

Abstract
Activated protein C (APC) down-regulates thrombin formation through proteolytic inactivation of factor Va (FVa) by cleavage at Arg506 and Arg306 and of factor VIIIa (FVIIIa) by cleavage at Arg336 and Arg562. To study substrate recognition by APC, active site-mutated APC (APC(S360A)) was used, which lacks proteolytic activity but exhibits anticoagulant activity. Experiments in model systems and in plasma show that APC(S360A), and not its zymogen protein C(S360A), expresses anticoagulant activities by competing with activated coagulation factors X and IX for binding to FVa and FVIIIa, respectively. APC(S360A) bound to FVa with a KD of 0.11 ± 0.05 nm and competed with active site-labeled Oregon Green activated coagulation factor X for binding to FVa. The binding of APC(S360A) to FVa was not affected by protein S but was inhibited by prothrombin. APC(S360A) binding to FVa was critically dependent upon the presence of Arg506 and not Arg306 and additionally required an active site accessible to substrates. Inhibition of FVIIIa activity by APC(S360A) was >100-fold less efficient than inhibition of FVa. Our results show that despite exosite interactions near the Arg506 cleavage site, binding of APC(S360A) to FVa is almost completely dependent on Arg506 interacting with APC(S360A) to form a nonproductive Michaelis complex. Because docking of APC to FVa and FVIIIa constitutes the first step in the inactivation of the cofactors, we hypothesize that the observed anticoagulant activity may be important for in vivo regulation of thrombin formation.
Keywords: Blood Coagulation Factors, Coagulation Factors, Enzyme Mechanisms, Serine Protease, Site-directed Mutagenesis, Thrombin, APC, Activated Protein C, Protein C Pathway, Prothrombinase
Introduction
Human activated protein C (APC)2 is formed after activation of zymogen (protein C) on endothelial cells by the thrombin·thrombomodulin (TM) complex. APC is a serine protease that, together with its cofactor, protein S, down-regulates thrombin formation by inactivating the activated forms of the coagulation factors V and VIII (FVa and FVIIIa) via limited proteolysis. The protein C pathway is vital to normal hemostasis, as is indicated by the severe thrombotic events observed in individuals with homozygous or compound homozygous protein C deficiency (1, 2). Heterozygous individuals have an increased risk for thromboembolic events (3, 4). Another commonly observed (∼5% of Caucasians) impairment of the anticoagulant properties of protein C is associated with the factor VLeiden missense mutation (FV(R506Q)), causing the ablation of one of two APC cleavage sites, the other being at Arg306. Carriership of the FVLeiden mutation is the most common hereditary risk factor of thromboembolic disease.
Interactions between APC and its substrates (FVa and FVIIIa) are largely dependent on three exposed surface loops (the 37-loop: Lys191(37)–Lys193(39); the 60-loop: Lys217(62) and Lys218(63); and the 70–80-loop: Arg229(74), Arg230(75), and Lys233(78)) that together form an electropositive exosite on APC (5–9). Protein C numbering is used throughout the text, followed by chymotrypsinogen numbering in parentheses. Several epitopes on the surface of FVa contribute to the interaction with APC at the Arg506 and the Arg306 cleavage sites (10, 11). Mutagenesis studies suggest the presence of an extended exosite on FVa near Arg506, which is much less prominent around the Arg306 cleavage site (10). These data are in line with three-dimensional molecular models for the complexes between FVa and APC (at Arg506 and Arg306), which suggest that exosite-mediated contacts are more important for docking of APC at Arg506 than for docking at Arg306, by virtue of the extended character of the surface loop in which Arg306 is located (12). Much less information is available about the interaction between APC and FVIIIa. Manithody et al. (13), however, have reported that the same basic residues in the 37-loop and the calcium-binding 70–80-loop of APC are also critical for recognition and inactivation of factor VIIIa.
The affinity of APC for FVa has not been measured directly but can be inferred from the apparent Kmfor the proteolysis at Arg506 in native FVa of between 2 and 20 nm and for cleavage at Arg306 in FVaLeiden, which is ∼10-fold higher (Km = 196 nm (12, 14). Note that the value of Km for both cleavage sites is influenced by the ionic strength and the source of APC, explaining the differences in reported Km values for Arg506. In the present study, we have investigated the interactions between APC and FVa or FVIIIa using an APC variant that lacks catalytic activity caused by a mutation of the active site Ser360(195) to Ala (APC(S360A)). Catalytically inactive S195A mutants of proteinases or proteinases with chemically modified active sites have been used to study substrate-proteinase complex formation (15–17). In recent studies of prothrombin substrate recognition by prothrombinase, S195A FXa in combination with prothrombin mutants containing Arg to Gln-substituted cleavage sites allowed discrimination of the roles of exosite engagement and active site docking in the mechanism of sequential Arg320 and Arg271 cleavage during prothrombin activation (18). A similar mechanism also applies to the extrinsic tenase complex (19) and to factor IX activation by factor XIa (20). Analysis of protein C substrate recognition by the thrombin·(soluble) TM complex, for which crystallographic and mutagenesis studies implicate exosite interactions, however, shows that protein C binding to thrombin depends strongly on interactions between protein C and the active site of thrombin when the latter is bound to TM (21). Here we report that an analogous situation occurs with APC(S360A) recognition of the cleavage site at Arg506 in FVa. The effects of this APC mutant on coagulation were quantitated, both in systems of purified components and plasma-based assay systems. Our results show that APC(S360A) binds with high affinity to a FXa binding site on FVa but not on FVaLeiden and as a result of this binding expresses proteolysis-independent anticoagulant activity in plasma.
EXPERIMENTAL PROCEDURES
Proteins and Reagents
Human FV and FVLeiden from plasma of a normal individual and an individual homozygous for the FV R506Q mutation, respectively, and FVa2 were prepared as described (14). Recombinant FV molecules were expressed as described (10). Two variants were expressed: FVa Q306R506Q679, in which Arg306 and Arg679 were replaced by glutamine and FVa R306Q506Q679, in which Arg506 and Arg679 were replaced by glutamine. FXa, α-thrombin, protein S, wild type APC (wt-APC), and prothrombin were purchased from Kordia Lab Supplies. All of the coagulation factors were of human origin, unless otherwise stated. Purified factor VIII and a polyclonal goat anti-human FVIII inhibitory antibody were kindly provided by Dr. P. Turecek (Baxter AG, Vienna, Austria). α-FXa, purified as described (22) in 5 mm MES, 150 mm NaCl, pH 6.0, was mixed with 0.24 volume of 1 m HEPES, 50 mm CaCl2, pH 7.2, to raise the pH and active site-labeled by inactivation of 12 μm FXa with a 10-fold excess of Nα-[(acetylthio)acetyl]-d-Phe-Pro-Arg-CH2Cl and incubation at 25 °C for 80 min, at which time inactivation was complete as monitored by the initial rate of methoxycarbonyl-d-cyclohexylglycyl-Gly-Arg-p-nitroanilide (Spectrozyme FXa) hydrolysis. FPR-CH2Cl (100 μm) was added to inhibit any traces of active FXa. The free thiol on the inhibitor was generated by the addition of 0.1 volume of 1 m NH2OH, 50 mm CaCl2, pH 7.0, in the presence of 540 μm 5- (and 6)-iodoacetamido-2′,7′-difluorofluorescein (Oregon Green 488-iodoacetamide) and incubation in the dark for 3 h. Free probe was removed by Sephadex G-25 chromatography and dialysis against 50 mm HEPES, 125 mm NaCl, pH 7.4. Probe incorporation was 1.4 mol of probe/mol of FXa, measured as described previously (23). Recombinant tissue factor (TF) (Innovin®) was purchased from Behring. Phospholipid vesicles were prepared as described (24). Synthetic lipids 1,2 dioleoyl-sn-glycero-3-phosphoserine (DOPS), 1,2 dioleoyl-sn-glycero-3-phosphocholine (DOPC), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were obtained from Avanti Polar Lipids (Alabaster, Alabama). The chromogenic substrates pyroGlu-Pro-Arg-p-nitroanilide (S-2366) and H-d-Phe-Pip-Arg-p-nitroanilide (S-2238) were obtained from Chromogenix (Milan, Italy).
Expression and Purification of Recombinant Human Protein C
Recombinant protein C(S360A) was obtained by PCR-based site-directed mutagenesis of the eukaryotic expression vector pRC/RSV (Invitrogen), expressed in HEK293 cells (CRL-1573 ATCC), purified, and characterized essentially as described earlier (25, 26). The purity and integrity of protein C was evaluated by SDS-PAGE. Protein C concentrations were quantified by measurement of absorbance at 280 nm using an absorption coefficient of 14.5 (280 nm, 1%, 1 cm). Protein C was further quantified using a protein C ELISA, employing the anti-human protein C polyclonal antibody Dako 0370 (Dako, Denmark) as a catcher antibody and the horseradish peroxidase-conjugated anti-human protein C polyclonal DAKO P0374 as a detecting antibody. Dilutions of normal pooled plasma were used to calibrate the assay, assuming that plasma contains 64.5 nm protein C (4).
Activation of Protein C and Catalytic Activity against Small Substrates
Protein C(S360A) was activated with bovine thrombin, and the thrombin was removed by chromatography, essentially as described previously (27). Complete removal of thrombin was ascertained by the measured absence of a difference in amidolytic activity between measurement with S-2238 of the APC preparation in the presence and absence of hirudin. APC concentrations were determined from the absorbance at 280 nm and by the above-described protein C ELISA.
Factor Va Assay
FVa activity was determined from the rate of FXa-catalyzed prothrombin activation, as described previously (14). Briefly, prothrombin activation in a reaction mixture containing 0.5 μm prothrombin, a limiting concentration of FVa (≤ 83 pm), 5 nm FXa, 40 μm phospholipid vesicles (10:90, DOPS:DOPC, mol:mol), 0.5 mg/ml ovalbumin, and 2 mm CaCl2 was incubated for 1 min at 37 °C, after which thrombin was measured from the rate of S-2238 hydrolysis.
APC-catalyzed Inactivation of FVa
FVa inactivation by APC was determined by following the loss of FXa cofactor activity of FVa in the prothrombinase complex as a function of time. Routinely, 0.8 nm of plasma-derived FVa or FVaLeiden was preincubated with 25 μm phospholipid vesicles (10:90, DOPS:DOPC, mol:mol) in 25 mm HEPES buffer, 150 mm NaCl, 5 mg/ml bovine serum albumin, pH 7.5 (Buffer I), to which 3 mm CaCl2 was added for 5 min at 37 °C. Inactivation was started by the addition of APC, and the progressive loss of FVa activity was monitored by transferring aliquots of the inactivation mixture into the FVa assay described above.
