

Abstract
The autosomal dominant peripheral sensory neuropathy HSAN1 results from mutations in the LCB1 subunit of serine palmitoyltransferase (SPT). Serum from patients and transgenic mice expressing a disease-causing mutation (C133W) contain elevated levels of 1-deoxysphinganine (1-deoxySa), which presumably arise from inappropriate condensation of alanine with palmitoyl-CoA. Mutant heterodimeric SPT is catalytically inactive. However, mutant heterotrimeric SPT has ∼10–20% of wild-type activity and supports growth of yeast cells lacking endogenous SPT. In addition, long chain base profiling revealed the synthesis of significantly more 1-deoxySa in yeast and mammalian cells expressing the heterotrimeric mutant enzyme than in cells expressing wild-type enzyme. Wild-type and mutant enzymes had similar affinities for serine. Surprisingly, the enzymes also had similar affinities for alanine, indicating that the major affect of the C133W mutation is to enhance activation of alanine for condensation with the acyl-CoA substrate. In vivo synthesis of 1-deoxySa by the mutant enzyme was proportional to the ratio of alanine to serine in the growth media, suggesting that this ratio can be used to modulate the relative synthesis of sphinganine and 1-deoxySa. By expressing SPT as a single-chain fusion protein to ensure stoichiometric expression of all three subunits, we showed that GADD153, a marker for endoplasmic reticulum stress, was significantly elevated in cells expressing mutant heterotrimers. GADD153 was also elevated in cells treated with 1-deoxySa. Taken together, these data indicate that the HSAN1 mutations perturb the active site of SPT resulting in a gain of function that is responsible for the HSAN1 phenotype.
Keywords: ER Stress, Lipid Synthesis, Neurological Diseases, Sphingolipid, Yeast, 1-Deoxysphinganine, HSAN1, Serine Palmitoyltransferase
Introduction
Serine palmitoyltransferase (SPT)3 catalyzes the committed and rate-limiting step in sphingolipid biosynthesis. Until recently, it was believed that there was a single isozyme of this enzyme containing two subunits, LCB1 and LCB2, with the catalytic domain located at the interface and composed of residues from each subunit. However, Hornemann et al. showed that there was a second isoform of LCB2 (1). More recently, we identified two highly related isoforms of a third subunit, ssSPTa and ssSPTb, which are essential for maximal enzymatic activity (2). Thus, there are at least four potential SPT isozymes, each containing a common hLCB1 subunit and one of each of the two hLCB2 and ssSPT subunits. Examination of the acyl-CoA preferences of the hLCB1/LCB2a and hLCB1/LCB2b heterodimers and the hLCB1/LCB2/ssSPT heterotrimers showed that both the LCB2 isoform and the ssSPT isoform conferred different acyl-CoA preferences (2, 3), revealing a previously unappreciated complexity in SPT substrate selectivity, and the corresponding long chain base (LCB) products.
Mutations in the hLCB1 subunit of SPT have been demonstrated to cause the autosomal dominant late-onset hereditary sensory neuropathy, type I (HSAN1) (4–6). In addition, an HSAN1-like phenotype is seen in transgenic mice overexpressing the LCB1C133W mutation (7). Because modeling studies placed the Cys-133 residue near the active site (8–10), reduced SPT activity was seen in transgenic mice (7), and in vitro characterization indicated that the mutant heterodimers were catalytically inactive (7, 8), it appeared that the HSAN1 phenotype might be the result of haploinsufficiency. However, several lines of evidence suggest that this is not the case. First, measurement of SPT activity in lymphoblastoid cell lines established from normal and affected members of HSAN1 families showed a wide range of SPT activities with some unaffected individuals having less SPT activity than some of the affected relatives (11). Second, despite the fact that heterozygous mLCB1 knock-out mice have a greater reduction in SPT activity than the transgenic mice overexpressing the LCB1C133W allele, they show no neurologic impairment (12). Third, the presence of deoxy-LCBs has been reported in both the serum of affected patients (13) and the transgenic mice expressing the mutant allele (12) suggesting that the mutant enzyme is active but has acquired the ability to condense alanine and glycine with acyl-CoAs. Thus, the disease-causing HSAN mutations may result in a gain-of-function. This hypothesis is entirely consistent with the fact that, although many mutations in hLCB1 would inactivate the subunit, only a few mutations have been identified in various HSAN1 kindreds (14). In addition, because transgenic mice co-overexpressing both mutant and wild-type LCB1 have no obvious phenotype (12), it does not appear that disease-causing mutations lead to misfolding of LCB1 resulting in a toxic protein analogous to those responsible for other neurodegenerative diseases (15, 16).
The most common HSAN1 mutation resides in a highly conserved domain. Previous studies of yeast SPT demonstrated that this mutation rendered it catalytically inactive (8). The same result was obtained when Chinese hamster LCB1C133W was cotransfected with LCB2a in CHO cells (7). However, the lack of activity of the mammalian LCB1133W/LCB2a heterodimers might simply reflect an extremely reduced basal activity of the mutant enzymes that lack an ssSPT subunit. Our recent discovery of the ssSPT subunits and our ability to express the human isozymes in yeast cells lacking endogenous SPT (2) has allowed us to directly test the possibility that the trimeric human SPT isozymes containing the most common HSAN1 mutant, hLCB1C133W, are in fact enzymatically active and capable of utilizing alanine to synthesize the deoxy-long-chain base, 1-deoxySa. Our results demonstrate that this is indeed the case and provide additional evidence that these unusual products may be causally related to the loss of neurons in the dorsal root ganglia of patients with HSAN1.
