

Abstract
This study used recombinant A1A2A3 tri-domain proteins to demonstrate that A domain association in von Willebrand factor (VWF) regulates the binding to platelet glycoprotein Ibα (GPIbα). We performed comparative studies between wild type (WT) A1 domain and the R1450E variant that dissociates the tri-domain complex by destabilizing the A1 domain. Using urea denaturation and differential scanning calorimetry, we demonstrated the destabilization of the A1 domain structure concomitantly results in a reduced interaction among the three A domains. This dissociation results in spontaneous binding of R1450E to GPIbα without ristocetin with an apparent KD of 85 ± 34 nm, comparable with that of WT (36 ± 12 nm) with ristocetin. The mutant blocked 100% ristocetin-induced platelet agglutination, whereas WT failed to inhibit. The mutant enhanced shear-induced platelet aggregation at 500 and 5000 s−1 shear rates, reaching 42 and 66%, respectively. Shear-induced platelet aggregation did not exceed 18% in the presence of WT. A1A2A3 variants were added before perfusion over a fibrin(ogen)-coated surface. At 1500 s−1, platelets from blood containing WT adhered <10% of the surface area, whereas the mutant induced platelets to rapidly bind, covering 100% of the fibrin(ogen) surface area. Comparable results were obtained with multimeric VWF when ristocetin (0.5 mg/ml) was added to blood before perfusion. EDTA or antibodies against GPIbα and αIIbβ3 blocked the effect of the mutant and ristocetin on platelet activation/adhesion. Therefore, the termination of A domain association within VWF in solution results in binding to GPIba and platelet activation under high shear stress.
Keywords: Adhesion, Mutant, Platelet, Protein Domains, von Willebrand Factor, Glycoprotein Ib, Flow Chamber
Introduction
Platelet adhesion at sites of vascular injury contributes to the arrest of bleeding as well as to the pathologic occlusion of diseased vessels under elevated shear stress. Under this high shear stress, the platelet-von Willebrand factor (VWF)3 interaction is essential for platelet adhesion. The interaction between VWF and the exposed subendothelium permits the VWF to interact with circulating platelets via the receptor glycoprotein (GP)Ib/IX/V complex (1).
Mature VWF consists of a 2050-residue subunit that contains domains that are arranged in the order D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK (cystine knot) (2–4). The VWF contains a triplicate repeat sequence or A domains in the central portion of the subunit. Binding sites for platelet GPIbα, heparin, sulfatides, and collagen (5–9) are within the A1 domain, whereas its homologous A3 domain only binds to collagen, and the A2 domain contains the cleavage site for the metalloprotease ADAMTS-13 (10–12). The functions of these A domains relevant to the biology of VWF have been characterized by individual recombinant expression of each of the A domains (9, 10, 13).
The interaction between VWF and GPIbα occurs when the binding site for GPIbα in the A1 domain of VWF has been exposed by the influence of high fluid forces or when the multimeric protein has been immobilized (14). This activation can also be induced by naturally occurring gain-of-function mutations in the A1 domain that causes Type 2B von Willebrand disease, which increases the binding affinity for platelet GPIbα (15). VWF activation can also be induced artificially with the modulator, ristocetin (16).
Previously, we described that isolated A2 domain binds to A1 domain and blocks the interaction with platelet GPIbα (17). Based on this observation, we proposed that an interdomain interaction between A1 and A2 domains may be a regulatory mechanism for the activation of VWF in solution. We have identified the gain-of-function mutation R1450E for which GPIbα binding activity has been characterized with a low binding activity for purified A2 domain (8, 18). Here, we have used recombinant A1A2A3 tri-domains to demonstrate that the R1450E mutation alters both the stability of A1 and its interdomain interaction with both the A2 and A3 domains, and like Type 2B VWF, it induces spontaneous binding to GPIbα. Moreover, this spontaneous binding of the monomeric A1A2A3 mutant potentiated platelet activation in a shear stress-dependent manner. The results described here show that the destabilization of A1 by the R1450E mutation alters domain association within the A1A2A3 tri-domain and enhances the binding to GPIbα, demonstrating a novel coupling between A1 domain stability and tri-domain association. In addition, the A1A2A3 segment used in this study appears to be the smallest size of VWF that contains this regulatory mechanism for the activation of VWF in solution.
EXPERIMENTAL PROCEDURES
Antibodies and Proteins
Antibodies 6D1, 7E3, and 10E5 were gifts from Dr. Barry Coller (The Rockefeller University, New York, NY). Monoclonal antibody 5D2 was obtained from Dr. Michael C. Berndt (University College Cork, Ireland). Monoclonal antibody VP-1 (19) against human VWF-A2 domain was a gift from Dr. Z. M. Ruggeri (Scripps Research Institute, La Jolla, CA). Human fibrinogen was from Calbiochem. The recombinant A domain proteins (WT-A1, R1450E-A1 mutant, -A2, and -A3) were obtained as previously described (8, 10, 17). The recombinant A1A2A3 (WT and R1450E mutant) proteins were expressed in mammalian (HEK293) cells and purified from the conditioned medium as we previously described (18, 20). The purity and the monomeric state of the recombinant proteins were verified by SDS-gel electrophoresis and gel filtration chromatography. The gel filtration analysis was conducted using an Amersham Biosciences Bioscience ÄKTA prime chromatography system equipped with two Superose 6TM 10/300 GL columns in tandem (GE Healthcare) at a constant flow rate of 0.5 ml/min. The samples were diluted in 25 mm Tris-HCl, 150 mm NaCl, pH 7.4 (TBS).
Binding Assays
The analyses of the interaction between the A1 variants and A2 protein were performed as we described elsewhere (17). Briefly, the microtiter wells were coated with A2 or A1 protein (5 μg/ml) in 65 mm sodium phosphate buffer, pH 6.5, and blocked with 3% (w/v) bovine serum albumin (BSA). The A1 or A2 protein bound to immobilized A2 or A1 protein, respectively, was detected by enzyme-linked immunosorbent assay with antibody 5D2 for A1 domain or anti-human A2 antibody for A2 domain. Previously, we characterized the A1-R1450E mutant and demonstrated that this mutation does not affect the binding capacity for conformation-specific monoclonal antibody 5D2 (8, 18).