Factor VIIIa Assay and FVIIIa Inactivation
FVIIIa activity was determined from the rate of FIXa-catalyzed FX activation. Briefly, 3 nm recombinant FVIII was activated with 2 nm thrombin in 50 mm HEPES buffer, 150 mm NaCl, 5 mm CaCl2, 0.2 mg/ml ovalbumin, pH 7.5 (Buffer II), for 60 s. Thrombin was inhibited with recombinant hirudin (20 nm), and the activation mixture was diluted 25-fold into a reaction mixture containing 40 μm phospholipid vesicles (10:90, DOPS:DOPC, mol:mol) in Buffer II. Because FVIIIa stability measurements showed that FVIIIa activity reached a stable value after ∼10 min of incubation at 37 °C, FX activation was started after 15 min of incubation by the addition of FIXa (1 nm) and FX (400 nm). At varying time points, aliquots were taken from the reaction mixture, and the concentration of FXa was determined from the rate of Z-d-Arg-Gly-Arg-p-nitroanilide (S-2765) hydrolysis. FVIIIa inactivation by APC was determined by following the loss of FIXa cofactor activity of FVIIIa as a function of time by transferring aliquots into the FVIIIa assay described above.
Calibrated Automated Thrombinography
Thrombin generation in clotting plasma was monitored by calibrated automated thrombinography (CAT), where conversion of a low affinity fluorogenic substrate (Z-Gly-Gly-Arg-7-amido-4-methylcoumarin (Bachem, Bubendorf, Switzerland) by thrombin was followed with time in a Fluoroskan Ascent® microtiter plate reader (Thermo Labsystems, Helsinki, Finland) with 390-nm excitation and 460-nm emission (28). Thrombin generation curves and the area under the curve, the endogenous thrombin potential, were calculated using Thrombinoscope software (Thrombinoscope B.V., Maastricht, The Netherlands). Thrombin formation was routinely initiated in the absence or presence of APC by the addition of various concentrations of recombinant TF (0.4–13.6 pm), 16 mm CaCl2, and 30 μm phospholipid vesicles (DOPS:DOPC:DOPE, 20:60:20, mol:mol:mol), and 50 μg/ml corn trypsin inhibitor. Likewise, intrinsic activation of coagulation was triggered in the absence of corn trypsin inhibitor by the addition of 1 volume of activated partial thromboplastin time (APTT) reagent (Chromogenix) that was diluted 2-fold in Buffer I to 1 volume of plasma as described by the manufacturer. Thrombin formation was initiated in the presence or absence of APC by the addition of 1 volume of CaCl2 (final concentration, 8.3 mm; Chromogenix), as advised by the manufacturer. PMS-APC was prepared by incubation of 2 μm APC in 25 mm HEPES, 150 mm NaCl, pH 7.5 (Buffer III) with 3.8 mm freshly prepared PMSF at room temperature. The amidolytic activity of the solution was monitored by subsampling of aliquots into 1 ml of 189 μm S-2366 and measurement of the initial rate of hydrolysis at 405 nm. The incubation was continued until amidolytic activity had decreased to that of a blank measurement. Excess inhibitor was removed with a ZebaTM spin column (Pierce), and the PMS-APC was dialyzed against Buffer III overnight. The concentration of PMS-APC was determined with the protein C ELISA. FPR-APC was prepared by incubating 2.4 μm APC for 20 min at room temperature followed by 10 min at 37 °C with a 10-fold excess of PPACK (FPR-CH2Cl; Calbiochem) in Buffer III, and the progress of inhibition was monitored as described for PMS-APC. Free inhibitor was removed with a ZebaTM column. FPR-APC was prepared freshly prior to the experiment, and the concentration was determined using the ELISA.
Fluorescence Studies
Fluorescence was measured with an SLM 8100 spectrofluorometer in the ratio mode, using acrylic cuvettes coated with polyethylene glycol 20,000. All of the experiments were performed in 50 mm HEPES, 110 mm NaCl, 5 mm CaCl2, and 1 mg/ml polyethylene glycol 8000, pH 7.4, at 37 °C. Equilibrium binding studies of Oregon Green-labeled FXa (OG-FXa) binding to FVa were performed by titrating OG-FXa with human FVa and measuring the fluorescence change at 520 nm with excitation at 491 nm (8-nm band passes). Fluorescence changes, expressed as (Fobs − Fo)/Fo = ΔF/Fo, as a function of total FVa concentration were fit by the quadratic binding equation to obtain the maximum fluorescence change ((Fmax − Fo)/Fo = ΔFmax/Fo), the dissociation constant for the probe (OG-FXa) interaction (KP), and the stoichiometric factor (n) (29). Binding experiments for APC(S360A) competing with OG-FXa for FVa were performed by measurement of the OG-FXa fluorescence change at different fixed concentrations of FVa, followed by titration with APC(S360A). The direct titrations of FVa binding to OG-FXa and the competitive displacement curves at various FVa concentrations were analyzed simultaneously by fitting of the cubic binding equation for tight competitive binding to obtain KC and the stoichiometric factor (m) for the competitor binding to FVa. KP and/or ΔFmax/Fo for FVa binding to OG-FXa were allowed in some cases to vary to optimize the fit, with n fixed at its determined value. Nonlinear least square fitting was performed with SCIENTIST software (MicroMath). Fluorescence measurements were corrected for dilution and for background (≤10%) by subtraction of a buffer blank. Experimental error in the parameters represents ± 2 S.D.
Structural Analysis of APC Variants
The Protein Data Bank coordinate file 1AUT for γ-carboxyglutamic acid domainless human APC (15) was retrieved. This APC structure was energy-minimized, using the What If-Yasara twinset (Yasara Biosciences, Graz, Austria) employing the Yamber3 force field. Given that Protein Data Bank code 1AUT contains FPR-CH2Cl covalently attached to the active site, this structure was subsequently described as FPR-APC. Removal of the inhibitor from the molecule and subsequent energy minimization yielded a free APC structure. The free APC structure was used as a template to build a model for PMS-APC. To this end, the structural coordinates of PMSF were retrieved from the deposited structure of the PMSF-inhibited serine protease, human chymase (Protein Data Bank code 1KLT). After structural overlay of the catalytic domains of free APC and human chymase, the PMSF molecule was grafted onto Ser360(195) of APC. The resulting structure was energy-minimized and was subsequently described as PMS-APC. The free APC structure was likewise used as a template for creating a model of the active site-mutated APC(S360A). The free APC structure was mutated in silico at position Ser360(195) into Ala. The mutated structure was energy-minimized to calculate the optimal structure after mutation. The resulting structures of APC variants were analyzed using the What If-Yasara Twinset, CONCOORD (30), and the ICM-Pro package (Molsoft, La Jolla, CA). The volume of the active site region for the different APC forms tested was calculated essentially as described previously (31), using a cut-off distance of 8 Å from the catalytic site residues.
RESULTS
Expression and Purification of Recombinant Protein C Variants
Protein C variants were expressed under serum-free conditions at levels of ∼5 mg/liter and purified with overall recoveries of ∼40%. On SDS-PAGE, protein C(S360A) migrated with a molecular mass of ∼60 kDa. After reduction, the light chain as well as the α, β, and γ isoforms of the heavy chain were seen (data not shown). Upon activation by thrombin, the heavy chain isoforms shifted to slightly lower migration positions, consistent with full activation (>95%) as judged by SDS-PAGE. The purified APC(S360A) preparation contained no measurable (<0.03%) amidolytic activity toward the chromogenic substrates S-2366 or S-2238. To establish further the functional integrity of the APC(S360A) mutant, its capacity to compete with wt-APC for inactivation of FVa was evaluated in a reaction system containing purified proteins and phospholipid vesicles. APC(S360A) competed with wt-APC in a dose-dependent manner, with inactivation of 0.5 nm FVa by 0.16 nm wt-APC half-maximally inhibited by 1.1 nm APC(S360A) (data not shown).
Effect of APC(S360A) on Prothrombinase Activity
The effect of APC(S360A) on the activity of the prothrombinase complex was tested by incubation of APC(S360A) (0–9.5 nm) with FVa, Ca2+, and phospholipid vesicles in the presence of varying concentrations of FXa (25 pm to 5 nm) before initiation of thrombin generation by the addition of prothrombin. In the presence of 5 nm FXa, 0–9.5 nm APC(S360A) only had a marginal effect on prothrombin activation (Fig. 1A). In contrast, at a low FXa concentration (25 pm) prothrombinase activity was reduced to ∼50% by 1.0 nm APC(S360A). Under these conditions, protein S (0–200 nm) had no effect on the inhibition of prothrombin activation by APC(S360A). Upon variation of the prothrombin concentration at a fixed concentration of FXa (50 pm), the effect of APC(S360A) was most prominent at low prothrombin concentrations (Fig. 1B), with a calculated half-maximal inhibition of prothrombin activation at 1.9–7.2 nm APC(S360A) in the presence of 0.5 and 10 μm prothrombin, respectively. At the near physiological concentration of 1.0 μm prothrombin, the concentration of APC(S360A) required to inhibit prothrombin activation by 50% was 2.5 nm.
FIGURE 1.