EXPERIMENTAL PROCEDURES
Yeast Strains
The yeast strains used were TDY4116, Matα lcb1α tsc3Δ ura3 leu2 lys2 his 3 met15, and TDY8055, Mata lcb1Δ lcb2Δ leu2 ura3 his3 met 15 lys2 trp1::His3 (2). The mutants were cultured in medium containing 15 μm phytosphingosine and 0.2% tergitol.
Plasmids
Construction of plasmids used to express the human SPT subunits in yeast and mammalian cells has been previously described (2). The C133W mutation was introduced into the yeast hLCB1 expression plasmid using QuikChange mutagenesis (Stratagene, La Jolla, CA), and the AvrII restriction fragment from this plasmid was substituted for the corresponding fragment in the mammalian expression plasmid. Yeast plasmids expressing HA-ssSPTa and HA-ssSPTb were constructed by PCR amplifying the ssSPTa or ssSPTb open reading frame to generate a fragment flanked by NheI-XbaI sites, which was ligated into pGH316 (2), followed by insertion of an SpeI-ended HA epitope. The plasmid expressing the hLCB2a-ssSPTa-LCB1 triple fusion protein was constructed by ligating an XbaI-NheI-ended PCR fragment containing hLCB2a into the NheI site of pGH316, followed by insertion of an NheI-XbaI-ended PCR fragment containing hLCB1 into the NheI site to create an hLCB2a-hLCB1 fusion with an in-frame NheI site at the junction. Finally, an XbaI-Nhel-ended ssSPTa PCR fragment was ligated into the Nhe1 between hLCB2a-LCB1 to generate the yeast expression plasmid. To create the mammalian expression plasmid, a BglII/SalI-ended PCR fragment containing hLCB2a-ssSPTa-LCB1 was ligated between the BglII and XhoI sites of pDual GFP-CCM (Vector Biolabs, Philadelphia, PA).
CHO Cell Culture and Transfection
The CHO cell line CHO-LyB (RIKEN Bioresource Center, Japan) deficient in LCB1 expression (17) was maintained in 95:5 Ham's F-12/Dulbecco's modified Eagle's medium containing 4.5 g/liter glucose (Invitrogen) supplemented with 10% fetal bovine serum (JRH Biosciences, Lenexa, KS) and penicillin and streptomycin. Cells were transfected using Lipofectamine 2000 (Invitrogen) as previously described (18).
Microsome Preparation and Immunoblotting
Microsomes were prepared from yeast and mammalian cells as previously described (18). hLCB1, hLCB2a, HA-ssSPTa, and HA-ssSPTb were resolved by SDS-PAGE and visualized by immunoblotting using antibodies directed against LCB1, LCB2a, and the HA epitope as previously described (2). GADD153 was detected using the SC-7351 mouse monoclonal IgG (Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:250 and glyceraldehyde-3-phosphate dehydrogenase was detected using mouse monoclonal antibody MA B374 (Millipore, Billerica, MA) at a dilution of 1:10,000.
SPT Assays
Microsomal SPT activity was assayed by measuring acyl-CoA-dependent incorporation of [3H]serine into long-chain bases as previously described (2). For determination of kinetic constants, microsomes were incubated for 10 min at the indicated serine or alanine concentrations, with 18 μCi/ml [3H]serine or [3H]alanine. For determination of the alanine Ki, the apparent Km values for serine were determined using 0.1 mm serine with 18 μCi/ml [3H]serine at varying alanine concentrations.
LCB Analysis
LCBs were extracted, derivatized, and analyzed by reversed-phase high-performance liquid chromatography on a C18 column (4 μm, Grace Vydac, Hesperia, CA) using an HP Series II 1090 liquid chromatograph with HP Chemstation coupled to an Agilent 1100 series fluorescence detector as previously described (19). The 1-deoxySa standards were either purchased (C18) from Avanti Polar Lipids (Alabaster, Al) or were synthesized (C20) using purified Sphingomonas paucimobilis SPT enzyme (20) with alanine and stearoyl-CoA as substrates to produce C20-3-keto-deoxysphinganine followed by sodium borohydride reduction.
RESULTS
Expression and Activities of Wild-type and Mutant SPT Heterodimers
It was previously shown that the yeast Lcb1pC180W mutant, which is analogous to the hLCB1C133W mutant, heterodimerizes with yeast Lcb2p, but that the mutant heterodimers are catalytically inactive (8). To determine whether the hLCB1C133W mutation abolishes the activity of hLCB1/hLCB2 heterodimers, hLCB1 or hLCB1C133W was coexpressed with hLCB2a in CHO-LyB cells, and the activities of the human mutant and wild-type heterodimers were compared. Immunoblotting revealed that the hLCB1C133W mutant and wild-type hLCB1 proteins were expressed at similar levels, and both stabilized cotransfected hLCB2a (Fig. 1A). Because hLCB2a stability is dependent upon its ability to form a heterodimer with hLCB1 (21–22) (Fig. 1A), this result indicates that hLCB1C133W forms stable heterodimers with hLCB2a. Measurement of SPT activity in microsomes prepared from transfected cells showed that, unlike wild-type hLCB1, which restored some SPT activity that was further increased by the co-expression of hLCB2a, the hLCB1C133W subunit did not restore SPT activity with endogenous CHO LCB2 or cotransfected hLCB2a (Fig. 1B). Indeed, cotransfection of increasing ratios of the hLCB1C133W to wild-type hLCB1 showed that SPT activity was inversely proportional to the ratio of mutant/wild-type hLCB1 (Fig. 1C). Thus, although wild-type hLCB1 and hLCB1C133W have similar affinities for the hLCB2a subunit, the mutant hLCB1 subunit does not form a catalytically active heterodimer with hLCB2a, and behaves as a classic dominant negative mutant.
FIGURE 1.