For competition assays, microtiter wells containing immobilized A2 protein (5 μg/ml) were incubated with increasing concentrations of the WT or R1450E mutant mixed with a fixed concentration of biotinylated WT-A1 protein (0.25 μm). Bound biotinylated WT-A1 was detected using neutravidin-horseradish peroxidase conjugate (Pierce).
Monoclonal antibody VP-1 was diluted to 5 μg/ml with 50 mm carbonate buffer, pH 9.6, and used to coat the wells of a microtiter plate. Coating was carried out overnight at 4 °C. The wells were washed with TBS-Tween and blocked with 3% BSA, 0.1% Tween 20 for 60 min at 37 °C. Increasing concentrations of each WT or mutant A1A2A3 were incubated for 60 min at 37 °C. After incubation, wells were then washed with TBS-Tween, and the bound A1A2A3 was detected by enzyme-linked immunosorbent assay using polyclonal anti-VWF-horseradish peroxidase conjugate (Dako). The wells were again washed, and the substrate (o-phenylenediamine, Sigma) was added. Substrate conversion reactions were stopped with 0.025 ml of 2 n H2SO4, and the plates were read at 490 nm. Net specific binding was determined by subtracting optical density values from wells coated only with BSA from the total binding values obtained.
Platelets Binding Assay
This assay was similarly performed as we described elsewhere (17). Briefly, microtiter wells were coated with 75 μl of a suspension of fixed platelets. After 1 h of incubation at 37 °C, the wells were washed 1 time and then incubated with 1% paraformaldehyde. After 15 min of incubation, the wells were washed with TBS, and the wells were blocked by the addition of 3% (w/v) BSA for 1 h at 37 °C. To examine the binding of A1A2A3 proteins to GPIbα, increasing concentrations of each of the protein were mixed with ristocetin (0.5 mg/ml) or TBS and incubated in the platelet-coated wells for 1 h at 37 °C. A polyclonal anti-VWF-horseradish peroxidase conjugated (Dako, Carpinteria, CA) was used for detection of the A1A2A3 proteins. Net specific binding was determined by subtracting optical density values from wells coated only with BSA from the total binding values obtained in the wells coated with the corresponding protein. The software KaleidaGraph 4.0 was used for curve-fitting and to determine the approximate half-maximal binding values using the equation y = Bmax × x/(Kd + x). These values were reported as the mean ± S.E.
Ristocetin-induced Platelet Agglutination
Platelet agglutination was carried out in siliconized glass cuvettes at 37 °C with constant stirring at 1200 rpm in an eight-channel aggregometer (Bio/Data Corp. Horsham, PA). A suspension of platelet-rich plasma (PRP) containing different concentrations of each WT or mutant A1A3A3 protein was prepared. After 5 min of incubation at 37 °C, agglutination was initiated by the addition of ristocetin (Helena Laboratories, Beaumont, TX) to a final concentration of 1 mg/ml.
Shear-induced Platelet Aggregation
PRP was mixed with either WT or mutant A1A2A3 protein (250 nm) or buffer and incubated for 2 min at room temperature. Then the mixture was loaded onto a cone-and-plate viscometer (RS1, HAAKE Instrument Inc., Paramus, NJ) and exposed to shear stress of 500 or 5000 s−1 for 1 min. In some experiments the antibody 7E3 (20 μg/ml) was added to PRP. Shear-induced platelet aggregation was determined as the percentage reduction of single platelets as compared with the unsheared controls from the same donors as described elsewhere (21). Briefly, 10 μl of sample was fixed in 0.5% glutaraldehyde, and the platelet count was determined by a Coulter Epics XL-MCL flow cytometer (Beckman-Coulter).
Preparation of Protein-coated Coverslips
The plates coated with fibrinogen or A1A2A3 proteins were prepared as we previously described. Human fibrinogen was diluted to 100 μg/ml in 65 mm sodium phosphate buffer, pH 6.5, and added to 35-mm culture dishes and incubated for 1 h at 37 °C. After washing twice with phosphate-buffered saline, the plates were blocked with 3% BSA in phosphate-buffered saline.
Flow Assays
To obtain the blood, approval was obtained from the Baylor College of Medicine institutional review board for these studies. Informed consent was provided according to the Declaration of Helsinki. Perfusion assays were carried out as we described elsewhere (17, 22). One ml of citrated whole blood containing WT, mutant, or buffer (TBS) was perfused over the analyte-surface-coated plate followed by TBS or phosphate-buffered saline. Tethered platelets were observed with phase contrast objectives and recorded by videomicroscopy. In some assays the number of platelets tethered to the surface was determined by overlaying a 36-square grid on 6 frames and counting and averaging the number of platelets in 12 randomly selected squares. In others the surface area covered by deposited platelets was determined by a computer-assisted program. In some experiments whole blood mixed with the mutant protein or buffer was incubated with 10 units/ml apyrase (grade III, Sigma), 5 mm EDTA, 10 μm prostacyclin (prostaglandin I2), and 20 μg/ml concentrations of antibodies 10E5 or 6D1 for 5 min before perfusion. In another experiments ristocetin (0.5 mg/ml) was added to whole blood right before to initiate perfusion. Some experiments were performed in triplicate or quadruplicate using different blood donors.
Protein Biotinylation and Quantitation
Protein biotinylation was performed as directed by the manufacturer, and concentration was determined by the BCA method (Pierce). Coomassie Blue staining of SDS-PAGE gel was used to assess the purity of the VWF-A domain proteins.
Protein Unfolding
Urea denaturation of the WT and R1450E A1 single domain was monitored by circular dichroism spectroscopy as previously described (23, 24). Differential scanning calorimetry (DSC) measurements were performed on a VP-DSC instrument (MicroCal, GE Healthcare) with a 1°/min scan rate at 2 atmospheres using 0.5 mg/ml WT, R1450E A1, and A3 single domains and 0.2 mg/ml WT and R1450E A1A2A3 tri-domains in Tris-buffered saline at pH 7.4. Thermal transitions were calorimetrically irreversible, as no thermal transition was observed in subsequent temperature scans. All DSC traces were corrected for an irreversible scan that was used as the base line. Analysis of the excess heat capacity DSC transitions for the single domains was accomplished on the basis of a simple two-state irreversible model, N → F, as described by us previously considering only two significantly populated macroscopic states, the initial native state (N) and the final irreversible state (F) (24). The A1A2A3 tri-domain DSC transitions were analyzed according to the model by Lyubarev and Kurganov (26) involving two consecutive irreversible steps of reaction N → I → F, where N, I, and F are the native, intermediate, and irreversible state, as described by Sedlák et al. (25).