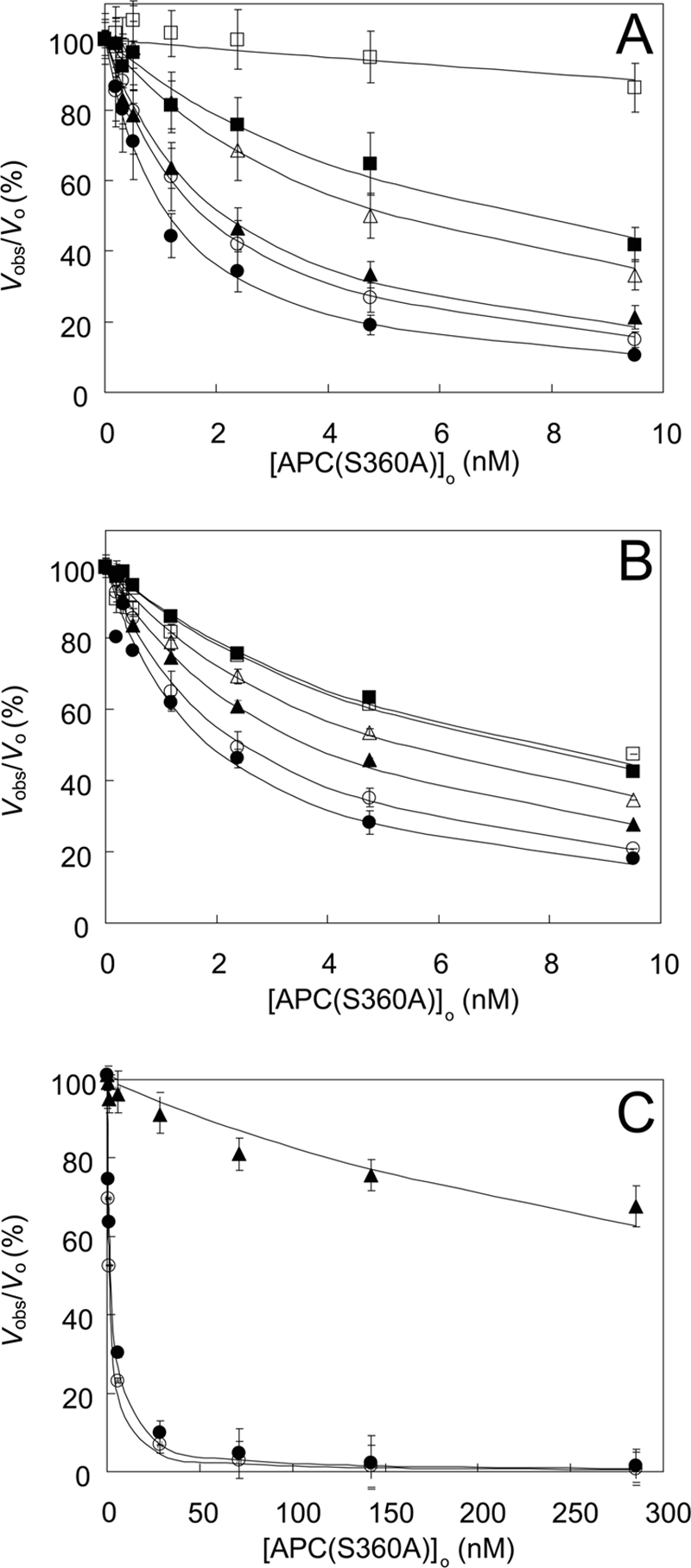
Inhibition of prothrombin activation by APC(S360A). Shown are the effects of FXa and prothrombin in the presence of Arg506 and Arg306 cleavage sites in FVa. A, effect of varying APC(S360A) concentrations on prothrombin activation by 7.2 pm FVa, 40 μm phospholipid vesicles (10:90, DOPS:DOPC), 3 mm CaCl2, 1 μm prothrombin, and 25 (●), 50 (○), 100 (▴), 250 (▵), 500 (■), and 5,000 (□) pm FXa. B, effect of varying APC(S360A) concentrations on prothrombin activation by 7.2 pm FVa, 40 μm phospholipid vesicles (10:90, DOPS:DOPC), 3 mm CaCl2, 50 pm FXa, and 0.5 (●), 1.0 (○), 2.5 (▴), 5 (▵), 10 (■), and 20 (□) μm prothrombin. C, effect of varying APC(S360A) concentrations on prothrombin activation by 7.2 pm wild type FVa (●), FVa Q306R506Q679 (○), or FVa R306Q506Q679 (▴), 40 μm phospholipid vesicles (10:90, DOPS:DOPC), 3 mm CaCl2, 1 μm prothrombin, and 50 pm FXa. Initial rates of prothrombin activation at varying APC(S360A) concentrations are presented as fractions (%) of the rate in the absence of APC(S360A) (vobs/vo). Indicated are the averages ± S.E. from two different experiments. The solid lines represent the hyperbolic fits of the data. Further experimental conditions are described under “Experimental Procedures.”
Because proteolytic inactivation of FVa by APC mainly proceeds through cleavage of two peptide bonds in FVa (Arg506 and Arg306), we used recombinant FV variants that can only be cleaved at one site to determine whether the effects observed with native (plasma-derived) FVa could be attributed specifically to one of these two cleavage sites. To this end, 7.2 pm of recombinant wild type FVa, FVa Q306R506Q679 (a recombinant FVaCambridge-like molecule) or FVa R306Q506Q679 (a recombinant FVaLeiden-like molecule) were incubated with phospholipid vesicles, 50 pm FXa, and increasing concentrations of APC(S360A). Residual FVa activity was determined by thrombin generation following the addition of 1 μm prothrombin. Inhibition of the cofactor activity of FVa Q306R506Q679 by APC(S360A) was virtually the same as that of wild type FVa (Fig. 1C). In contrast, inhibition of FVa R306Q506Q679 required APC(S360A) concentrations that were 2 orders of magnitude higher, indicating that the expression of the proteolysis-independent anticoagulant effect of APC(S360A) required the presence of Arg506.
Competitive Binding of APC(S360A) and OG-FXa to FVa
To quantitate the effect of APC(S360A) on the stability of the FVa·FXa complex, we performed fluorescence experiments with FXa that was active site-labeled with Oregon Green, linked by FPR-CH2Cl (OG-FXa). Analysis of data pooled from six titrations of 1.2 nm OG-FXa with native FVa gave KP of 0.14 ± 0.10 nm for binding to 0.82 ± 0.18 mol of FVa/mol of OG-FXa, accompanied by a 36 ± 2% enhancement in fluorescence (Fig. 2A). Competitive binding titrations with varying concentrations of APC(S360A) at fixed OG-FXa concentration and four fixed concentrations of FVa were analyzed using the cubic equation for tight competitive binding (Fig. 2B). For this analysis, the stoichiometric factor for OG-FXa binding to FVa (n) was fixed at the determined value (0.82), and the fitted parameters were KP for OG-FXa binding, the stoichiometric factor (m), KC for APC(S360A) binding to FVa, and the maximum fluorescence change. The results showed a good fit of the competitive binding model with the same maximum change in fluorescence of 37 ± 2%, KP for OG-FXa binding to FVa of 0.14 ± 0.06 nm, a stoichiometric factor m = 0.92 ± 0.13 mol APC(S360A)/mol FVa, and KC of 0.11 ± 0.05 nm for APC(S360A) binding to FVa. Protein C(S360A) zymogen had no detectable influence on the stability of the FVa·FXa complex at concentrations as high as 74 nm (Fig. 2C).
FIGURE 2.
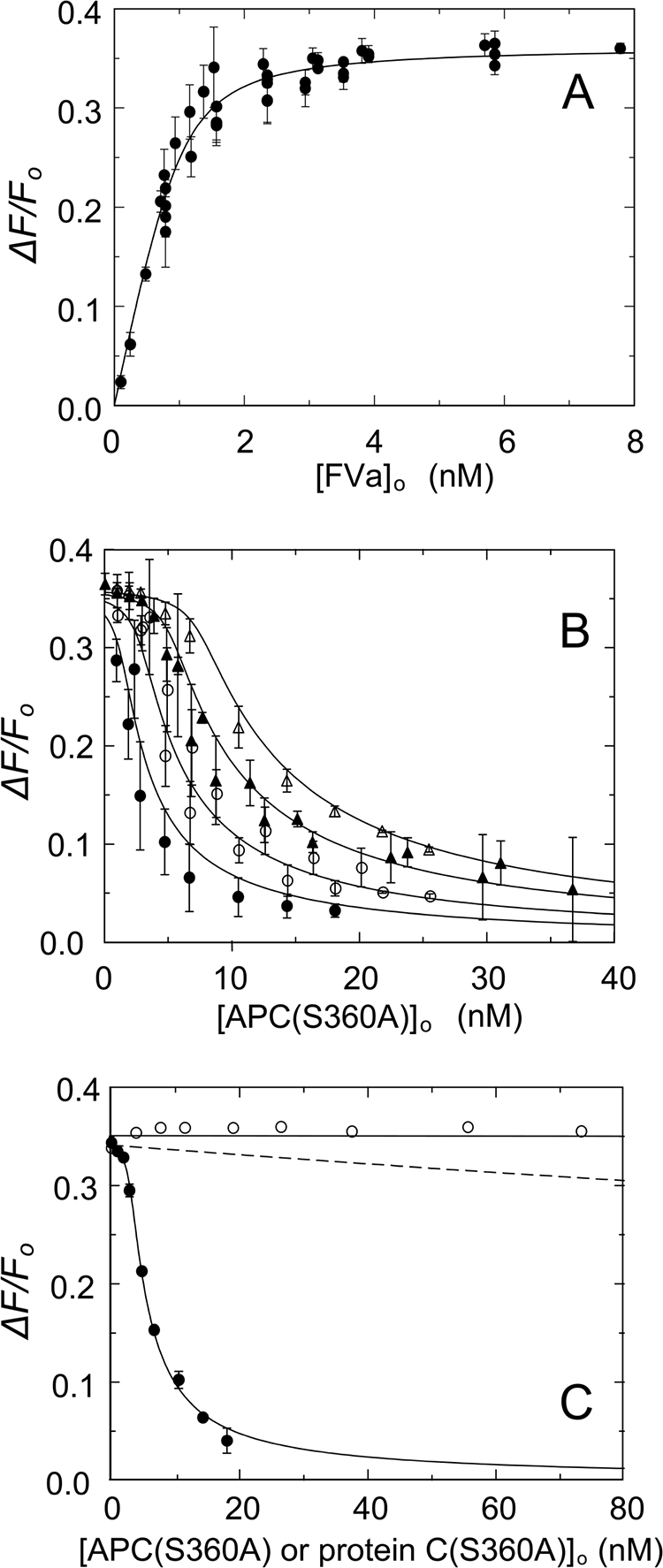
Equilibrium binding of native FVa to OG-FXa and dissociation of the OG-FXa·FVa complex by APC(S360A) and protein C(S360A). A, the fractional increase in fluorescence (ΔF/Fo) of 1.2 nm OG-FXa as a function of the total concentration of FVa ([FVa]o) in the presence of 50 μm phospholipid vesicles (20:60:20, DOPS:DOPC:DOPE) shown for data pooled from six independent titrations (●). B, effect of varying APC(S360A) concentrations on OG-FXa·FVa complexes preformed after incubating 1.2 nm OG-FXa with, 2.35 (●), 3.72 (○), 5.85 (▴), and 7.8 (▵) nm FVa. The solid lines in A and B represent nonlinear least square fits of the combined data with the cubic competitive binding equation with n = 0.82 ± 0.18 FVa/OG-FXa (mol/mol) fixed, and fitted parameters of KP = 0.14 ± 0.06 nm, m = 0.92 ± 0.13 APC(S360A)/FVa (mol/mol), KC = 0.11 ± 0.05 nm, and ΔFmax/Fo = 37 ± 2%. C, effect of varying APC(S360A) (●) or protein C(S360A) (○) concentrations of preformed OG-FXa·FVa complexes (1.2 nm OG-FXa and 5.8 nm FVa). The solid line for APC(S360A) displacement of FVa from OG-FXa represents the fit by the cubic equation with the parameters given in B, except the fitted values of m = 1.3 ± 0.2 APC(S360A)/FVa (mol/mol), KC = 0.08 ± 0.02 nm, and ΔFmax/Fo = 35 ± 1%. The flat solid line was drawn through the protein C(S360A) data at ΔF/Fo = 0.35. The error bars represent the 95% confidence interval. The dashed line represents a simulation with KC = 28 nm (200 × KP). Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
Effect of APC(S360A) on the Stability of the FVLeiden·OG-FXa Complex
Given the importance of Arg506 in FVa for proteolytic inactivation by APC, similar experiments were performed using FVaLeiden (Fig. 3A). In the absence of APC(S360A), native FVa and FVaLeiden bound to OG-FXa with similar fluorescence increases of 40 ± 2 and 44 ± 1%. Simultaneous analysis of the direct titrations of OG-FXa with FVa and FVaLeiden and the competitive binding titrations with APC(S360A) showed displacement of native FVa, with a KC value of 0.25 ± 0.07 nm but very little effect on the OG-FXa·FVaLeiden complex (Fig. 3B). Analysis of the small, linear decrease in fluorescence gave a KC value of 38 ± 27 nm for APC(S360A) binding to FVaLeiden, representing an affinity that is ∼150-fold lower than for native FVa. These results suggest that direct binding of the active site of APC(S360A) to Arg506, which is not possible for the Gln506 mutant, dominates the exosite-mediated binding of APC(S360A) to FVa. To test this hypothesis further, binding of APC covalently inactivated with the transition state analog, FPR-CH2Cl (FPR-APC) to FVa, was examined. By its dimensions and interactions, this inhibitor blocks the active site and engages the S1–S3 specificity subsites,3 forms a hemiketal with Ser360(195), and alkylates the side chain imidazole ring of the catalytic His residue. Comparison of APC(S360A) to FPR-APC showed that APC(S360A) bound to FVa with KC of 0.17 ± 0.09 nm, whereas at concentrations up to 128 nm, FPR-APC produced a small linear decrease in fluorescence with an apparent KC of 36 ± 9 nm (Fig. 4), which is similar to the KC for binding of APC(S360A) to FVLeiden (38 nm).