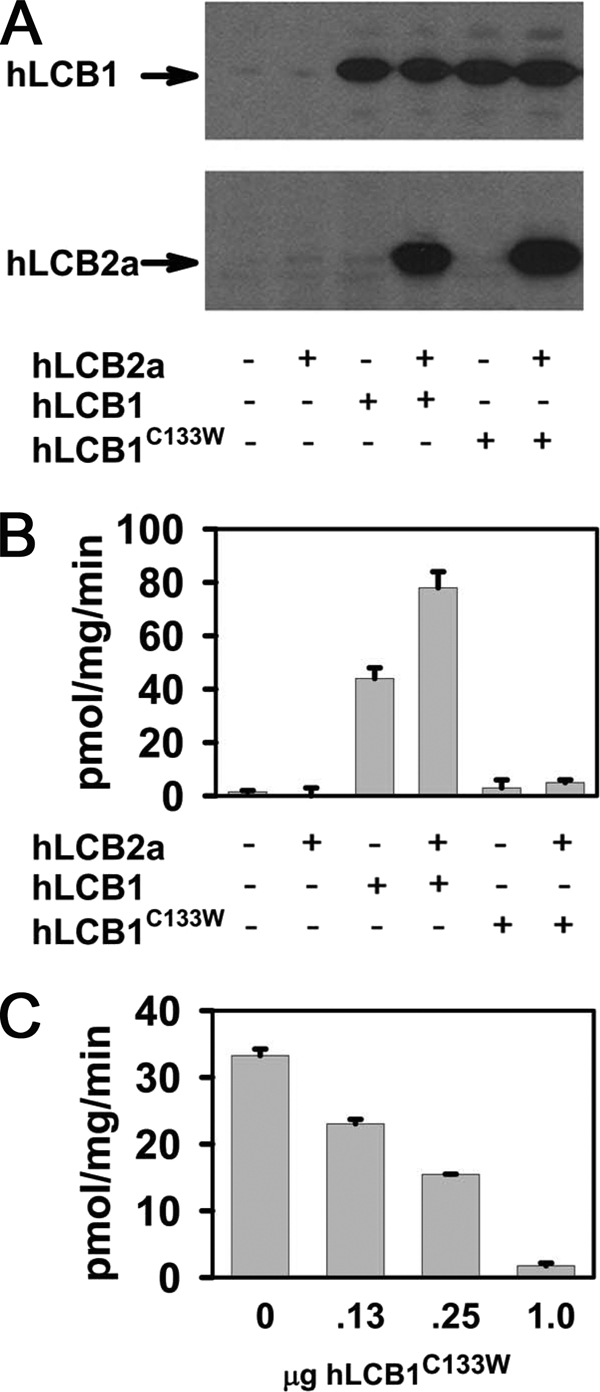
Activity of SPT containing the hLCB1C133W mutant subunit. hLCB1 or hLCB1C133W were expressed with or without hLCB2a in CHO-LyB cells, and microsomes were prepared for immunoblotting to ascertain the expression of transfected genes (A) or used to assay SPT activity (B). Alternatively, a constant amount of hLCB1 (0.25 μg) was expressed with increasing amounts of hLCB1C133W and microsomal SPT activity was determined (C).
Expression and Activity of Mutant SPT Heterotrimers
Because even wild-type human SPT heterodimers have only modest activity in yeast and transfected CHO-LyB cells (2), the possibility that the disease-causing mutation simply reduces SPT activity below the level of detection could not be excluded. We therefore investigated the effect of the highly homologous small proteins (ssSPTa and ssSPTb) recently shown to increase SPT activity 50- to 100-fold (2) on the activity of the hLCB1C133W/LCB2 mutant heterodimers. For these experiments, we took advantage of the ability to express active human SPT isozymes in yeast cells lacking any endogenous SPT activity. As previously reported (2), coexpression of ssSPTa or ssSPTb with hLCB1 and hLCB2a resulted in sufficient increase in activity of the wild-type heterodimer to support growth of the lcb1Δlcb2Δ mutant at 37 °C (Fig. 2A). More significantly, coexpression of either ssSPTa or ssSPTb with hLCB1C133W/LCB2a also provided sufficient SPT activity to support growth at both 26 °C and 37 °C (Fig. 2A). Measurement of SPT activity in microsomes prepared from yeast cells expressing mutant isozymes containing hLCB2a with either ssSPTa or ssSPTb showed that the mutant enzymes had 10–20% of wild-type activity (Fig. 2C). This is not a result of differential expression of wild-type and mutant hLCB1 or differential stabilization of hLCB2a, because both mutant and wild-type proteins were expressed to the same level, and equivalent amounts of hLCB2a were seen in the presence of either hLCB1 or hLCB1C133W (Fig. 2B). A similar activation of hLCB1C133W/LCB2a by ssSPTa or ssSPTb was also observed in CHO-LyB cells (Fig. 2E). Thus, the C133W disease-causing mutant forms a catalytically active enzyme when either of the small subunits is present.
FIGURE 2.