RESULTS
Mutation R1450E Decreases the Binding of A1 to A2 Domain
The A1 domain R1450E mutation has a significant decreased binding to immobilized A2 domain in comparison to WT A1 (Fig. 1A). This negative effect of the mutation was tested by the capacity of the R1450E mutant to block the interaction between WT and immobilized A2 domain. In this experiment both biotinylated-WT and non-biotinylated WT A1 domain or R1450E mutant were mixed. Fig. 1B shows that the mutant required >10.0 μm to block 50% binding of biotinylated WT, whereas non-biotinylated WT effectively blocked with <0.1 μm. These results confirm that mutation R1450E affects the contact site for A2 domain in A1 domain. This mutation was also expressed in the context of the A1A2A3 tri-domain protein and purified to homogeneity as we previously described. Both WT and mutant tri-domains proteins migrated under reduced conditions slower than their respective non-reduced forms, confirming the reduction of the disulfide bonds located in each A1 and A3 domains. According to non-reducing gel electrophoresis and gel filtration column chromatography, these proteins had an apparent molecular mass corresponding to 95 kDa (Fig. 2). Each protein eluted as a single homogeneous peak with an elution position consistent with the molecular weights determined by SDS-PAGE under non-reducing conditions. Neither of the proteins formed dimers or any higher order aggregates.
FIGURE 1.

Mutation R1450E impaired the interaction between A1 and A2 domain. A, increasing concentrations of recombinant A1 domain proteins were incubated with immobilized A2 domain. The bound A1 protein was determined by enzyme-linked immunosorbent assay as described under “Experimental Procedures” using monoclonal antibody 5D2. The graph is representative of two separated experiments. Each point represents the mean ± S.D. of values obtained from a triplicate assay. B, the binding of the biotinylated WT-A1 domain (250 nm) to immobilized A2 was measured in the presence of purified non-biotinylated WT-A1 or R1450E mutant at the indicated concentrations or the same volume of TBS in the control mixture. 100% is defined as the fraction of added A1 domain polypeptide bound with no A1 variants. Each point represents the mean ± S.E. of two independent sets of triplicate determinations.
FIGURE 2.

SDS-PAGE and gel filtration analyses of recombinant A1A2A3 proteins. Inset, a Coomassie Blue-stained SDS-PAGE (10%) verified the purity of the recombinant WT (1) and mutant (2) proteins under non-reducing conditions (MW, molecular mass). Both purified A1A2A3 proteins were subjected to gel filtration to analyze their native conformation. Using gel filtration column with a TBS mobile phase, the proteins were detected by their absorbance at 280 nm. The graph represents the elution profile for both A1A2A3 proteins (∼92 kDa), and the arrows above represent the elution volume for β-amylase (200 kDa) and human serum albumin (68 kDa). mAu, milliabsorbance units.
Mutation R1450E Alters the Structural Conformation of the A1A2A3 Tri-domain and Induces the Exposure of Cryptic Binding Sites
To assess the effect of R1450E on the structural stability of the A1 single domain and A1 in the context of the A1A2A3 tri-domain, we performed both urea denaturation and differential scanning calorimetry (Fig. 3). In panel A, the thermal DSC transitions of WT and R1450E A1 single domains have a single irreversible transition temperature of 55.5 and 47 °C, respectively, demonstrating that the mutation destabilizes the A1 domain. The A3 domain has a much higher stability than A1, with a 67.5 °C irreversible transition temperature. We also did DSC on the A2 domain; however, it did not yield a thermal transition due to its high degree of disorder in the absence of neighboring domains. In panel B, the destabilizing effect of the mutation was also confirmed by isothermal urea denaturation at 25 °C in which the midpoint of urea transition decreased from ∼3 to 2 m urea for R1450E relative to WT A1. Panels C and D show the thermal DSC transitions of the WT and R1450E tri-domains, which are composed of two sequential irreversible transitions. The first transition is predominantly the A1 domain but could also have some contributions from A2 if it is more structured within the tri-domain. The second transition is due primarily to the A3 domain. This interpretation of the sequential unfolding of A1 followed by A3 is corroborated by both the DSC transitions of the single domains in panel A as well as the urea denaturation results we previously reported for both single A domains and the WT A1A2A3 tri-domain (23). In WT A1A2A3, the first transition occurs at 58 °C followed by the second transition, which occurs at 60 °C. Relative to the single A1 and A3 domains, this corresponds to a net stabilization of A1 by 2.5 °C and a net destabilization of A3 by 7.5 °C as a result of the association of the domains within the tri-domain complex. For R1450E A1A2A3, the first transition occurs at 56.5 °C followed by the second transition that occurs at 63 °C. Relative to the single A1 and A3 domains, this corresponds to a net stabilization of R1450E A1 by 9.5 °C and a net destabilization of A3 by only 4.5 °C; however, relative to the WT tri-domain, the mutation destabilizes the A1 domain by 1.5 °C, and the A3 domain is stabilized by 3 °C. Another feature of these DSC transitions is that the R1450E mutation both separates and broadens the transitions, indicating that not only is the A1 domain destabilized, but this also results in a concomitant loss of interaction between A1 and its neighboring A2 and A3 domains within the tri-domain complex. This conclusion is also supported by the activation energy, a parameter derived from the curve-fitting of the excess heat capacity profiles that co-determine the energetic barrier of the irreversible transition. Comparing the activation energies in panel E, both the WT and R1450E single A1 domains and the first transition of the R1450E tri-domain have an average of 88.3 ± 5.5 kcal/mol, whereas the activation energy of the WT tri-domain is 120 kcal/mol. This indicates that the A1 domain in the R1450E tri-domain is essentially dissociated from the other domains because its activation energy is equivalent to that of the single A1 domain. However, in the WT tri-domain, the activation energy of the A1 domain is significantly greater than for the single A1 domain, indicating that an association between A1 and the neighboring A2 and A3 domains stabilizes the tri-domain complex. The A3 domain activation energies in panel F further support the idea that the stability of the tri-domain complex is predominantly influenced by the high stability of the A3 domain. As a single domain, the activation energy of A3 is a highly stable 130 kcal/mol that decreases to 46 kcal/mol in the WT tri-domain complex. In contrast, the activation energy of A3 in the R1450E tri-domain is only reduced to 74 kcal/mol. These results indicate that the A3 domain imparts stability to the A1 and A2 domains to enable stable contact interactions between domains, and when the A1 domain is destabilized, the stability of the A3 domain is partially restored due to perturbed domain interactions that are coupled to the stability of A1.
