming the homopropargylic sulfonamide. For instance, reactions with p-toluenesulfonamide, benzenesulfonamide and 4-nitrobenzenesulfonamide all afforded the desired propargylation products in moderate to high yield, with a single diastereomer observed for branched systems and moderate diastereoselectivity for unbranched aliphatic systems (Table 2). Reactions with cyclohexanecarboxaldehyde and hydrocinnamaldehyde gave higher yields when conducted at lower temperature (−78 °C), while experiments with trimethylacetaldehyde needed to be run at higher temperatures to help drive the reactions to completion.
Table 2.
Enantioselective Propargylations with Other Sulfonamides
 | |||||
|---|---|---|---|---|---|
| entry | aldehyde(R1) | R2 | yield(%)a | drb | product |
| 1 | Cyclohexanecarboxaldehyde | p-Tolyl | 80 | >20:1 | 4a |
| 2 | Trimethylacetaldehyde | p-Tolyl | 76c | >20:1 | 4b |
| 3 | Hydrocinnamaldehyde | p-Tolyl | 68 | 5:1 | 4c |
| 4 | Cyclohexanecarboxaldehyde | Ph | 72 | >20:1 | 4d |
| 5 | Trimethylacetaldehyde | Ph | 83c | >20:1 | 4e |
| 6 | Hydrocinnamaldehyde | Ph | 78 | 5:1 | 4f |
| 7 | Cyclohexanecarboxaldehyde | 4-No2Ph | 69 | >20:1 | 4g |
| 8 | Trimethylacetaldehyde | 4-No2Ph | 74c | >20:1 | 4h |
| 9 | Hydrocinnamaldehyde | 4-No2Ph | 71 | 6:1 | 4i |
Isolated yields after purification over silica gel.
Diastereomeric ratios determined by 1H NMR analysis on crude material.
Reaction run at −45 °C in MeCN.
The observed syn stereochemistry can be rationalized by open transition states using antiperiplanar or synclinal orientations of reaction partners (Figure 1), where allenylsilane (Ra)-1 is shown adding to the Re face of the iminium ion. While either transition state nicely illustrates the stereochemical course of the reaction, the synclinal transition state, where the destabilizing interaction between the R group on the iminium ion and the methylene and ester groups of the allene is minimized, may be the best illustration.12
Figure 1.
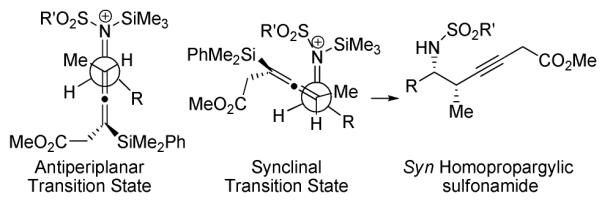
Transition States
Chiral HPLC analysis was used to confirm that the axial chirality in the allenylsilane was transferred into point chirality in the homopropargylic sulfonamide products. In that context, select products have been used to show how the chirality of either enantiomer of allenylsilane 1 is fully transferred to the propargylation products (Scheme 2). All of the homopropargylic sulfonamides were found to have >97% ee. The relative and absolute stereochemistry of the homopropargylic amines was confirmed by comparison to known compounds.12
Scheme 2.
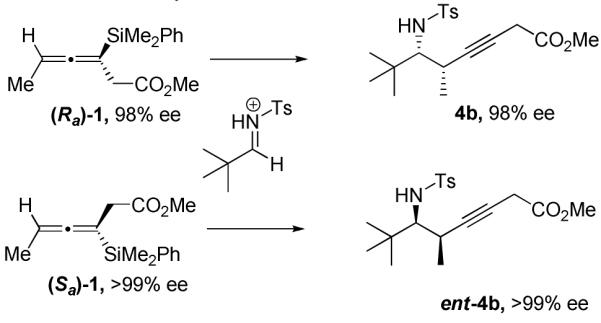
Ee analysis of each enantiomer of product 4b
While the propargylation reactions with aliphatic aldehydes were very effective, aromatic aldehydes have proven to be more difficult, with diminished reaction rate and selectivity. For instance, when many aromatic aldehydes were treated with TMSOTf in the presence of a sulfonamide, a large amount of an unidentified precipitate was observed, and product formation was sluggish or not observed. While small amounts of the desired products could be obtained when by increasing the reaction temperature, this typically led to decomposition of the starting materials. Despite these setbacks, we were able to isolate the desired homopropargylic sulfonamide products in moderate yields and diastereoselectivities (Table 3). The lower yields in the propargylations with aromatic aldehydes may be a result of several factors, ranging from sluggish reactions, extensive decomposition of reactants and products, and a competing reaction, which resulted in the formation of lactone 6.