Coexpression of the ssSPT subunits with the hLCB1C133W/hLCB2a heterodimer results in significant catalytic activity. A, yeast lcb1Δlcb2Δ mutants were transformed with plasmids expressing hLCB2a, wild-type, or mutant hLCB1, and empty vector (top row), HA-tagged ssSPTa (middle row), or HA-ssSPTb (bottom row). Transformants were tested for growth on YPD plates at 26° or 37 °C or with phytosphingosine. B, microsomal proteins obtained from the lcb1Δlcb2Δ yeast cells expressing hLCB2a with hLCB1 or hLCB1C133W without (lanes 1 and 4) or with HA-ssSPTa (lanes 2 and 5) or HA-ssSPTb (lanes 3 and 6) were fractionated by SDS-PAGE, and subunit expression was visualized by immunoblotting using anti-hLCB1, anti-hLCB2a, and anti-HA antibodies. The lower of the two bands seen in immunoblots of yeast microsomal proteins using the anti-hLCB2a antibody is a nonspecific protein. C, microsomal SPT activities from yeast cells expressing various combinations of human subunits as in panel B were determined according to “Experimental Procedures.” D, microsomal protein obtained from CHO-LyB cells expressing hLCB2a, wild-type, or C133W mutant hLCB1, and HA-ssSPTa (lanes 1 and 3) or HA-ssSPTb (lanes 2 and 4) were fractionated by SDS-PAGE, and subunit expression was visualized by immunoblotting using anti-hLCB1, anti-hLCB2a, and anti-HA. E, microsomal SPT activities from CHO-LyB cells expressing various combinations of human subunits as in panel D were determined.
Altered Substrate Specificity of Mutant SPT Isozyme
We have recently shown that SPT isozymes containing ssSPTb have an increased ability to condense stearoyl-CoA with serine (2). In addition, although serine is its preferred amino acid substrate, SPT can also condense alanine or glycine with palmitoyl-CoA to generate 1-deoxysphinganine (1-deoxySa) and 1-amino-2-deoxy-n-heptadecane (23–24). Furthermore, the presence of these LCB species has been reported in HEK 293 cells expressing hLCB1C133W in Epstein-Barr virus-transformed lymphoblastoid cell lines and plasma derived from HSAN patients (13), and in transgenic mice expressing CHO LCB1C133W (12). To determine whether SPT heterotrimers containing hLCB1C133W have an increased ability to utilize alanine, wild-type and mutant heterotrimers were expressed in yeast, and formation of C18-1-deoxySa and C20-1-deoxySa was determined. The results showed that wild-type SPT isozymes synthesized neither C18-1-deoxySa nor C20-1-deoxySa at detectable levels. In contrast, yeast cells expressing the hLCB1C133W mutant along with hLCB2a and ssSPTa synthesized significant amounts of C18-1-deoxySa, but no C20-1-deoxySa (Fig. 3A), whereas cells expressing the hLCB1C133W and hLCB2a along with ssSPTb produced both C18-1-deoxySa and C20-1-deoxySa (Fig. 3B). As we have previously shown that ssSPTa confers a preference for C16-CoA, whereas ssSPTb confers a preference for C18-CoA (2), we conclude that the C133W mutation allows the mutant SPT isozymes to react with alanine without altering their acyl-CoA preferences. Cells expressing the SPT isozymes containing hLCB2b instead of hLCB2a also accumulated 1-deoxySa.4 It is also interesting to note that, although the products of SPT are 3-ketosphinganine or 3-ketodeoxysphinganine, it appears that both are efficiently reduced by Tsc10p and FVT1 in yeast and mammalian cells, respectively, because the 3-keto products were not detected.
FIGURE 3.

Mutant human SPT heterotrimers synthesize 1-deoxySa (doxSA). Long-chain bases were analyzed from yeast lcb1Δlcb2Δ mutants expressing wild-type (broken line) or mutant (solid line) hLCB1/hLCB2a/ssSPTa (A) or hLCB1/hLCB2a/ssSPTb (B) heterotrimers or from CHO-LyB cells expressing wild-type (broken line) or mutant (solid line) hLCB1/hLCB2a/ssSPTa (C) or hLCB1/hLCB2a/ssSPTb (D) heterotrimers. The C18-1-deoxySa (C18 DoxSa) eluted at ∼36 min and the C20 DoxSa at ∼73 min.
Similar results were obtained when the mutant SPT isozymes were expressed in CHO-LyB cells. Long-chain base analysis of CHO-LyB cells expressing SPT isozymes containing mutant hLCB1C133W revealed the presence of substantial amounts of C18-1-deoxySa not found in cells expressing any wild-type isozyme (Fig. 3C). In addition, in contrast to the hLCB1/LCB2a/ssSPTa heterotrimers, expression of mutant hLCB1C133W/LCB2a/ssSPTb heterotrimers also produced low but detectable amounts of C20-1-deoxySa (Fig. 3D). Taken together, these results indicate that the mutant heterotrimeric SPT enzymes are active in both yeast and mammalian cells and have an enhanced ability to condense alanine with the preferred acyl-CoA.
Kinetic Properties of the Mutant SPT
To investigate the effect of the HSAN1 mutation on amino acid selectivity, microsomes prepared from yeast expressing the wild-type hLCB1/hLCB2a/ssSPTa and mutant hLCB1C133W/hLCB2a/ssSPTa heterotrimeric SPT isozymes were used to measure the Km and Vmax for serine. The results showed that the wild-type enzyme had a Km for serine of ∼0.75 mm and a Vmax of ∼1350 pmol/mg/min (Fig. 4A). By comparison the Km and Vmax for serine of the mutant enzyme were ∼1.4 mm and ∼275 pmol/mg/min, respectively (Fig. 4B). Thus, the major affect of the HSAN1 mutation is on Vmax. With alanine as substrate, the HSAN1 mutant enzyme had a Km of ∼9.6 mm and a Vmax of ∼110 pmol/mg/min (Fig. 4C). The wild-type SPT did not efficiently utilize alanine, precluding the direct measurement of a Km. However, inhibition by alanine of serine utilization by the wild-type and mutant enzymes revealed a similar relative affinity for alanine with Ki values of 2 mm and 5 mm, respectively (Fig. 4D). Combined with the similar affinities of the mutant and wild-type enzymes for serine and the significant rate of reaction of the mutant enzyme with alanine, these data suggest that the major effect of the HSAN1 mutation is to perturb the active site in such a manner as to allow bound alanine to be activated to react with the acyl-CoA substrate.