FIGURE 3.

Differential scanning calorimetry and urea denaturation of A1 and A3 single and A1A2A3 tri-domains. A, DSC of the WT and R1450E A1 domain and the A3 domain is shown. B, urea denaturation of WT and R1450E A1 domain at 25 °C via circular dichroism at 222 nm is shown. C and D, DSC of the WT and R1450E A1A2A3 tri-domains is shown. E and F, shown is activation energy of the single domains and of the first and second transition of the tri-domain corresponding to the A1 domain and A3 domain, respectively.
We analyzed the effect of the R1450E mutation on the binding of A1A2A3 to ligands of A1 and A2 domains such as GPIbα and monoclonal antibody VP-1, respectively. We assessed the binding of WT (open squares) or mutant protein (open diamonds) to GPIbα without the modulator ristocetin (Fig. 4A). Although WT had poor binding activity, the mutant R1450E protein bound spontaneously to immobilized fixed platelets in a concentration-dependent and saturable manner with half-maximal binding observed at 85 ± 34 nm. This apparent dissociation constant was comparable with that obtained for WT protein (apparent KD ∼ 36 ± 12 nm) in the presence of ristocetin (Fig. 4A, closed squares). The binding of the mutant A1A2A3 protein to VP-1 was also markedly higher than that of WT as shown in Fig. 4B, indicating that the epitope recognized by the anti-A2 domain antibody (VP-1) in the mutant A1A2A3 protein is more accessible than in WT. These results indicate that the cryptic recognition sites for each GPIbα and VP-1 are exposed as a consequence of structural instability caused by the mutation R1450E in the A1 domain and the resulting perturbed domain interactions within the tri-domain.
FIGURE 4.

Binding of recombinant A1A2A3 proteins to platelet GPIbα and antibody VP-1. Increasing concentrations of the recombinant A domain proteins were incubated with immobilized fixed platelets (A) or monoclonal antibody VP-1 (B). The bound protein was determined by enzyme-linked immunosorbent assay as described under “Experimental Procedures.” A, the mutant A1A2A3 (closed square) had a markedly different binding activity for GPIbα than WT A1A2A3 (closed diamond) in the absence of ristocetin. In the presence of ristocetin, the WT bound to GPIbα similarly to the mutant A1A2A3 without ristocetin. B, mutation R1450E also induced the exposure of the recognition site for the antibody in A2 domain, increasing the binding capacity for VP-1. The graphs are representative of two separated experiments. Each point represents the mean ± S.D. of values obtained from a triplicate assay.
The Mutant R1450E A1A2A3 Tri-domain Potentiates the Activation of Platelets under Shear Conditions
The ability of the mutant A1A2A3 in binding instantly to platelet GPIbα suggested that it should compete with multimeric VWF for binding to GPIbα. To test this speculation, we used ristocetin-induced platelet agglutination to analyze the capacity of the mutant tri-domain in blocking platelet agglutination (9). As demonstrated in the binding assay, R1450E blocked 100% ristocetin-induced platelet agglutination at a concentration of 250 nm, whereas in sharp contrast, WT failed to impair ristocetin-induced platelet agglutination even at concentration of 1.0 μm (Fig. 5A). The strong inhibitory capacity of the R1450E tri-domain in ristocetin-induced platelet agglutination led us to test whether it could also function as an inhibitor in shear-induced platelet aggregation. The same PRP used in ristocetin-induced platelet agglutination was incubated with WT or mutant tri-domain (250 nm) for 1–2 min and sheared for 60 s at low (500 s−1) or high (5000 s−1) shear rates. Interestingly, R1450E significantly increased platelet aggregation (48% at low shear and 66% at high shear) in comparison with WT (9% at low and 18% at high shear rates) (Fig. 5B). The platelet-platelet interaction required the involvement of integrin αIIbβ3, as blocking the receptor with antibody 7E3 (20 μg/ml) completely prevented platelet aggregation.
FIGURE 5.

Effect of the A1A2A3 proteins in ristocetin-induced platelet agglutination and shear-induced platelet aggregation. A, the mutant (250 nm) or WT protein (1000 nm) was incubated with PRP for 2 min at 25 °C. The platelet agglutination was then initiated with the addition of ristocetin (1.0 mg/ml). The figure represents four separated experiments using different donors. B, the mutant (250 nm) was incubated with the same PRP used in ristocetin-induced platelet agglutination for 2 min at 25 °C. The mixture was then sheared at 500 or 5000s−1 for 1 min, and platelet aggregation was determined by the reduction of single platelets. The graph depicts the average of percentage of inhibition of platelet aggregation from three separated experiments (mean ± S.D.).