FIGURE 3.
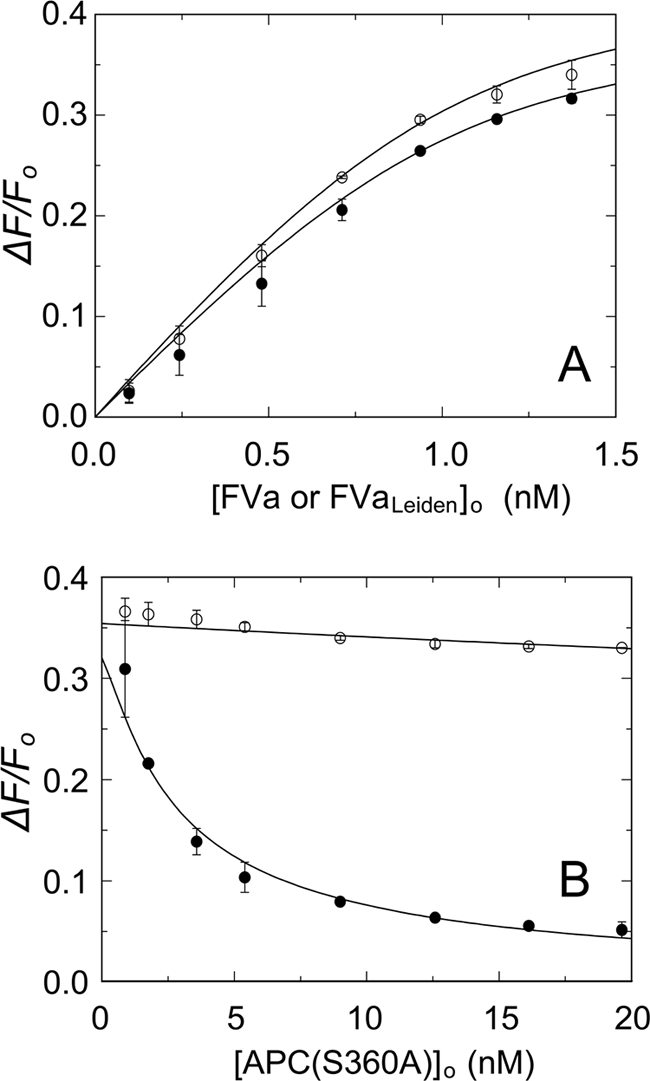
Binding of OG-FXa and APC(S360A) to native FVa and FVaLeiden. A, fractional fluorescence increase (ΔF/Fo) after binding of varying concentrations native FVa (●) or FVaLeiden (○) to 1.2 nm OG-FXa in the presence of 50 μm phospholipid vesicles (20:60:20, DOPS:DOPC:DOPE). B, effect of varying APC(S360A) concentrations of preformed complexes of 1.4 nm native FVa (●) or FVaLeiden (○) with 1.2 nm OG-FXa. Fitting of the combined data of A and B by the cubic competitive binding equation with fixed parameters n = 0.82 FVa/OG-FXa (mol/mol), KP = 0.14 nm, and m = 1, yielded KC = 0.25 ± 0.07 nm, and ΔFmax/Fo = 40 ± 2% for native FVa and KC = 38 ± 27 nm and ΔFmax/Fo = 44 ± 1% for FVaLeiden. The error bars represent the 95% confidence interval. Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
FIGURE 4.
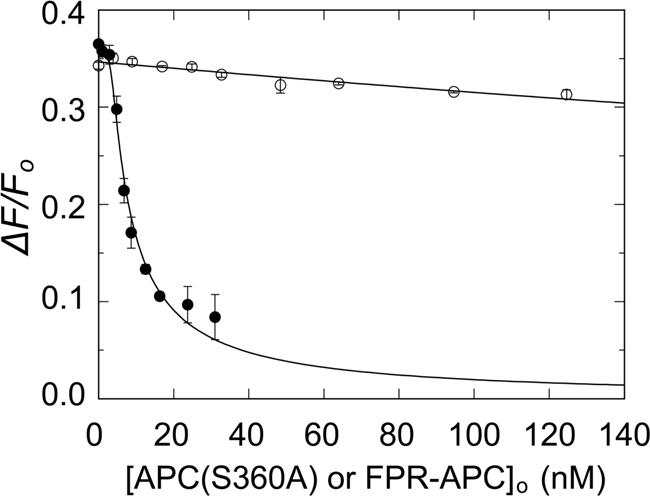
Effect of active site labeling of APC with FPR-CH2Cl on competitive binding of APC and OG-FXa to native FVa. The fractional fluorescence change (ΔF/Fo) of a mixture of 1.2 nm OG-FXa and 5.8 nm FVa as a function of the total concentration of APC(S360A) (●) or FPR-APC (○) in the presence of 50 μm phospholipid vesicles (20:60:20, DOPS:DOPC:DOPE) is shown. The solid lines represent fits of the data by the cubic competitive binding equation with fixed parameters of n = 0.82 ± 0.18 FVa/OG-FXa (mol/mol), KP = 0.14 nm, and fitted parameters, m = 1.4 ± 0.4 APC(S360A)/FVa (mol/mol), KC = 0.17 ± 0.09 nm, and ΔFmax/Fo = 38 ± 2%. For FPR-APC, m was fixed at 1, KC = 36 ± 9 nm, and ΔFmax/Fo = 36 ± 1%. The error bars represent the 95% confidence interval. Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
Influence of Protein S and Prothrombin on Binding of APC(S360A) to FVa
Because both protein S and prothrombin affect APC-catalyzed FVa inactivation (32–34), we investigated whether these proteins also affect the APC(S360A)-mediated dissociation of the OG-FXa·FVa complex. In the presence of 150 nm protein S (Fig. 5A), competitive binding of FVa to APC(S360A) and OG-FXa was indistinguishable from that observed in the absence of protein S, indicating that plasma levels of protein S do not affect the binding of APC(S360A) to FVa. The addition of 0.6 μm prothrombin to the OG-FXa·FVa complex resulted in an initial 18% decrease in fluorescence, which is due to formation of a ternary OG-FXa·FVa·prothrombin complex.4 Prothrombin abolished the effect of APC(S360A) on the stability of the OG-FXa·FVa complex (Fig. 5B), and no significant decrease in fluorescence was observed upon the addition of up to 37 nm APC(S360A).
FIGURE 5.