FIGURE 4.
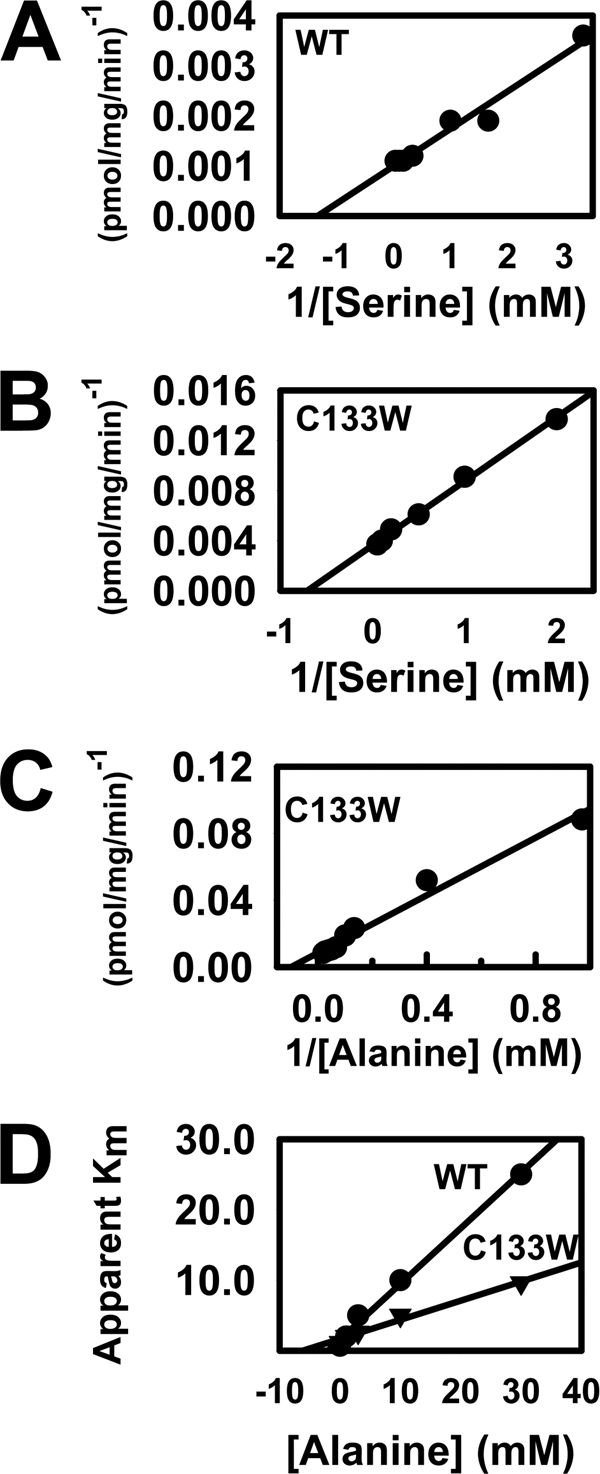
Properties of SPT enzymes containing wild-type and mutant LCB1 subunits. The serine Km and the Vmax of the wild-type (A) and HSAN1 C133W mutant (B) SPT enzymes were determined using [3H]serine as described under “Experimental Procedures.” The alanine Km and Vmax of the mutant enzyme was determined using [3H]alanine (C). The Ki values for alanine inhibition of serine utilization by the mutant and wild-type enzymes were determined by measuring the apparent Km for serine in the presence of varying concentrations of unlabeled alanine (D).
Effects of Varying Extracellular Alanine and Serine on 1-DeoxySa Accumulation
Because the normal extracellular concentrations of serine and alanine are well below the Km values of SPT, the possibility that altering the ratio of serine to alanine would affect the amount of 1-deoxySa synthesized was investigated. It is clear from the data in Figs. 3 and 5B that yeast and mammalian cells expressing the mutant HSAN1 enzyme produce a small but significant amount of 1-deoxySa even when no additional alanine is added to the growth medium. Based on the serum concentration of alanine, ∼0.12 mm alanine was present in the growth medium of the CHO-LyB cells. Addition of as little as 1 mm alanine elicited a large increase in 1-deoxySa production (Fig. 5C). Perhaps more importantly, increasing the serine concentration from 0.12 mm in normal medium to 1.12 mm reversed the effect of added alanine (Fig. 5D), and addition of 10 mm serine completely abolished 1-deoxySa synthesis (Fig. 5E). Thus, relatively small increases in the ratio of serine to alanine have the potential to inhibit 1-deoxySa synthesis by the HSAN1 enzyme containing the C133W mutation in hLCB1.
FIGURE 5.