These results obtained in shear-induced platelet aggregation suggested that the interaction of the mutant tri-domain with GPIbα was sufficient to activate αIIbβ3 to bind soluble fibrinogen. We further examined the effect of the mutant on platelet adhesion to fibrinogen under hydrodynamic conditions, reasoning that platelets activated by the mutant will adhere more efficiently to immobilized fibrinogen. One ml of whole blood mixed with WT or mutant protein (250 nm) was perfused over a surface coated with fibrinogen at flow rates of 750, 1500, and 2500 s−1. As expected, the control experiments (blood mixed with WT) demonstrated that adhesion of platelets onto fibrin(ogen) surface decreases as shear rate increases (Figs. 6, A, top panels, and B). In addition, the lack of rolling platelets in the control assays confirmed that plasma VWF did not bind to fibrin(ogen) from the flowing blood (27). At medium (750 s−1) shear rate, ∼1100 ± 300 platelets/field were observed in the control experiment, whereas a remarkably innumerable platelet deposition and thrombus formation was observed when blood was incubated with the R1450E tri-domain (Figs. 6, A and B). Furthermore, both platelet deposition and thrombus formation was greater as the shear rate increased to 1500 s−1. Although the effect of R1450E on platelet adhesion to fibrinogen was significantly attenuated at shear rate of 2500 s−1, the number of platelets firmly adhered, and microaggregates remained higher than control (Fig. 6A). The mutant was capable of enhancing platelet adhesion to fibrinogen at concentrations of 125 and 50 nm at a shear rate of 1500s−1 (not shown). This enhanced platelet adhesion and thrombus formation onto a fibrin(ogen) surface promoted by the mutant protein was entirely eliminated with the monoclonal antibody 10E5 (Fig. 6C). Furthermore, the anti-GPIbα antibody 6D1 effectively blocked the interaction of the mutant with GPIbα, inhibiting notably platelet adhesion (Fig. 6C).
FIGURE 6.

Effect of the mutant A1A2A3 in flow-dependent platelet adhesion to immobilized fibrin(ogen). A, whole blood mixed with WT (top panels) or mutant A1A2A3 (250 nm, bottom panels) was perfused over a surface coated with fibrinogen at different shear rates as indicated. After a 2-min perfusion, the plates were washed with TBS, and several frames of attached platelets were recorded. The photomicrographs represent three separated assays. B, the bar graph shows the percent of surface covered by firmly adhered platelets (mean ± S.E.) after a 2-min perfusion of the whole blood as described in A. The columns represent three separate experiments. C, whole blood containing the mutant A1A2A3 (250 nm) was incubated with either anti-αIIbβ3 (10E5), anti-GPIbα (6D1) (20 μg/ml), EDTA (5 mm), prostaglandin E1 (PGE1) (10 μm), or apyrase (10 units/ml). The blood was perfused over a surface coated with fibrinogen at shear rates of 1500 s−1. After a 2-min perfusion, the plates were washed with TBS, and several frames of attached platelets were recorded and quantified.
These results provide compelling evidence that the interaction between the R1450E tri-domain and GPIbα activates αIIbβ3, exposing a binding site by which fibrin(ogen) was able to interact and immediately arrest the platelets from the flowing blood. The results were identical when performed in the presence of apyrase, an ADP scavenger, indicating that the signaling pathway was not a consequence of either stimulated release of granules and secondary ADP-mediated integrin activation (Fig. 6C). In addition, when tested in the presence of the platelet activation inhibitor prostaglandin E1, the number of stationary platelets was markedly reduced, and the thrombus growth was completely inhibited (Fig. 6C). Because Ca2+ flux is required for αIIbβ3 activation in response to VWF-GPIbα binding (28–30), we also examined the effect of EDTA on mutant-induced platelet adhesion to fibrinogen. EDTA, like the antibody 10E5, completely inhibited the interaction between platelets and fibrin(ogen) (Fig. 6C).
Based on these results, we reasoned that we could obtain the same results observed with the mutant A1A2A3 if plasma VWF exposes the GPIbα site under the same flow conditions. We tested this speculation by activating plasma VWF with the modulator ristocetin at 0.5 mg/ml. As shown in the middle panel of Fig. 7, platelet deposition and thrombus formation was observed when blood was treated with ristocetin just before perfusion. As described for the mutant protein, the antibody 10E5 blocked effectively the platelet deposition onto fibrin(ogen) (Fig. 7, bottom panel).
FIGURE 7.
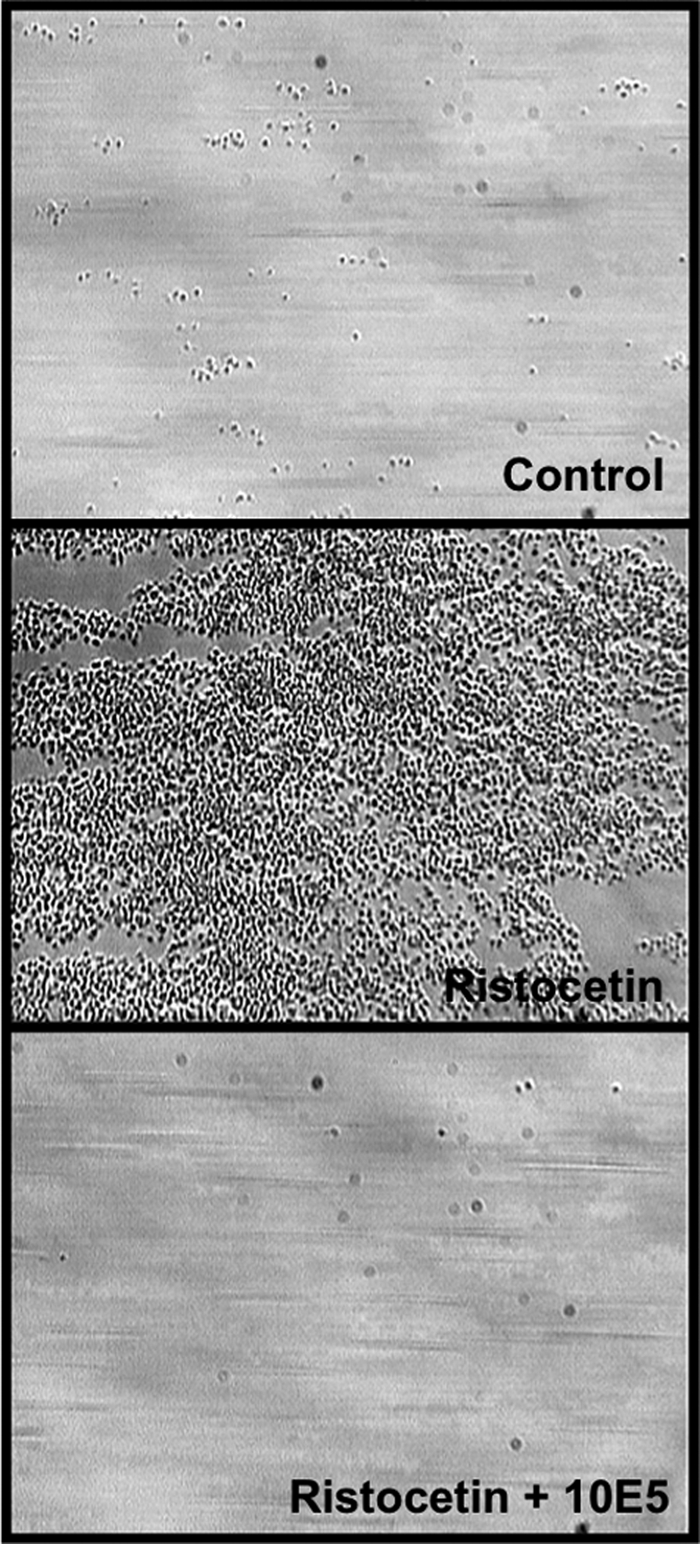
Effect of ristocetin on flow-dependent platelet adhesion to immobilized fibrin(ogen). Whole blood mixed with buffer (control, top panel) or ristocetin (0.5 mg/ml, middle panel) was perfused over a surface coated with fibrinogen at a shear rate of 1500 s−1. After a 2-min perfusion, the plates were washed with TBS, and several frames of attached platelets were recorded, (control <1% surface coverage and with ristocetin, ∼68 ± 14% surface coverage). The effect of ristocetin was completely inhibited by the anti-αIIbβ3 antibody 10E5 (bottom panel, <1% surface coverage). The photomicrographs represent three separated assays.