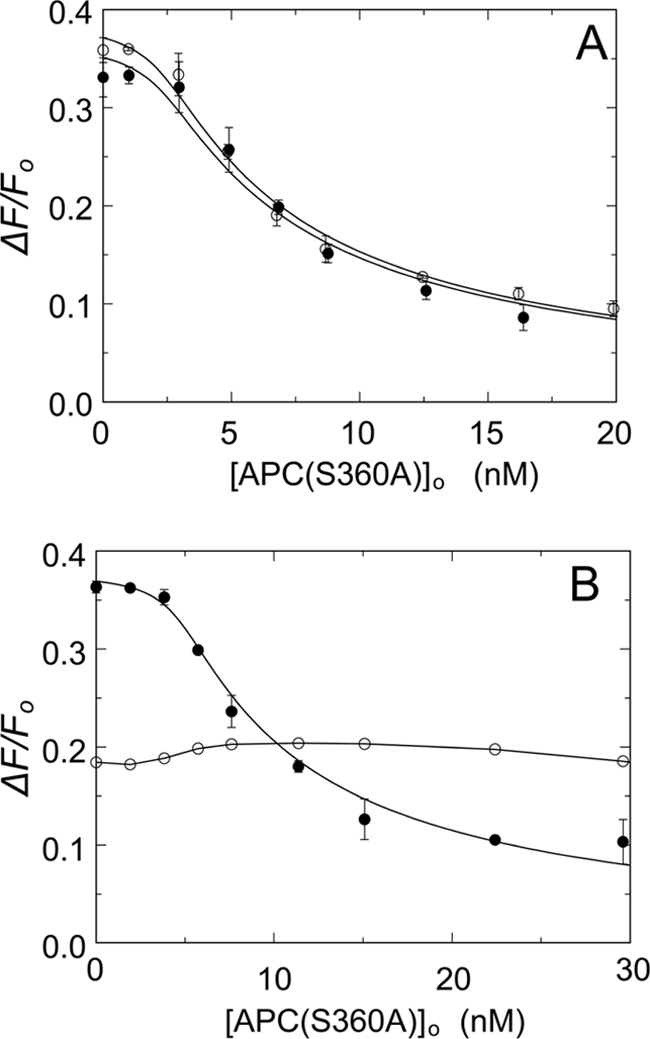
Effect of protein S or prothrombin on the binding of APC(S360A) and OG-FXa to FVa. A, titrations of mixtures of 1.2 nm OG-FXa and 3.5 nm native FVa in the presence of 50 μm phospholipid vesicles (20:60:20, DOPS:DOPC:DOPE) with APC(S360A) in the absence (●) and presence of 150 nm protein S (○). The solid lines represent fits of the data by the cubic competitive binding equation with fixed parameters of n = 0.82 ± 0.18 FVa/OG-FXa (mol/mol), KP = 0.14 ± 0.06 nm, m = 1, and fitted parameters in the absence of protein S, KC = 0.21 ± 0.05 nm and ΔFmax/Fo = 37 ± 2%, and the presence of protein S, KC = 0.21 ± 0.04 nm and ΔFmax/Fo = 39 ± 2%. B, titrations as in A of 1.2 nm OG-FXa and 5.7 nm native FVa in the absence (●) and presence of 0.6 μm prothrombin (○). The titration in the absence of prothrombin was analyzed with the fixed parameters as in A and fitted KC = 0.17 ± 0.03 nm and ΔFmax/Fo = 38 ± 1%. The error bars represent the 95% confidence interval. The solid line for the titration in the presence of prothrombin was arbitrarily drawn through the data. Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
Effect of APC(S360A) on Thrombin Generation in Plasma
Because inhibition of prothrombin activation by APC(S360A) in the model system was reduced by prothrombin and high FXa concentrations, the effect of APC(S360A) on thrombin generation in human plasma was investigated. At a fixed concentration of TF (3.4 pm) and 30 μm phospholipid vesicles, APC(S360A) inhibited thrombin generation in a concentration-dependent manner (Fig. 6A). APC(S360A) had, however, a minor effect on thrombin generation in the plasma of homozygous carriers of the FVLeiden mutation (Fig. 6B). For comparison, the effect of wild type APC on thrombin generation was tested under the same experimental conditions. Similar to the fluorescence experiments, the addition of FPR-APC to normal plasma verified that at concentrations as high as 143 nm, there was very little effect on thrombin formation in FVLeiden plasma, confirming that this APC derivative has greatly reduced nonproteolytic anticoagulant activity on FVLeiden.
FIGURE 6.
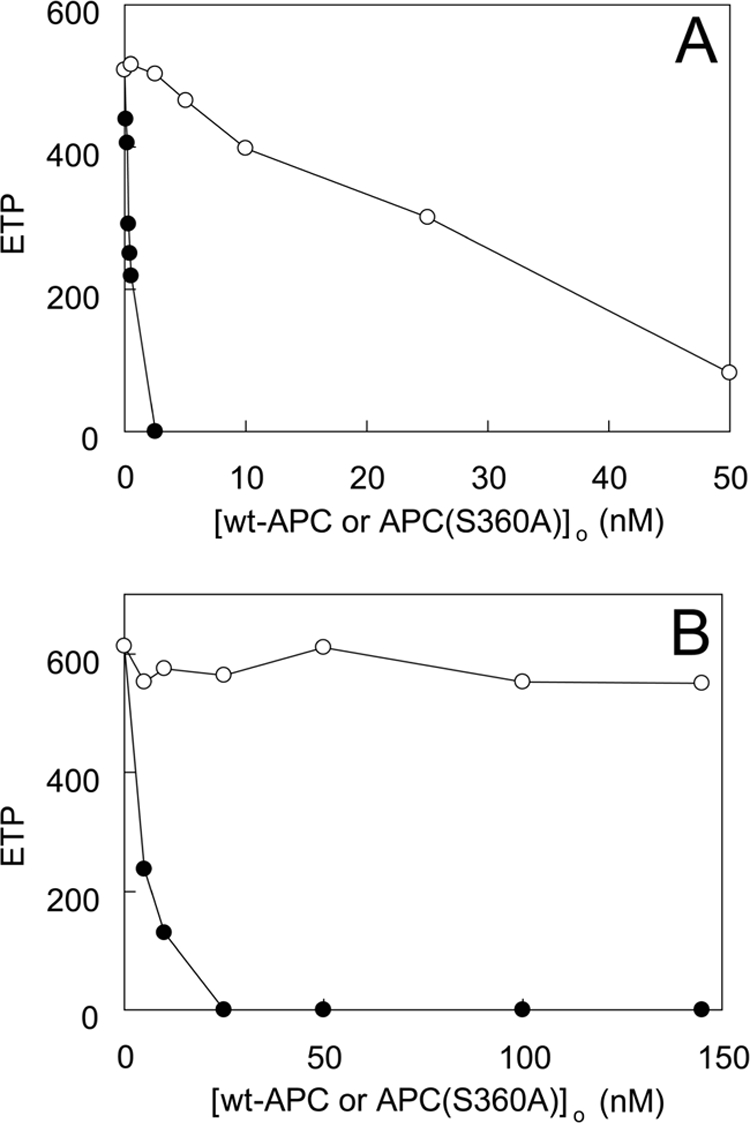
Effect of APC(S360A) on extrinsic thrombin generation in normal plasma and in plasma from FV R506Q carriers. Thrombin generation was measured by CAT as described under “Experimental Procedures” using 3.4 pm TF and 30 μm phospholipid vesicles (20:60:20, DOPS:DOPC:DOPE) and varying total concentrations of wt-APC (●) or APC(S360A) (○). Shown are the average endogenous thrombin potential (ETP) values from titrations (n = 3) performed in normal plasma (A) or in a plasma pool from three different donors carrying the R506Q mutation (B) with the standard errors of the mean/point not exceeding 12% of the measurement.
To examine the influence of FXa on the anticoagulant activity of APC(S360A), experiments were performed in which thrombin generation was measured in either the absence or the presence of a fixed amount of APC(S360A), whereas the concentration of TF used to trigger the extrinsic coagulation pathway was varied to generate varying amounts of FXa (Fig. 7). In normal plasma, inhibition of thrombin generation by APC(S360A) was most pronounced at low TF concentrations (<2 pm) and absent at [TF] > 10 pm. In contrast, in the plasma of a homozygous carrier of the FVLeiden mutation, APC(S360A) had no effect on thrombin generation over the complete range of TF concentration tested (data not shown). These observations indicated that inhibition of thrombin formation by APC(S360A) was dependent on the concentration of FXa generated in plasma and required the presence of Arg506 in FVa.
FIGURE 7.

Effect of PMS-APC and APC(S360A) on thrombin generation in normal plasma as a function of TF concentration. Thrombin generation was measured by CAT as described under “Experimental Procedures” at varying TF concentrations and a fixed concentration of 30 μm phospholipid vesicles (20:60:20, DOPS:DOPC:DOPE) in the absence of APC (○) or in the presence of either 4.2 nm APC(S360A) (●) or PMS-APC (▴). The percentage of the thrombin peak at each TF concentration, relative to that in the absence of APC (Relative thrombin peak) is shown. The inset shows the height of the thrombin peak in the absence of APC as a function of the TF concentration. Shown are the averages ± S.E. from three different experiments.
Comparison of FPR-APC and PMS-APC as Inhibitors of Thrombin Generation
It was hypothesized that FPR-APC had greatly reduced anticoagulant properties because of the extensive interactions of the inhibitor with the S1–S3 specificity subsites, engagement of Ser360(195) in a hemiketal, and alkylation of His211(57), as well as the relatively bulky inhibitor blocking access the APC active site. Hence, we prepared an active site-inhibited APC derivative with predicted intermediate properties, PMS-APC, in which PMSF sulfonates Ser360(195). Under the conditions described for Fig. 7, the addition of 4.2 nm PMS-APC to plasma produced an inhibitory effect on thrombin generation in plasma that was less than that of APC(S360A) but still clearly discernable.
Effect of the Intrinsic and Extrinsic Pathways on APC(S360A) Inhibition of Thrombin Formation
The effects of APC(S360A) on thrombin generation in normal and FVLeiden plasma were also studied when coagulation was triggered via the intrinsic pathway with APTT reagent (Fig. 8). When APTT reagent was used to initiate intrinsic coagulation, normal plasma was approximately as sensitive to APC(S360A) as when the extrinsic pathway was triggered (Fig. 8A), but the FVLeiden plasma was more sensitive to APC(S360A) when the intrinsic pathway was activated (Fig. 8B). To estimate the relative potency of APC(S360A), the effect of wt-APC was examined in the same experimental setting. Under all conditions, wt-APC showed more anticoagulant activity than APC(S360A), a difference that was greatest for the extrinsically triggered plasmas, particularly for FVLeiden plasma.
FIGURE 8.