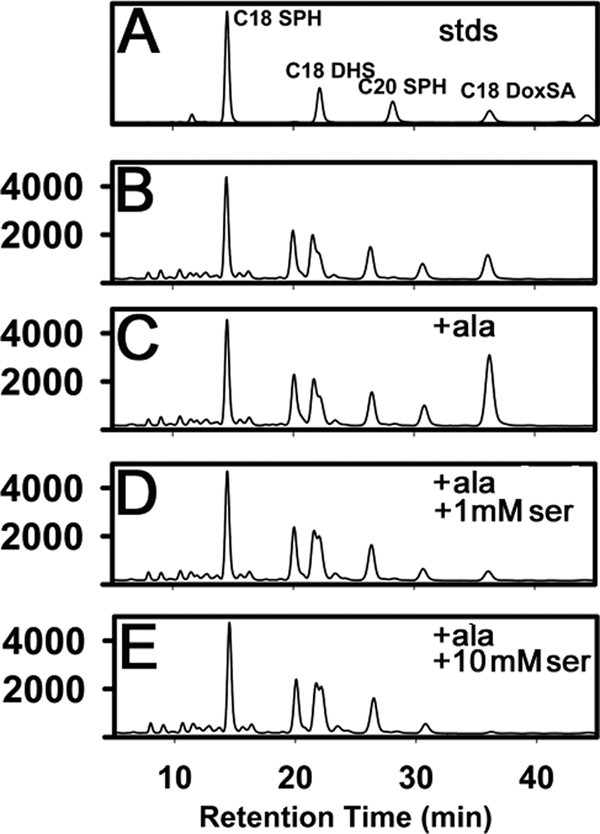
Synthesis of 1-deoxySa is modulated by the extracellular concentrations of the amino acid substrates. A, the indicated LCB standards (stds) were analyzed by high-performance liquid chromatography as described under “Experimental Procedures.” CHO-LyB cells expressing hLCB1C133W/hLCB2a/ssSPTa were grown in Ham's F-12 with 5% Dulbecco's modified Eagle's medium (B), or in the same media supplemented with 1 mm alanine (C), 1 mm alanine and 1 mm serine (D), or 1 mm alanine and 10 mm serine (E), and long-chain bases were extracted and analyzed as described under “Experimental Procedures.”
1-DeoxySa Induces ER Stress Response
1-deoxySa has been implicated as the potentially neurotoxic agent in HSAN1 (12–13). However, the basis for this toxicity has not been determined. Because 1-deoxySa is synthesized in the ER, we examined the possibility that it induces ER stress. Initial experiments expressing the three SPT subunits from three cotransfected expression plasmids provided some indication that the HSAN1 mutant heterotrimer increased GADD153 immunoreactivity, a marker for ER stress and the unfolded protein response (25). However, even the wild-type enzyme, though to a lesser extent, also increased GADD153 expression (data not shown). This is likely due to non-stoichiometric expression of the three subunits and the consequent misfolding of monomeric subunits. To obtain stoichiometric expression, we extended our previous observation that a functional SPT enzyme could be synthesized as an LCB2-LCB1 fusion protein (26) and created a triple fusion containing all three subunits (Fig. 6A). Their arrangement was dictated by the observations that : 1) the C terminus of LCB2 is cytoplasmic5; 2) split ubiquitin two-hybrid experiments place the N termini of the ssSPTs in the cytoplasm and the ssSPTs contain a single predicted transmembrane domain; and 3) the N terminus of LCB1 is luminal and the C terminus is cytoplasmic (18). This arrangement is analogous to the known organization of the coccolithoviral single-chain SPT, in which the Lcb2-like domain precedes the Lcb1-like domain with the two separated by a short spacer (26). Thus, we reasoned that an hLCB2-ssSPT-LCB1 triple fusion protein would adopt a topology that recapitulates that of the native SPT heterotrimer.
FIGURE 6.

1-deoxySa induces ER stress. A, model of the hLCB2-ssSPT-LCB1 triple fusion SPT in which ssSPT (stippled) is inserted between hLCB2 and hLCB1. Topologies are based on hydropathy analyses as well as preliminary experimental data as described in the text. Cys-133, which when mutated to tryptophan causes HSAN, is indicated (C). The pyridoxal phosphate-binding lysine in hLCB2, which in the crystal structure of S. paucimobilis lies directly across the dimer interface from Cys-133 (9), is also indicated (K). B, CHO-LyB cells expressing mutant human LCB2a-ssSPTa-LCB1C133W (bottom panel), but not wild-type LCB2-ssSPTa-LCB1 (top panel), fusion SPT accumulate 1-deoxySa (DoxSA). C, cells transfected with vector (lanes 1–3), or plasmids expressing hLCB2a-ssSPTa-LCB1 (WT; lanes 4–6), or hLCB2a-ssSPTa-LCB1C133W (C133W; lanes 7–9) were grown in Ham's F-12 with or without 10 mm serine or 10 mm alanine for 48 h. As controls, untransfected cells were treated with vehicle (lane 10) or 20 μg/ml tunicamycin for 5 h (lane 11). LCB2a-ssSPTa-LCB1, GADD153, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were visualized by immunoblotting following fractionation of total cellular proteins by SDS-PAGE. D, untransfected CHO-LyB cells were treated with vehicle (0.4% bovine serum albumin/phosphate-buffered saline/ethanol) (lane 1), 1 μm 1-deoxySa (lane 2), 1 μm dihydrosphinganine (lane 3), 1 μm sphingosine (lane 4), 0.25% DMSO (lane 5), or 20 μg/ml tunicamycin in DMSO (lane 6). GADD153 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were visualized by immunoblotting as above. Results are representative of data obtained in three independent experiments.
Indeed, the triple fusion protein had activity equal or greater to, and biochemical properties indistinguishable from, the normal heterotrimer.5 In addition, introduction of the HSAN1 mutation into the triple fusion results in 1-deoxySa synthesis comparable to that obtained by coexpression of mutant hLCB1C133W, hLCB2a, and ssSPTa (compare Figs. 5B and 6B).
Using the triple fusion protein, it is clear that the wild-type enzyme has virtually no affect on GADD153 expression, whereas the mutant fusion protein significantly elevates GADD153 immunoreactivity (Fig. 6C). This increase is greater when cells are grown in 10 mm alanine and is blocked by inclusion of 10 mm serine (Fig. 6C), suggesting that 1-deoxySa, or a metabolite, induces ER stress. Indeed, the level of GADD153 was substantially elevated in cells grown in 1 μm deoxySa (Fig. 6D). Thus, there is a clear correlation between 1-deoxySa production and the ER stress response.