DISCUSSION
Previously we demonstrated the interaction between A1 and A2 domains and the capacity of isolated A2 domain to inhibit the binding of VWF to platelet GPIbα (17). Based on those results, we proposed that the relationship between A1 and A2 domains in VWF may be part of the regulatory mechanism for the VWF-GPIbα binding. Although we have not demonstrated a direct association between the two vicinal domains in the context of full-length VWF, in this study we used recombinant proteins encompassing the natural sequence of the A1A2A3 domains (amino acid sequence Gln-1238—Gly-1874) to test this hypothesis. The WT A1A2A3 protein, like full-length VWF, bound to GPIbα in a ristocetin- and immobilization-dependent manner and did not effectively bind to platelet GPIbα under the influence of the shear forces used in both shear-induced platelet aggregation and flow chamber experiments in this study. These similarities between the monomeric WT A1A2A3 protein and full-length VWF suggest that our recombinant tri-domain fragment of VWF retains the regulatory structural elements that modulate the activation of VWF in solution.
Among the repertoire of single A1 mutants in our laboratory, we found that the non-naturally occurring gain-of-function R1450E mutation decreases the interaction with A2 domain (Fig. 1) without impairing the binding to GPIbα or collagen (8). The specific negative effect on binding to A2 domain may be a consequence of changing an amino acid residue that forms part of the contact site for A2 domain and/or this mutation lowers the intrinsic conformational stability of the A1 domain, perturbing the association with A2 domain. The former possible explanation cannot be proved until crystallographic studies of the A1-A2 complex is completed; however, the biophysical approaches such as urea denaturation and DSC (Figs. 3, A and B) strongly support the latter proposition because we described the same effect for other Type 2B von Willebrand disease gain-of-function mutations expressed in the single A1 domain (24).
Comparative analyses between WT and A1A2A3 protein expressing the R1450E mutation demonstrate that the mutation alters the structural conformation of the complex. The results obtained from DSC imply that there is a close relationship between the three A domains in the complex stability (Figs. 3, C and D). Relative to the single domains, the intrinsic conformation stability of A1 domain increases when it is expressed in the context of the tri-domain, whereas in contrast, the stability of the A3 domain decreases when it is expressed in the tri-domain protein. In other words, A3 imparts stability to the association of domains in the A1A2A3 complex. The DSC analysis for the R1450E tri-domain suggests that the structural destabilization of the A1 domain decouples A1 from domains A2 and A3, thereby impairing proper domain association in the complex. Furthermore, this decoupling of domains may explain the observations described for the same protein using atomic force microscopy (20). In that study structural destabilization of A1A2A3 was induced by applying mechanical forces. While pulling the protein, an abrupt molecular length increased two times, a result that based on the results obtained with DSC, was probably because of A domain dissociation followed by the unfolding of A2.
We previously proposed that VWF containing a mutation that decreases the stability of the A1 structure could preferentially adopt a conformation that exposes the binding site for GPIbα faster than WT VWF under low hydrodynamic forces and, therefore, spontaneously interacts with platelets (24). This hypothesis was tested in this study. First, destabilization of the A1 domain structure by mutation R1450E induces the spontaneous binding of A1A2A3 mutant to platelet GPIbα without the need of the modulator ristocetin (Fig. 4A). The functional consequences of impairing domain association in A1A2A3 support the notion that this interaction is important to inhibit VWF-GPIbα binding in solution because the spontaneous binding of the mutant A1A2A3 to GPIbα enhanced shear-induced platelet aggregation in the presence of shear forces. Our monomeric A1A2A3 mutant was capable of reproducing the same effect observed by others using multimeric VWF expressing naturally occurring gain-of-function mutations (32, 33). In addition, the binding of the mutant A1A2A3 to GPIbα, like ristocetin-induced plasma VWF binding to GPIbα (Fig. 7), resulted in platelet adhesion to fibrin(ogen) under high flow conditions (Fig. 6). Second, the increased binding activity of the mutant A1A2A3 tri-domain for monoclonal antibody VP-1 indicates that impairing the domain association exposes the cryptic site recognized by VP-1 in A2 domain. The reason is because VP-1, which effectively blocks cleavage by ADAMTS-13 (34), recognizes the sequence Leu-1591—Tyr-1605 that is mostly buried within the structure of the A2 domain (35). In fact, we have demonstrated that ADAMTS-13 cleaved the mutant A1A2A3 faster than WT (18). Consistent with this, we have used A1A2A3 protein and atomic force microscopy to demonstrate that mechanical forces cause structural destabilization of A1A2A3, exposing the A2 domain for cleavage by ADAMTS-13 (20). Therefore, A domain interactions within the tri-domain may regulate the GPIbα-binding and the susceptibility of the A2 domain for cleavage by ADAMTS-13. Interactions among all of the domains may also be terminated mechanically by hydrodynamic forces as it occurs in flowing blood.