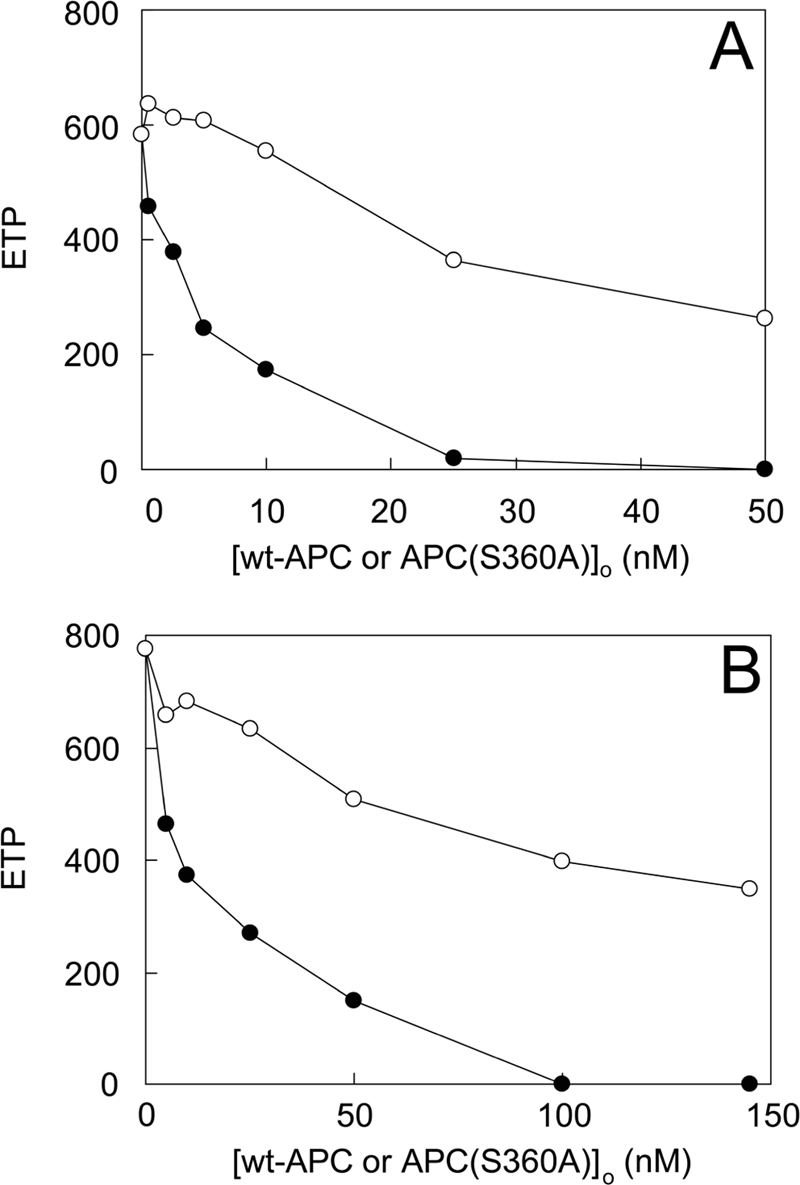
Effect of APC(S360A) on intrinsic thrombin generation in normal plasma and in FVLeiden plasma. Thrombin generation was measured by CAT as described under “Experimental Procedures” in normal plasma (A) or a plasma pool from three donors carrying the R506Q mutation (B) triggered via the intrinsic pathway in the presence of varying concentrations of wt-APC (●) or APC(S360A) (○). Averaged endogenous thrombin potential (ETP) values from three titrations are shown with standard errors of the mean/point not exceeding 15% of the measurement.
Role of the Intrinsic Tenase Complex in APC(S360A) Inhibition of Thrombin Generation
To verify that in extrinsically triggered plasma, regulation by APC(S360A) proceeded through inhibition of the prothrombinase and/or the tenase complexes, blocking antibodies to FVIII were used to assess the involvement of the tenase complex in thrombin formation under the conditions tested. At the TF concentration used, formation of thrombin was dependent on FX activation by the tenase complex (35, 36). Regulation through the prothrombinase complex was excluded by using FVLeiden plasma, based on the observation that FVLeiden was not influenced by APC(S360A). Other than an overall lowering of thrombin peak height, the addition of FVIII antibodies resulted in only a slightly altered inhibition by APC(S360A) (Fig. 9). From the slope of this curve and from the corresponding data for Figs. 6 and 8, it was possible to calculate the effect of APC(S360A) on the different physiological targets of APC (i.e. FVa and FVIIIa). The relative contributions to inhibition of the tenase complex by APC(S360A) in plasma did not exceed 10% of the total nonproteolytic activity of APC(S360A). As compared with the proteolytic activity of wt-APC, the nonproteolytic activity of APC(S360A) was 6% of the total activity of the active protease. The addition of antibodies to protein S in plasma did not result in an altered inhibition by APC(S360A), as was seen in the purified reaction systems (data not shown).
FIGURE 9.
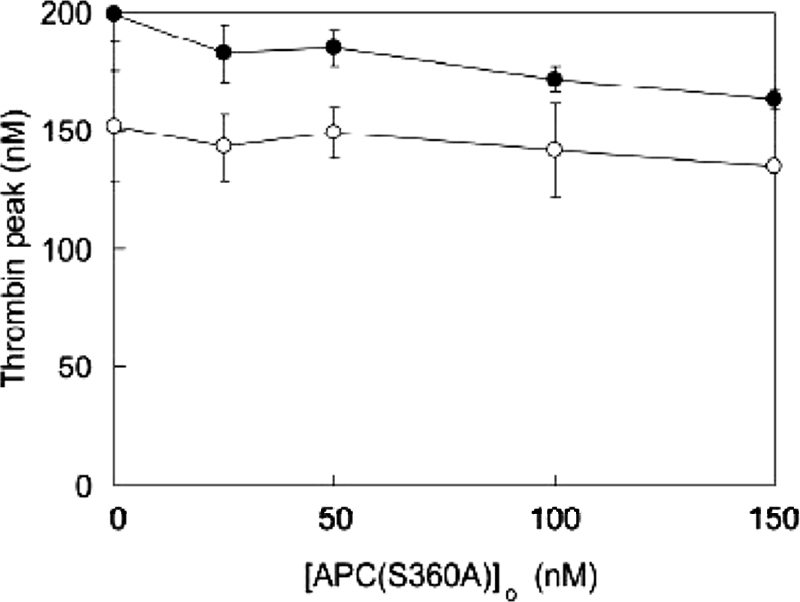
Effect of APC(S360A) on thrombin generation in extrinsically triggered FVLeiden plasma in the presence and absence of FVIII antibodies. Thrombin generation was triggered by the addition of 3.4 pm TF and 30 μm phospholipid vesicles (20:60:20, DOPS:DOPC:DOPE), and CAT was performed as described under “Experimental Procedures.” Varying total concentrations of APC(S360A) ([APC(S360A]o) were added to plasma from a carrier of the FV R506Q mutation in the absence (●) or presence (○) of a FVIII inhibitory antibody. Either 1.5 μl of polyclonal goat anti-human FVIII (final concentration, 54 BU/ml) or the same volume of buffer was added in a total reaction volume of 125 μl, and the plasma was incubated for 15 min at 37 °C prior to initiating thrombin generation. The average thrombin peak concentration ± S.E. (Thrombin peak) from two different experiments is plotted as a function of the total concentration of APC(S360A).
Effects of APC(S360A) on FVIIIa
Inhibition of FVIIIa activity by APC(S360A) was investigated in model systems of purified components. APC(S360A) inhibited activation of FX by the FIXa·FVIIIa complex in a dose-dependent manner (Fig. 10). Remarkably, half-maximal inhibition occurred at APC(S360A) concentrations that were 1 or 2 orders of magnitude higher than required for FVa inhibition (Fig. 1A). When FPR-APC was used, no significant inhibition was observed over the concentration range tested (0–950 nm). It was verified that in the model system, APC(S360A) also competed with wt-APC and inhibited APC-dependent inactivation of FVIIIa (data not shown).
FIGURE 10.
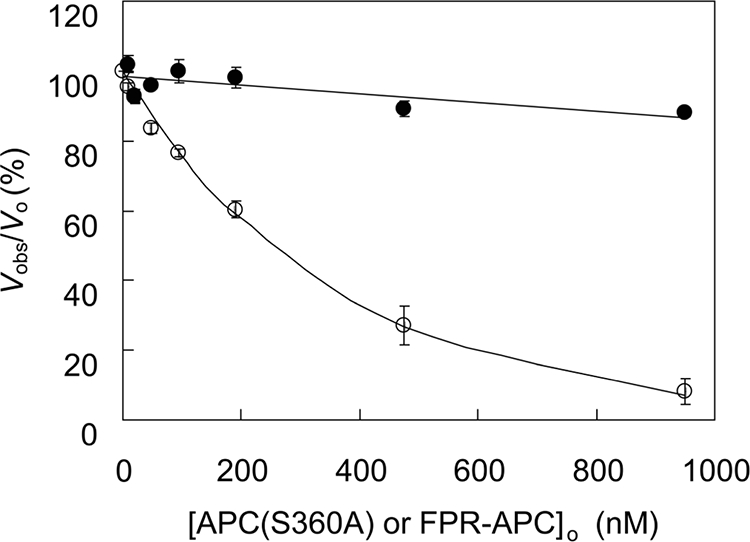
Effect of APC(S360A) and FPR-APC on FX activation by the intrinsic tenase complex. FVIIIa (140 pm) was incubated with 40 μm phospholipid vesicles (10:90, DOPS:DOPC) and varying total concentrations of FPR-APC (●) or APC(S360A) (○) in Buffer II for 15 min. FIXa (1 nm) was added, and after 15 s, FX activation was started by the addition of 400 nm FX. Samples from the reaction mixture were taken at 5 and 8 min, diluted in EDTA buffer, and assayed for FXa with the chromogenic substrate S-2765. Initial rates of FX activation were expressed as percentages of the rate of FX activation determined in the absence of APC (vobs/vo). The experiments were performed as described under “Experimental Procedures.” The average initial rate of FXa formation ± S.E. from two different experiments is plotted as a function of the total concentration of APC(S360A) ([APC(S360A)]o or FPR-APC [FPR-APC]o).
Molecular Modeling of APC and Its Derivatives
A set of molecular models was created to interpret the effects seen with the different active site-inhibited derivatives of APC. From superimposing of the four APC structures modeled, we conclude that the change in the active site caused by either covalent modification or mutagenesis did not alter the overall shape characteristics of the APC serine protease domain. The observed difference in inhibitory functions between these structures is caused by occupation of critical sites necessary for substrate binding and the accessible volume of the active site. In the case of FPR-APC, the entrance to the active site is obstructed by the inhibitor, which also engages the S1–S3 substrate-binding subsites, forms a hemiketal with Ser360(195), and alkylates His211(57), leaving the exosites of APC accessible. For PMS-APC, this is much less the case, with interactions of the region around Glu357(192) possibly hampered. The calculated volumes of the active site for each APC derivative: wt-APC, APC(S360A), FPR-APC, and PMS-APC were 1830, 1841, 1046, and 1402 Å2, respectively, quantitating the dominant obstructing effect of FPR on the volume of the APC active site (43% reduction of calculated volume). CONCOORD analysis of the PMS-APC structure, which probes structural flexibility, revealed that the PMS moiety has some conformational freedom that is not observed in FPR-APC, and consequently, PMS-APC could accommodate an inserted FVa 506-loop after a modest conformational change of the bound inhibitor. The fact that the active site volumes of APC(S360A) and PMS-APC are not identical adds to the explanation for the difference in inhibitory properties of these two APC derivatives (Fig. 7).