DISCUSSION
Until recently, SPT was thought to be a simple heterodimer composed of two similarly sized subunits, hLCB1 and hLCB2, with each subunit contributing residues that form a catalytic site at their interface for the condensation of serine and palmitoyl-CoA. The identification a second isoform (hLCB2b) of the pyridoxal phosphate-binding subunit, LCB2, added some complexity, because this isoform appears to confer a broader acyl-CoA preference facilitating the incorporation of shorter acyl-CoAs (2, 3). This is similar to what was observed for the single-chain SPT we previously characterized from Emiliania huxleyi (26). However, the activity of either heterodimer is negligible in the absence of a third subunit (ssSPT) that increases SPT activity 50- to l00-fold (2). There are two isoforms of this subunit. Both increase SPT activity of hLCB1/hLCB2a and hLCB1/hLCB2b heterodimers. The hLCB1/hLCB2a/ssSPTa isozyme has a clear preference for C-16 acyl-CoA, and the hLCB1/hLCB2b/ssSPTa prefers C-14 acyl-CoAs. In contrast, the hLCB1/hLCB2a/ssSPTb and hLCB1/hLCB2b/ssSPTb isozymes have a clear preference for longer rather than shorter acyl-CoAs (2). Indeed, the hLCB1/hLCB2b/ssSPTb isozyme prefers C-18 and C-20 acyl-CoAs. Thus, the acyl-CoA preferences ascribed to the hLCB1/hLCB2 heterodimers are likely a consequence of the relative amounts of ssSPTa and ssSPTb in transfected cells and the complexities introduced by the transient overexpression of individual subunits of a multimeric enzyme.
The activation of the hLCB1/LCB2a heterodimer by the ssSPT subunits allowed us to investigate whether the unusual LCB, 1-deoxySa, suggested to underlie the pathophysiology of HSAN1, is generated by the mutant SPT enzymes containing the C133W mutation in hLCB1. Our results show that, although the mutant heterotrimeric SPTs have the same acyl-CoA preferences as their wild-type counterparts, the mutant enzymes have acquired the ability to activate and condense alanine with acyl-CoAs to produce the 1-deoxySas. Although it has been suggested that the HSAN1 mutations cause a higher affinity for alanine (13), two lines of evidence suggest that the C133W mutation does not significantly alter the amino acid binding site of the mutant SPT enzyme. First, the mutant enzyme has only a 2-fold greater Km for serine than the wild-type enzyme. Second, the Ki for alanine inhibition of serine utilization by the mutant enzyme is similar to that of the wild-type SPT. The more likely effect of the mutation is to perturb the catalytic site in such a way as to facilitate the formation of the external aldimine with alanine and/or allow the external aldimine to be more efficiently deprotonated to form and stabilize the reactive quininoid intermediate. This is consistent with the crystal structures of the wild-type and mutant soluble bacterial enzymes, which show that the HSAN1-like mutation alters the relative positions of multiple hydrophobic residues contributed by both subunits at the dimer interface (9). Further studies of the HSAN1-like mutations of the bacterial SPT enzyme are likely to reveal the precise effect of these amino acid substitutions on catalysis.
Interestingly, Hanada et al. (27) reported that alanine did not inhibit SPT purified from CHO cells. In preliminary studies we have observed that the ssSPT subunits dissociate during purification. This raises the possibility that the ssSPT subunit is necessary for alanine binding. Resolution of this question will require the purification of sufficient heterodimeric and heterotrimeric SPT for kinetic and structural studies. The SPT fusion proteins should facilitate these studies.
It has been suggested that 1-deoxySa contributes to the pathophysiology of HSAN1 (12, 14). The observation that GADD153, an ER stress response protein, is induced by growth in 1-deoxySa as well as the fact that GADD153 levels are increased in cells expressing the disease-causing SPT isozyme support this hypothesis. The ability of physiological extracellular concentrations of alanine to increase both 1-deoxySa production and the ER stress response, combined with the finding that serine blocks both of these effects, suggests that it may be possible to directly test this hypothesis by modulating the ratios of serine to alanine in the diet of HSAN1 transgenic mice. A positive result would suggest a potentially simple therapeutic strategy for treating affected individuals. A negative result would be harder to interpret. It could indicate that modification of dietary serine does not alter the serine concentration in the DRG neurons that are most affected in HSAN1 patients. Alternatively, it could indicate that the catalytic promiscuity reflects a more subtle alteration in the enzyme, which over time elicits the late onset peripheral neuropathy. It will therefore be important to examine the enzymatic properties of the other HSAN1 disease-causing mutations.
Acknowledgments
We are grateful to Dominic Campopiano, Marine Raman, and Jonathan Lowther for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant NS47717 (to T. M. D.) from the USPHS, National Institute of Neurological Disorders and Stroke.
K. Gable, S. D. Gupta, J. M. Harmon, and T. M. Dunn, unpublished results.
G. Han, K. Gable, S. D. Gupta, J. M. Harmon, and T. M. Dunn, in preparation.
- SPT
- serine palmitoyltransferase
- 1-deoxySa
- 1-deoxysphinganine
- HSAN1
- human sensory neuropathy type 1
- LCB
- long-chain base
- HA
- hemagglutinin
- CHO
- Chinese hamster ovary.