In addition to the A2 domain, the D′D3 domains have been described to regulate the binding of A1 domain to GPIbα (36). However, our WT A1A2A3 protein excludes D′D3 domains, and the binding site for GPIbα remained cryptic. One possible explanation is that the amino acid sequence 1238–1247 from the C terminal end of the D3 domain, which is included in our A1A2A3 construct, forms part of the molecular mechanism that masks the binding site for GPIbα. In fact, others have proposed that segment as a regulatory element of VWF-GPIbα binding (37, 38).
It is well established that the binding of GPIbα to multimeric VWF and the concurrently integrin-αIIbβ3-VWF interaction are both necessary to mediate platelet aggregation in shear-induced platelet aggregation and platelet adhesion to fibrinogen at high shear rates (1, 40, 41). However, this study has conclusively demonstrated that the interaction of monomeric A1A2A3 mutant with GPIbα in the presence of shear force is sufficient to trigger the reactions in platelets that activate αIIbβ3. The platelet aggregation and adhesion to fibrin(ogen) induced by the mutant A1A2A3 was αIIbβ3-fibrinogen-mediated as demonstrated by using inhibitors of αIIbβ3, and moreover, the A1A2A3 protein does not include the C1C2 domains, which contain the binding sites for both αIIbβ3 and fibrinogen (41). Furthermore, the mutant A1A2A3 did not induce spontaneous platelet aggregation under the stirring conditions in ristocetin-induced platelet agglutination but potentiated platelet activation in shear-induced platelet aggregation. The main difference between ristocetin-induced platelet agglutination and shear-induced platelet aggregation assays was the hydrodynamic forces applied in the latter. This indicates that platelet activation via GPIbα in solution requires both binding of A1 domain and a minimum threshold of shear force. Moreover, the differential effect of the mutant A1A2A3 in ristocetin-induced platelet agglutination and shear-induced platelet aggregation contrasts previous studies reporting that single monomeric or homodimeric A1 domain proteins effectively inhibited both ristocetin-induced platelet agglutination and shear-induced platelet aggregation (9, 14, 42). A possible explanation for this dissimilarity is that other structural features present in the A1A2A3 sequence influence on the A1-GPIbα binding. Finally, this study has demonstrated αIIbβ3-mediated platelet adhesion to fibrin(ogen) at high shear rates. Our observations contrast previous reports that described platelet adhesion to fibrinogen only at low shear rates (1, 31, 43, 44). The quantity of immediately arrested platelets and the magnitude of thrombus formation on fibrin(ogen)-coated surface observed from blood incubated with the mutant A1A2A3 or ristocetin were comparable with that of whole blood perfused over a collagen-coated surface under high shear rates. Consistent with this, the strong platelet activation induced by the mutant A1A2A3 parallels a recent study describing that VWF-GPIbα interaction may trigger signaling events comparable with those induced by collagen receptors (39). The possible participation of plasma VWF or secreted VWF from activated platelets is excluded because rolling platelets were not seen during the flow of blood with inhibitors of αIIbβ3. In fact, it has been established that perfusion of whole blood does not promote the binding of plasma VWF to immobilized fibrin(ogen) (27).
In summary, the results obtained using a recombinant A1A2A3 domains proteins indicate that interactions between the A domains are coupled to the structural stability of the A1 domain. This association regulates the binding of VWF to GPIbα in solution, which if terminated either through mutation or via rheological shear, causes platelet activation and promotes thrombus formation.
This work was supported, in whole or in part, by National Institutes of Health Grant HL72886 (to M. A. C.). This work was also supported by the Mary R. Gibson Foundation and Alkek Foundation.
- VWF
- von Willebrand factor
- GP
- glycoprotein
- TBS
- Tris-buffered saline
- BSA
- bovine serum albumin
- PRP
- platelet-rich plasma
- DSC
- differential scanning Calorimetry
- WT
- wild type.
REFERENCES
- 1.Savage B., Saldívar E., Ruggeri Z. M. (1996) Cell 84, 289–297 [DOI] [PubMed] [Google Scholar]
- 2.Sadler J. E. (1998) Annu. Rev. Biochem. 67, 395–424 [DOI] [PubMed] [Google Scholar]
- 3.Verweij C. L., Diergaarde P. J., Hart M., Pannekoek H. (1986) EMBO J. 5, 1839–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shelton-Inloes B. B., Titani K., Sadler J. E. (1986) Biochemistry 25, 3164–3171 [DOI] [PubMed] [Google Scholar]
- 5.Mohri H., Yoshioka A., Zimmerman T. S., Ruggeri Z. M. (1989) J. Biol. Chem. 264, 17361–17367 [PubMed] [Google Scholar]
- 6.Borthakur G., Cruz M. A., Dong J. F., McIntire L., Li F., López J. A., Thiagarajan P. (2003) J. Thromb. Haemost. 1, 1288–1295 [DOI] [PubMed] [Google Scholar]
- 7.Mazzucato M., Spessotto P., Masotti A., De Appollonia L., Cozzi M. R., Yoshioka A., Perris R., Colombatti A., De Marco L. (1999) J. Biol. Chem. 274, 3033–3041 [DOI] [PubMed] [Google Scholar]
- 8.Morales L. D., Martin C., Cruz M. A. (2006) J. Thromb. Haemost. 4, 417–425 [DOI] [PubMed] [Google Scholar]