DISCUSSION
The present study supports the conclusion that APC(S360A) efficiently and completely down-regulates FXa and thrombin formation in the absence of proteolysis of its cofactor substrates, FVa and FVIIIa, both in model systems and in human plasma (37). Binding of APC to FVa or FVIIIa, the first step in their proteolytic inactivation, is sufficient to inhibit thrombin and FXa formation. Subsequent proteolysis of the cofactors ensures the irreversible loss of activity and presumably liberates APC for subsequent anticoagulant action toward other FVa and FVIIIa molecules. Inhibition of FVa cofactor activity by APC(S360A) requires the presence of Arg506 but not Arg306. The expression of nonproteolytic anticoagulant activity and the binding affinity of APC(S360A) for FVa is dependent on proteolytic activation of the protein C(S360A) mutant zymogen and almost totally dependent on the accessible active site in APC(S360A). FPR-labeled APC has greatly reduced anticoagulant activity and binding affinity for FVa, whereas PMS-labeled APC exhibits more modestly reduced anticoagulant activity. The strong dependence of the anticoagulant activity and FVa binding affinity on the intact substrate-binding site in APC(S360A) and on active site docking of the Arg506 side chain, which does not occur in FVaLeiden, shows that the binding of APC to FVa that precedes cleavage at Arg506 is largely determined by active site interactions rather than exosite-exosite interactions.
Mutagenesis studies (10, 12) indicate that patches of charged residues on FVa near Arg506 constitute an exosite for APC binding mediated by its electropositive exosite, which in the catalytic domain of APC is in a topologically similar position as exosite I of thrombin (38). If Arg506 active site docking is primarily responsible for the majority of the affinity of APC(S360A) for FVa, this may not be easily accommodated in the exosite-dependent mechanism of substrate recognition described by Krishnaswamy and co-workers (18, 39–42) for prothrombin activation. In this two-step mechanism, prothrombin initially binds to prothrombinase through exosites expressed on FXa. The active site of FXa in the exosite-bound substrate complex remains accessible to low molecular mass substrates and inhibitors. In the second, unimolecular step, the exosite-bound substrate docks into the active site of FXa and engages the scissile bond for cleavage.
The dissociation constant of the APC(S360A)·FVa complex obtained in the present studies (0.11 ± 0.05 nm) is ∼18-fold lower than the Km for APC cleavage of FVa at Arg506 of 2 nm (12). Active site docking interactions are of major importance for the formation of the APC(S360A)·FVa complex, as shown by its dependence on accessibility of the active site and the loss of high affinity binding for the R506Q P1-substituted cleavage site. This shows that a two-step exosite-dependent binding mechanism does not apply to APC-FVa interaction at Arg506 but that the binding mechanism is similar to that described for the interaction between protein C and the thrombin·(soluble) TM complex. As is the case for APC and FVa, there is mutagenesis and structural evidence for exosite-mediated substrate recognition for protein C activation by thrombin·TM, but kinetic analysis demonstrated that substrate affinity was primarily dependent on active site interactions. A P1-Arg to Gln mutant of protein C that was not cleaved by thrombin·TM was a very weak inhibitor of protein C activation, providing further evidence for the importance of active site interactions rather than exosite interactions in the protein C system. Similarly, the mutation in FVaLeiden did not demonstrably affect FXa binding but greatly decreased the affinity of APC(S360A) binding. Our binding studies show, however, detectable affinities of FPR-APC for FVa and APC(S360A) for FVaLeiden with apparent KD values of 36 ± 9 and 38 ± 27 nm for the FPR-APC·FVa and APC(S360A)·FVaLeiden complexes, respectively, which could represent residual exosite-mediated contributions to affinity.
Our findings are in agreement with those reported earlier for APC(S360A) by Gale et al. (37); although the assay conditions and experimental approaches used in the present study are different, inhibition of prothrombinase was also reported (37). The modifying effects that FXa, protein S, and prothrombin have on the expression of the anticoagulant properties of APC(S360A) were not studied, however, and all effects were attributed to FVa. In a more recent publication Yegneswaran et al. (11) propose that the mode of action of APC(S360A) is through competition between APC and FXa, which is in agreement with our observations. It was also observed previously that APC(S360A) has much less effect on the clotting (APTT) of plasma of a homozygous carrier of the FVLeiden mutation (37). Using purified proteins, we show that APC(S360A) lacks high affinity and nonproteolytic anticoagulant activity in the case of FVaLeiden, because it does not compete with the binding of FXa to FVaLeiden. We conclude that Arg506 and not Arg306 is critical to the inhibition by APC(S360A) of the FXa·FVa complex by competitive displacement of FXa from FVa.
The absence of nonproteolytic inhibition of the FXa·FVaLeiden interaction by APC(S360A) may represent an previously unrecognized contribution to the APC resistance phenotype. The R506Q mutation in FVaLeiden impairs the down-regulation of thrombin formation by APC in three ways: 1) the absence of the APC cleavage site at Arg506 results in slow inactivation of FVLeiden by cleavage at Arg306; 2) the absence of a proper binding site for APC at Arg506 blocks the ability of APC to directly inhibit thrombin formation by dissociation of FXa·FVa complex formation; and 3) FVaLeiden does not enhance APC-catalyzed FVIIIa inactivation (43, 44). As to the relative importance of the proteolysis-independent or -dependent activities of APC, we estimate in normal plasma that ∼6% of APC activity can be attributed to the proteolysis-independent activity. However, expression of this activity (binding to FVa) is a prerequisite for expression of the proteolytic activity, because in the absence of high affinity FVa binding (like in FVaLeiden), virtually no inhibition of FVa is observed with APC(S360A).
Several studies provided evidence for overlap between binding sites of FXa and APC on FVa that explain how FXa protects FVa against inactivation by APC (11, 12, 32, 45, 46). A similar protection has been reported for the APC-catalyzed proteolysis of FVIIIa by FIXa (47). Inhibition of prothrombinase by APC(S360A) is enhanced at low FXa concentrations. Prothrombin effectively abolished the anticoagulant effect APC(S360A) in the model system, which is in agreement with recent results (34). These observations are in line with an anticoagulant mechanism in which APC(S360A) inhibits the cofactor activity of FVa by competing with FXa and prothrombin for binding to FVa. The binding regions on FVa for prothrombin have been less well described. However, the fact that prothrombin inhibits APC-catalyzed FVa inactivation in the absence of FXa (33–34) suggests that APC and prothrombin also share binding sites on FVa. The protective effect of prothrombin in the presence of OG-FXa can be explained by formation of a nonproductive ternary OG-FXa·FVa·prothrombin complex.4 The established thermodynamic linkage between protein-membrane and protein-protein interactions in facilitating the high affinity formation of prothrombinase (48), which may also be enhanced by prothrombin binding through sites on FVa and FXa, may be involved in the resistance of FXa sequestered in the ternary complex from inhibition through binding to APC(S360A). The observations that prothrombin effectively counteracts the APC(S360A)-mediated dissociation of the FXa-FVa complex, whereas APC(S360A) is an inhibitor of prothrombin activation and thrombin formation in model systems and in plasma, need an explanation. The fluorescence experiments were performed with active site-labeled FXa, which binds to FVa to form an inactive complex, whereas the prothrombin activation experiments in model systems and thrombin generation in plasma involve an active FXa·FVa complex that rapidly activates prothrombin, and which, after subsequent dissociation of the products (thrombin and fragment 1.2), becomes available to bind new ligands. Because of the conversion of prothrombin, the local concentration of prothrombin in the neighborhood of the FXa·FVa complex may be considerably lower than bulk concentration. This suggests that continuous conversion of prothrombin may explain why prothrombin is a less effective competitor in the kinetic than in the (static) fluorescence experiments. The possibility that occupation of membrane-binding sites by APC(S360A) is the cause of the inhibition of prothrombinase in the model system was excluded by the observation that the addition of up to 4.1 μm prothrombin fragment 1 had no effect on the activity of the prothrombinase complex.
Inhibition by APC(S360A) was investigated primarily in the context of the prothrombinase complex, whereas in the plasma environment, it is likely that APC(S360A) targets not only FVa but also FVIIIa. In a model system, FVIIIa cofactor activity was also inhibited by APC(S360A), but ∼100-fold higher concentrations of APC(S360A) were required for inhibition of intrinsic tenase than prothrombinase. On this basis, it can be concluded that regulation of procoagulant activities by APC(S360A) is much more efficient in the case of prothrombinase. This was also seen in plasma of a homozygous carrier of the FVLeiden phenotype, where inhibition of FVIII only marginally altered the susceptibility to inhibition by APC(S360A). This suggests that FVa and to a much lesser extent FVIIIa are targets for APC(S360A). The effects of APC(S360A) in plasma experiments were equal to or larger than those seen in model systems. A potential explanation for the more pronounced effect of APC(S360A) in plasma being due to the formation of minute amounts of wt-APC from protein C in the activating plasma was ruled out because, in a control experiment, the effects of APC(S360A) were the same in protein C-deficient plasma and its parent plasma.
Given its activities in plasma and its inertness toward inhibition by serpins, APC(S360A) has strong potential to evolve into an anticoagulant that can be clinically applied. It has a long half-life in plasma (37) and may potentially have less bleeding side effects than other anticoagulants because its anticoagulant properties are only expressed during the initiation phase of coagulation, when low concentrations of TF and FXa are present. When higher concentrations of TF become exposed, such as in more pronounced damage to blood vessels, APC(S360A) hardly inhibits thrombin formation, and its anticoagulant activities can be bypassed.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 HL038779 (to P. E. B.). This work was also supported by VIDI Grant 916-046-330 from the Netherlands Organization for Scientific Research (to G. A. F. N.).
Schechter-Berger (49) notation refers to the residues of a substrate (from the NH2-terminal end) as …P4-P3-P2-P1-P1′-P2′…, with the scissile bond at P1-P1′, which interact with complementary specificity sites …S4-S3-S2-S1-S1′-S2′… on the proteinase.