Table 3.
Propargylations with Aryl Sulfonyl Imines
 | |||||
|---|---|---|---|---|---|
| entry | aldehyde (R1) | R2 | yield (%)a | drb | product |
| 1 |
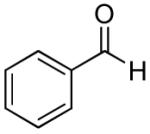
|
Me | 47 | 10:1 | 5a |
| 2 |
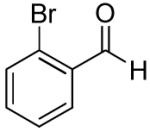
|
Me | 59 | 13:1 | 5b |
| 3 |
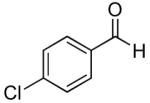
|
Me | 48 | 10:1 | 5c |
| 4 |
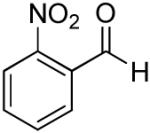
|
Me | 28 | 10:1 | 5d |
| 5 |
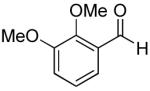
|
Me | 33 | 6:1 | 5e |
| 6 |
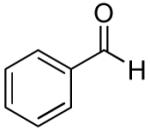
|
p-Tolyl | 57 | 7:1 | 5f |
| 7 |
|
p-Tolyl | 38 | 7:1 | 5g |
Isolated yields after purification over silica gel.
Diastereomeric ratios determined by 1H NMR analysis on crude material.
When MeCN or EtCN were used as solvents, lactone 6 was obtained as a minor byproduct, but it became the predominant product when the solvent was switched to DCM. The use of pre-formed aromatic imines resulted in the formation of lactone 6 in moderate to high yields (Table 4).13 The structure of this material was assigned based on 2-D NMR studies, and confirmed by x-ray crystallography of compound 6h (Scheme 3).12
Table 4.
Reactions with Pre-Formed Aryl Sulfonyl Imines
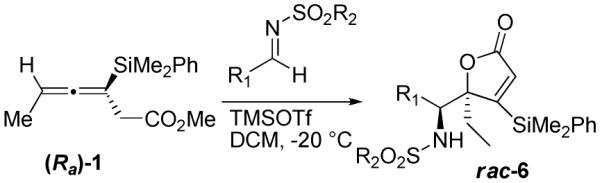 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | yield (%)a | productb |
| 1 | Ph | Me | 47 | 6a |
| 2 | 2-BrPh | Me | 72 | 6b |
| 3 | Ph | p-Tolyl | 82 | 6c |
| 4 | 2-BrPh | p-Tolyl | 77 | 6d |
| 5 | 4-MePh | p-Tolyl | 69 | 6e |
| 6 | 2,3-OMePh | p-Tolyl | 55 | 6f |
| 7 | 2-NO2Ph | p-Tolyl | 47 | 6g |
| 8 | 2-BrPh | p-NO2Ph | 49 | 6h |
| 9 | t-Bu | p-Tolyl | 0 | -- |
Isolated yields after purification over silica gel.
Scheme 3.
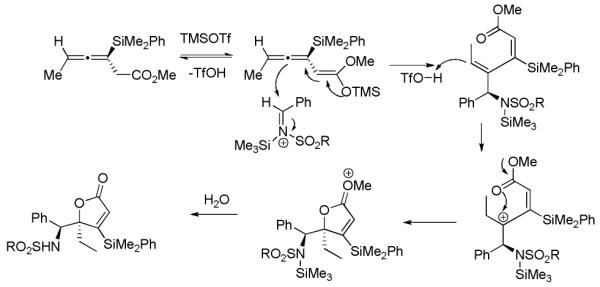
Proposed Mechanism for γ-Lactone Formation
Interestingly, when a pre-formed aliphatic imine was used under identical reaction conditions, no reaction was observed, and the starting materials were recovered (Table 4, entry 9).
A proposed mechanism for the formation of lactone 6 begins with silyl ketene acetal formation, promoted by TMSOTf (Scheme 3). The resulting silyl ketene acetal then attacks the iminium ion, which explains the addition to the central allene carbon, which has not been observed in alleylmetal chemistry to the best of our knowledge. Protonation of the trisubstituted olefin results in a tertiary carbocation, which is trapped by the ester, resulting in the quaternary center and formation of the observed lactone upon aqueous workup. For these examples transfer of chirality is lost, as racemic products are observed, which most likely arises from a steric argument, as the silyl ketene acetal can not differentiate between the π-faces of the iminium ion.