- 9.Cruz M. A., Diacovo T. G., Emsley J., Liddington R., Handin R. I. (2000) J. Biol. Chem. 275, 19098–19105 [DOI] [PubMed] [Google Scholar]
- 10.Cruz M. A., Yuan H., Lee J. R., Wise R. J., Handin R. I. (1995) J. Biol. Chem. 270, 10822–10827 [DOI] [PubMed] [Google Scholar]
- 11.Lankhof H., van Hoeij M., Schiphorst M. E., Bracke M., Wu Y. P., Ijsseldijk M. J., Vink T., de Groot P. G., Sixma J. J. (1996) Thromb. Haemost. 75, 950–958 [PubMed] [Google Scholar]
- 12.Cruz M. A., Whitelock J., Dong J. F. (2003) Thromb. Haemost. 90, 1204–1209 [DOI] [PubMed] [Google Scholar]
- 13.Whitelock J. L., Nolasco L., Bernardo A., Moake J., Dong J. F., Cruz M. A. (2004) J. Thromb. Haemost. 2, 485–491 [DOI] [PubMed] [Google Scholar]
- 14.Shankaran H., Alexandridis P., Neelamegham S. (2003) Blood 101, 2637–2645 [DOI] [PubMed] [Google Scholar]
- 15.Federici A. B., Mannucci P. M., Castaman G., Baronciani L., Bucciarelli P., Canciani M. T., Pecci A., Lenting P. J., De Groot P. G. (2009) Blood 113, 526–534 [DOI] [PubMed] [Google Scholar]
- 16.Azuma H., Sugimoto M., Ruggeri Z. M., Ware J. (1993) Thromb. Haemost. 69, 192–196 [PubMed] [Google Scholar]
- 17.Martin C., Morales L. D., Cruz M. A. (2007) J. Thromb. Haemost. 5, 1363–1370 [DOI] [PubMed] [Google Scholar]
- 18.Yago T., Lou J., Wu T., Yang J., Miner J. J., Coburn L., López J. A., Cruz M. A., Dong J. F., McIntire L. V., McEver R. P., Zhu C. (2008) J. Clin. Invest. 118, 3195–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dent J. A., Berkowitz S. D., Ware J., Kasper C. K., Ruggeri Z. M. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 6306–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu T., Lin J., Cruz M. A., Dong J. F., Zhu C. (2010) Blood 115, 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J. N., Bergeron A. L., Yu Q., Sun C., McBride L., Bray P. F., Dong J. F. (2003) Thromb. Haemost. 90, 672–678 [DOI] [PubMed] [Google Scholar]
- 22.Cruz M. A., Chen J., Whitelock J. L., Morales L. D., López J. A. (2005) Blood 105, 1986–1991 [DOI] [PubMed] [Google Scholar]
- 23.Auton M., Cruz M. A., Moake J. (2007) J. Mol. Biol. 366, 986–1000 [DOI] [PubMed] [Google Scholar]
- 24.Auton M., Sedlák E., Marek J., Wu T., Zhu C., Cruz M. A. (2009) Biophys. J. 97, 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sedlák E., Zoldák G., Wittung-Stafshede P. (2008) Biophys. J. 94, 1384–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyubarev A. E., Kurganov B. I. (1998) Biochemistry 63, 434–440 [PubMed] [Google Scholar]
- 27.Endenburg S. C., Hantgan R. R., Lindeboom-Blokzijl L., Lankhof H., Jerome W. G., Lewis J. C., Sixma J. J., de Groot P. G. (1995) Blood 86, 4158–4165 [PubMed] [Google Scholar]
- 28.Chow T. W., Hellums J. D., Moake J. L., Kroll M. H. (1992) Blood 80, 113–120 [PubMed] [Google Scholar]
- 29.Ikeda Y., Handa M., Kamata T., Kawano K., Kawai Y., Watanabe K., Kawakami K., Sakai K., Fukuyama M., Itagaki I. (1993) Thromb. Haemost. 69, 496–502 [PubMed] [Google Scholar]
- 30.Mazzucato M., Pradella P., Cozzi M. R., De Marco L., Ruggeri Z. M. (2002) Blood 100, 2793–2800 [DOI] [PubMed] [Google Scholar]
- 31.Savage B., Almus-Jacobs F., Ruggeri Z. M. (1998) Cell 94, 657–666 [DOI] [PubMed] [Google Scholar]
- 32.de Romeuf C., Hilbert L., Mazurier C. (1998) Thromb. Haemost. 79, 211–216 [PubMed] [Google Scholar]
- 33.Ajzenberg N., Ribba A. S., Rastegar-Lari G., Meyer D., Baruch D. (2000) Blood 95, 3796–3803 [PubMed] [Google Scholar]
- 34.Tsai H. M., Sussman I. I., Ginsburg D., Lankhof H., Sixma J. J., Nagel R. L. (1997) Blood 89, 1954–1962 [PubMed] [Google Scholar]
- 35.Zhang Q., Zhou Y. F., Zhang C. Z., Zhang X., Lu C., Springer T. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9226–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulrichts H., Udvardy M., Lenting P. J., Pareyn I., Vandeputte N., Vanhoorelbeke K., Deckmyn H. (2006) J. Biol. Chem. 281, 4699–4707 [DOI] [PubMed] [Google Scholar]
- 37.Mohri H., Fujimura Y., Shima M., Yoshioka A., Houghten R. A., Ruggeri Z. M., Zimmerman T. S. (1988) J. Biol. Chem. 263, 17901–17904 [PubMed] [Google Scholar]
- 38.Nakayama T., Matsushita T., Dong Z., Sadler J. E., Jorieux S., Mazurier C., Meyer D., Kojima T., Saito H. (2002) J. Biol. Chem. 277, 22063–22072 [DOI] [PubMed] [Google Scholar]
- 39.Gardiner E. E., Arthur J. F., Shen Y., Karunakaran D., Moore L. A., Am Esch J. S., 2nd, Andrews R. K., Berndt M. C. (2010) Platelets 4, 244–252 [DOI] [PubMed] [Google Scholar]
- 40.Goto S., Ikeda Y., Saldívar E., Ruggeri Z. M. (1998) J. Clin. Invest. 101, 479–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keuren J. F., Baruch D., Legendre P., Denis C. V., Lenting P. J., Girma J. P., Lindhout T. (2004) Blood 103, 1741–1746 [DOI] [PubMed] [Google Scholar]
- 42.Miura S., Sakurai Y., Takatsuka H., Yoshioka A., Matsumoto M., Yagi H., Kokubo T., Ikeda Y., Matsui T., Titani K., Fujimura Y. (1999) Br. J. Haematol. 105, 1092–1100 [DOI] [PubMed] [Google Scholar]
- 43.Endenburg S. C., Hantgan R. R., Sixma J. J., de Groot P. G., Zwaginga J. J. (1993) Blood Coagul. Fibrinolysis 4, 139–142 [PubMed] [Google Scholar]
- 44.Zaidi T. N., McIntire L. V., Farrell D. H., Thiagarajan P. (1996) Blood 88, 2967–2972 [PubMed] [Google Scholar]