REFERENCES
- 1.Hornemann T., Richard S., Rütti M. F., Wei Y., von Eckardstein A. (2006) J. Biol. Chem. 281, 37275–37281 [DOI] [PubMed] [Google Scholar]
- 2.Han G., Gupta S. D., Gable K., Niranjanakumari S., Moitra P., Eichler F., Brown R. H., Jr., Harmon J. M., Dunn T. M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 8186–8191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hornemann T., Penno A., Rütti M. F., Ernst D., Kivrak-Pfiffner F., Rohrer L., von Eckardstein A. (2009) J. Biol. Chem. 284, 26322–26330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bejaoui K., Wu C., Scheffler M. D., Haan G., Ashby P., Wu L., de Jong P., Brown R. H., Jr. (2001) Nat. Genet. 27, 261–262 [DOI] [PubMed] [Google Scholar]
- 5.Dawkins J. L., Hulme D. J., Brahmbhatt S. B., Auer-Grumbach M., Nicholson G. A. (2001) Nat. Genet. 27, 309–312 [DOI] [PubMed] [Google Scholar]
- 6.Nicholson G. A., Dawkins J. L., Blair I. P., Auer-Grumbach M., Brahmbhatt S. B., Hulme D. J. (2001) Am. J. Hum. Genet. 69, 655–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCampbell A., Truong D., Broom D. C., Allchorne A., Gable K., Cutler R. G., Mattson M. P., Woolf C. J., Frosch M. P., Harmon J. M., Dunn T. M., Brown R. H., Jr. (2005) Hum. Mol. Genet. 14, 3507–3521 [DOI] [PubMed] [Google Scholar]
- 8.Gable K., Han G., Monaghan E., Bacikova D., Natarajan M., Williams R., Dunn T. M. (2002) J. Biol. Chem. 277, 10194–10200 [DOI] [PubMed] [Google Scholar]
- 9.Raman M. C., Johnson K. A., Yard B. A., Lowther J., Carter L. G., Naismith J. H., Campopiano D. J. (2009) J. Biol. Chem. 284, 17328–17339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yard B. A., Carter L. G., Johnson K. A., Overton I. M., Dorward M., Liu H., McMahon S. A., Oke M., Puech D., Barton G. J., Naismith J. H., Campopiano D. J. (2007) J. Mol. Biol. 370, 870–886 [DOI] [PubMed] [Google Scholar]
- 11.Dedov V. N., Dedova I. V., Merrill A. H., Jr., Nicholson G. A. (2004) Biochim. Biophys. Acta 1688, 168–175 [DOI] [PubMed] [Google Scholar]
- 12.Eichler F. S., Hornemann T., McCampbell A., Kuljis D., Penno A., Vardeh D., Tamrazian E., Garofalo K., Lee H. J., Kini L., Selig M., Frosch M., Gable K., von Eckardstein A., Woolf C. J., Guan G., Harmon J. M., Dunn T. M., Brown R. H., Jr. (2009) J. Neurosci. 29, 14646–14651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penno A., Reilly M. M., Houlden H., Laura M., Rentsch K., Niederkofler V., Stoeckli E. T., Nicholson G., Eichler F., Brown R. H., Jr., von Eckardstein A., Hornemann T. (2010) J. Biol. Chem., 285, 11178–11187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hornemann T., Penno A., Richard S., Nicholson G., van Dijk F. S., Rotthier A., Timmerman V., von Eckardstein A. (2009) Neurogenetics 10, 135–143 [DOI] [PubMed] [Google Scholar]
- 15.Nakamura T., Lipton S. A. (2009) Apoptosis 14, 455–468 [DOI] [PubMed] [Google Scholar]
- 16.Luheshi L. M., Dobson C. M. (2009) FEBS Lett. 583, 2581–2586 [DOI] [PubMed] [Google Scholar]
- 17.Hanada K., Hara T., Fukasawa M., Yamaji A., Umeda M., Nishijima M. (1998) J. Biol. Chem. 273, 33787–33794 [DOI] [PubMed] [Google Scholar]
- 18.Han G., Gable K., Yan L., Natarajan M., Krishnamurthy J., Gupta S. D., Borovitskaya A., Harmon J. M., Dunn T. M. (2004) J. Biol. Chem. 279, 53707–53716 [DOI] [PubMed] [Google Scholar]
- 19.Tsegaye Y., Richardson C. G., Bravo J. E., Mulcahy B. J., Lynch D. V., Markham J. E., Jaworski J. G., Chen M., Cahoon E. B., Dunn T. M. (2007) J. Biol. Chem. 282, 28195–28206 [DOI] [PubMed] [Google Scholar]
- 20.Gupta S. D., Gable K., Han G., Borovitskaya A., Selby L., Dunn T. M., Harmon J. M. (2009) J. Lipid Res. 50, 1630–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gable K., Slife H., Bacikova D., Monaghan E., Dunn T. M. (2000) J. Biol. Chem. 275, 7597–7603 [DOI] [PubMed] [Google Scholar]
- 22.Hanada K., Hara T., Nishijima M. (2000) J. Biol. Chem. 275, 8409–8415 [DOI] [PubMed] [Google Scholar]
- 23.Pruett S. T., Bushnev A., Hagedorn K., Adiga M., Haynes C. A., Sullards M. C., Liotta D. C., Merrill A. H., Jr. (2008) J. Lipid Res. 49, 1621–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zitomer N. C., Mitchell T., Voss K. A., Bondy G. S., Pruett S. T., Garnier-Amblard E. C., Liebeskind L. S., Park H., Wang E., Sullards M. C., Merrill A. H., Jr., Riley R. T. (2009) J. Biol. Chem. 284, 4786–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oyadomari S., Mori M. (2004) Cell Death Differ. 11, 381–389 [DOI] [PubMed] [Google Scholar]
- 26.Han G., Gable K., Yan L., Allen M. J., Wilson W. H., Moitra P., Harmon J. M., Dunn T. M. (2006) J. Biol. Chem. 281, 39935–39942 [DOI] [PubMed] [Google Scholar]
- 27.Hanada K., Nishijima M. (2003) Methods Mol. Biol. 228, 163–174 [DOI] [PubMed] [Google Scholar]