P. E. Bock, unpublished results.
- APC
- activated protein C
- wt-APC
- recombinant wild type APC
- APC(S360A)
- recombinant APC with Ser360 substituted with Ala
- DOPS
- 1,2 dioleoyl-sn-glycero-3-phosphoserine
- DOPC
- 1,2 dioleoyl-sn-glycero-3-phosphocholine
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- FV
- coagulation factor V
- FVa
- activated FV
- FVIIIa
- activated FVIII
- FVLeiden
- FV(R506Q)
- FPR-CH2Cl
- d-Phe-Pro-Arg-CH2Cl
- FX
- coagulation factor X
- FXa
- activated coagulation factor X
- FIXa
- activated coagulation factor IX
- MES
- 4-morpholineethanesulfonic acid
- OG
- Oregon Green 488-iodoacetamide, 5- (and 6)-iodoacetamido-2′,7′-difluorofluorescein
- OG-FXa
- FXa inactivated with Nα-[(acetylthio)acetyl]-d-Phe-Pro-Arg-CH2Cl and labeled with OG
- PMSF
- phenylmethylsulfonyl fluoride
- S-2238
- H-d-Phe-Pip-Arg-p-nitroanilide
- S-2366
- l-pyroGlu-Pro-Arg-p-nitroanilide
- S-2765
- Z-d-Arg-Gly-Arg-p-nitroanilide
- TF
- tissue factor
- TM
- thrombomodulin
- ELISA
- enzyme-linked immunosorbent assay
- CAT
- calibrated automated thrombinography
- APTT
- activated partial thromboplastin time.
REFERENCES
- 1.Marlar R. A., Montgomery R. R., Broekmans A. W. (1989) J. Pediatr. 114, 528–534 [DOI] [PubMed] [Google Scholar]
- 2.Seligsohn U., Berger A., Abend M., Rubin L., Attias D., Zivelin A., Rapaport S. I. (1984) N. Engl. J. Med. 310, 559–562 [DOI] [PubMed] [Google Scholar]
- 3.Broekmans A. W., Veltkamp J. J., Bertina R. M. (1983) N. Engl. J. Med. 309, 340–344 [DOI] [PubMed] [Google Scholar]
- 4.Griffin J. H., Evatt B., Zimmerman T. S., Kleiss A. J., Wideman C. (1981) J. Clin. Invest. 68, 1370–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedrich U., Nicolaes G. A., Villoutreix B. O., Dahlbäck B. (2001) J. Biol. Chem. 276, 23105–23108 [DOI] [PubMed] [Google Scholar]
- 6.Gale A. J., Heeb M. J., Griffin J. H. (2000) Blood 96, 585–593 [PubMed] [Google Scholar]
- 7.Gale A. J., Tsavaler A., Griffin J. H. (2002) J. Biol. Chem. 277, 28836–28840 [DOI] [PubMed] [Google Scholar]
- 8.Nicolaes G. A., Dahlbäck B. (2003) Semin. Vasc. Med. 3, 33–46 [DOI] [PubMed] [Google Scholar]
- 9.Villoutreix B. O., Covell D. G., Blom A. M., Wallqvist A., Friedrich U., Dahlbäck B. (2001) J. Comput. Aided Mol. Des. 15, 13–27 [DOI] [PubMed] [Google Scholar]
- 10.Segers K., Dahlbäck B., Rosing J., Nicolaes G. A. (2008) J. Biol. Chem. 283, 22573–22581 [DOI] [PubMed] [Google Scholar]
- 11.Yegneswaran S., Kojima Y., Nguyen P. M., Gale A. J., Heeb M. J., Griffin J. H. (2007) J. Biol. Chem. 282, 28353–28361 [DOI] [PubMed] [Google Scholar]
- 12.Nicolaes G. A., Sørensen K. W., Friedrich U., Tans G., Rosing J., Autin L., Dahlbäck B., Villoutreix B. O. (2004) Eur. J. Biochem. 271, 2724–2736 [DOI] [PubMed] [Google Scholar]
- 13.Manithody C., Fay P. J., Rezaie A. R. (2003) Blood 101, 4802–4807 [DOI] [PubMed] [Google Scholar]
- 14.Nicolaes G. A., Tans G., Thomassen M. C., Hemker H. C., Pabinger I., Varadi K., Schwarz H. P., Rosing J. (1995) J. Biol. Chem. 270, 21158–21166 [DOI] [PubMed] [Google Scholar]
- 15.Mather T., Oganessyan V., Hof P., Huber R., Foundling S., Esmon C., Bode W. (1996) EMBO J. 15, 6822–6831 [PMC free article] [PubMed] [Google Scholar]
- 16.Baglin T. P., Carrell R. W., Church F. C., Esmon C. T., Huntington J. A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11079–11084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye S., Cech A. L., Belmares R., Bergstrom R. C., Tong Y., Corey D. R., Kanost M. R., Goldsmith E. J. (2001) Nat. Struct. Biol. 8, 979–983 [DOI] [PubMed] [Google Scholar]
- 18.Hacisalihoglu A., Panizzi P., Bock P. E., Camire R. M., Krishnaswamy S. (2007) J. Biol. Chem. 282, 32974–32982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baugh R. J., Dickinson C. D., Ruf W., Krishnaswamy S. (2000) J. Biol. Chem. 275, 28826–28833 [DOI] [PubMed] [Google Scholar]
- 20.Gailani D., Ho D., Sun M. F., Cheng Q., Walsh P. N. (2001) Blood 97, 3117–3122 [DOI] [PubMed] [Google Scholar]
- 21.Lu G., Chhum S., Krishnaswamy S. (2005) J. Biol. Chem. 280, 15471–15478 [DOI] [PubMed] [Google Scholar]
- 22.Bock P. E., Craig P. A., Olson S. T., Singh P. (1989) Arch. Biochem. Biophys 273, 375–388 [DOI] [PubMed] [Google Scholar]
- 23.Panizzi P., Friedrich R., Fuentes-Prior P., Kroh H. K., Briggs J., Tans G., Bode W., Bock P. E. (2006) J. Biol. Chem. 281, 1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicolaes G. A., Villoutreix B. O., Dahlbäck B. (1999) Biochemistry 38, 13584–13591 [DOI] [PubMed] [Google Scholar]
- 25.Sun Y. H., Shen L., Dahlbäck B. (2003) Blood 101, 2277–2284 [DOI] [PubMed] [Google Scholar]
- 26.Friedrich U., Pötzsch B., Preissner K. T., Müller-Berghaus G., Ehrlich H. (1994) Thromb. Haemost. 72, 567–572 [PubMed] [Google Scholar]
- 27.Shen L., Shah A. M., Dahlbäck B., Nelsestuen G. L. (1998) J. Biol. Chem. 273, 31086–31091 [DOI] [PubMed] [Google Scholar]
- 28.Hemker H. C., Giesen P., Al Dieri R., Regnault V., de Smedt E., Wagenvoord R., Lecompte T., Béguin S. (2003) Pathophysiol. Haemost. Thromb. 33, 4–15 [DOI] [PubMed] [Google Scholar]
- 29.Bock P. E., Olson S. T., Björk I. (1997) J. Biol. Chem. 272, 19837–19845 [DOI] [PubMed] [Google Scholar]
- 30.de Groot B. L., van Aalten D. M., Scheek R. M., Amadei A., Vriend G., Berendsen H. J. (1997) Proteins 29, 240–251 [DOI] [PubMed] [Google Scholar]
- 31.Segers K., Rosing J., Nicolaes G. A. (2006) Proteins 64, 968–984 [DOI] [PubMed] [Google Scholar]
- 32.Rosing J., Hoekema L., Nicolaes G. A., Thomassen M. C., Hemker H. C., Varadi K., Schwarz H. P., Tans G. (1995) J. Biol. Chem. 270, 27852–27858 [DOI] [PubMed] [Google Scholar]
- 33.Tran S., Norstrøm E., Dahlbäck B. (2008) J. Biol. Chem. 283, 6648–6655 [DOI] [PubMed] [Google Scholar]
- 34.Yegneswaran S., Nguyen P. M., Gale A. J., Griffin J. H. (2009) Thromb. Haemost. 101, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Josso F., Prou-Wartelle O. (1965) Thromb. Diath. Haemorrh. Suppl. 17, 35–44 [PubMed] [Google Scholar]
- 36.Cawthern K. M., van 't Veer C., Lock J. B., DiLorenzo M. E., Branda R. F., Mann K. G. (1998) Blood 91, 4581–4592 [PubMed] [Google Scholar]
- 37.Gale A. J., Sun X., Heeb M. J., Griffin J. H. (1997) Protein Sci. 6, 132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bock P. E., Panizzi P., Verhamme I. M. (2007) J Thromb. Haemost. 5, (Suppl. 1) 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bianchini E. P., Orcutt S. J., Panizzi P., Bock P. E., Krishnaswamy S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10099–10104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradford H. N., Micucci J. A., Krishnaswamy S. (2010) J. Biol. Chem. 285, 328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krishnaswamy S. (2005) J. Thromb. Haemost. 3, 54–67 [DOI] [PubMed] [Google Scholar]
- 42.Orcutt S. J., Krishnaswamy S. (2004) J. Biol. Chem. 279, 54927–54936 [DOI] [PubMed] [Google Scholar]
- 43.Shen L., Dahlbäck B. (1994) J. Biol. Chem. 269, 18735–18738 [PubMed] [Google Scholar]
- 44.Váradi K., Rosing J., Tans G., Schwarz H. P. (1995) Thromb. Haemost. 73, 730–731 [PubMed] [Google Scholar]
- 45.Kojima Y., Heeb M. J., Gale A. J., Hackeng T. M., Griffin J. H. (1998) J. Biol. Chem. 273, 14900–14905 [DOI] [PubMed] [Google Scholar]
- 46.Norstrøm E. A., Tran S., Steen M., Dahlbäck B. (2006) J. Biol. Chem. 281, 31486–31494 [DOI] [PubMed] [Google Scholar]
- 47.O'Brien L. M., Mastri M., Fay P. J. (2000) Blood 95, 1714–1720 [PubMed] [Google Scholar]
- 48.Krishnaswamy S. (1990) J. Biol. Chem. 265, 3708–3718 [PubMed] [Google Scholar]
- 49.Schechter I., Berger A. (1967) Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]