In summary, we have described a three component reaction of aldehydes, sulfonamides, and enantioenriched allenylsilanes, resulting in the selective formation of syn homopropargylic sulfonamides. The products were formed in high yield and moderate to high selectivity when aliphatic aldehydes were used. The formation of a γ-lactone is also described by a new reaction pathway for allenylmetal reagents. Current work is focused on expanding the functionality of the allenylsilane, which would expand the diversity of the resulting reaction products.
Supplementary Material
Acknowledgment
Financial support was obtained from NIH CA 53604. The authors are grateful to Will Youngsaye, Yun Zhang and Dr. Paul Ralifo at Boston University for helpful discussions and assistance with 2D NMR experiments and gratefully acknowledge Professor Karen Allen and Dr. Ezra Peisach (Boston University) for obtaining x-ray crystal data.
Footnotes
Supporting Information Available Experimental data and selected spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1).For recent reviews on allene chemistry see: Hassan HHAM. Curr. Org. Synth. 2007;4:413–439. Ma S. Chem. Rev. 2005;105:2829–2872. doi: 10.1021/cr020024j. Zimmer R, Dinesh CU, Nandanan E, Khan FA. Chem. Rev. 2000;100:3067–3125. doi: 10.1021/cr9902796.
- (2).(a) Danheiser RL, Carini DJ, Kwasigroch CA. J. Org. Chem. 1986;51:3870–3878. [Google Scholar]; (b) Marshall JA, Maxson K. J. Org. Chem. 2000;65:630–633. doi: 10.1021/jo991543y. [DOI] [PubMed] [Google Scholar]
- (3).Brawn RA, Panek JS. Org. Lett. 2007;9:2689–2692. doi: 10.1021/ol070936d. [DOI] [PubMed] [Google Scholar]
- (4).Danheiser RL, Stoner EJ, Koyama H, Yamashita DS, Klade CA. J. Am. Chem. Soc. 1989;111:4407–4413. [Google Scholar]
- (5).Danheiser RL, Kwasigroch CA, Tsai Y-M. J. Am. Chem. Soc. 1985;107:7233–7235. [Google Scholar]
- (6).Brawn RA, Panek JS. Org. Lett. 2009;11:473–476. doi: 10.1021/ol802618p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Fuchibe K, Hatemata R, Akiyama T. Tetrahedron Lett. 2005;46:8563–8566. [Google Scholar]; (b) Daidouji K, Fuchibe K, Akiyama T. Org. Lett. 2005;7:1051–1053. doi: 10.1021/ol047343c. [DOI] [PubMed] [Google Scholar]
- (8).(a) Yamamoto Y, Ito W, Maruyama K. J. Chem. Soc., Chem. Commun. 1984:1004–1005. [Google Scholar]; (b) Nikam SS, Wang KK. J. Org. Chem. 1985;50:2193–2195. [Google Scholar]; (c) Brown HC, Khire UR, Narla G, Racherla US. J. Org. Chem. 1995;60:544–549. [Google Scholar]
- (9).Gonzalez AZ, Soderquist JA. Org. Lett. 2007;9:1081–1084. doi: 10.1021/ol070074g. [DOI] [PubMed] [Google Scholar]
- (10).(a) Panek JS, Jain NF. J. Org. Chem. 1994;59:2674–2675. [Google Scholar]; (b) Schaus JV, Jain N, Panek JS. Tetrahedron. 2000;56:10263–10274. [Google Scholar]; (c) Lipomi DJ, Panek JS. Org. Lett. 2005;7:4701–4704. doi: 10.1021/ol051885s. [DOI] [PubMed] [Google Scholar]; (d) Berger R, Rabbat PMA, Leighton JL. J. Am. Chem. Soc. 2003;125:9596–9597. doi: 10.1021/ja035001g. [DOI] [PubMed] [Google Scholar]; (e) Shirakawa S, Berger R, Leighton JL. J. Am. Chem. Soc. 2004;127:2858–2859. doi: 10.1021/ja042522a. [DOI] [PubMed] [Google Scholar]; (f) Veenstra SJ, Schmid P. Tetrahedron Lett. 1997;38:997–1000. [Google Scholar]; (g) Phukan P. J. Org. Chem. 2004;69:4005–4006. doi: 10.1021/jo0498462. [DOI] [PubMed] [Google Scholar]
- (11).In addition to carbamates and sulfonamides, benzyl amines, allyl amines, acetamides, and anilines were screened, but all of these nitrogen sources showed little or no reactivity.
- (12).See supporting information for expanded transition state analysis, HPLC traces, ee analysis, crystal structure data, and assignment of the relative and absolute stereochemistry of the products.
- (13).Lee KY, Lee CG, Kim JN. Tetrahedron Lett. 2003;44:1231–1234. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.