

Abstract
The transcription factor nuclear factor E2-related factor 2 (Nrf2) induces the expression of antioxidant gene products that neutralize reactive oxygen species and restore redox homeostasis. Nrf2 is constitutively degraded by the ubiquitin proteolytic system in unperturbed cells, but this turnover is arrested in response to oxidative stress, thereby leading to Nrf2 accumulation. Yet, a mechanistic understanding of how Nrf2 stabilization and transcriptional activation are coupled remains to be determined. We have discovered that the ubiquitin-conjugating enzyme UbcM2 is a novel regulator of Nrf2. Recombinant Nrf2 and UbcM2 form a complex upon alkylation of a non-catalytic cysteine in UbcM2, Cys-136. Substitution of this cysteine with a phenylalanine (C136F) to mimic cysteine oxidation/alkylation results in constitutive binding of UbcM2 to Nrf2 and an increased half-life of the transcription factor in vivo. We provide evidence that UbcM2 and Nrf2 form a nuclear complex utilizing the DNA binding, Neh1 domain, of Nrf2. Finally, we demonstrate that UbcM2 can enhance the transcriptional activity of endogenous Nrf2 and that Cys-136 and the active-site cysteine, Cys-145, jointly contribute to this regulation. Collectively, these data identify UbcM2 as a novel component of the Nrf2 regulatory circuit and position cysteine 136 as a putative redox sensor in this signaling pathway. This work implicates UbcM2 in the restoration of redox homeostasis following oxidative stress.
Keywords: Oxidative Stress, Reactive Oxygen Species (ROS), Transcription Factors, Ubiquitin, Ubiquitin Conjugating Enzyme (Ubc), Nrf2
Introduction
Eukaryotic cells possess a host of mechanisms to neutralize reactive oxygen species produced by oxidative metabolism and xenobiotics. Nrf22 is a central component of the endogenous antioxidant defense system. Nrf2 is a basic leucine zipper transcription factor that heterodimerizes with the small Maf proteins (1, 2) to bind a cis-acting, antioxidant response element (ARE) in the promoters of phase 2 genes (3). Nrf2 activates both the basal and inducible expression of a battery of antioxidant gene products that function collectively to eliminate reactive oxygen species. Studies using knock-out mice have highlighted the diverse cytoprotective functions of Nrf2. These range from mitigating acetaminophen hepatotoxicity (4) to preventing acute pulmonary injury (5) and carcinogenesis (e.g. Ref. 6). Numerous natural (e.g. sulforaphane from broccoli) and synthetic (e.g. oltipraz) antioxidants confer their protective effects by stabilizing and activating Nrf2 (6, 7).
The stability and activity of Nrf2 are directly coupled to the cellular redox state. In homeostatic cells, Nrf2 is degraded by the UPS (8, 9). This degradation is mediated by a UPS E3 ligase called CUL3Keap1 (10, 11). CUL3Keap1 is comprised of three proteins: the cullin 3 (CUL3) scaffold protein, a really interesting new gene (RING)-finger containing protein, ROC1, and the substrate adaptor, Keap1. Keap1 recruits Nrf2 to a CUL3-ROC1 complex for polyubiquitylation. The tagged transcription factor is subsequently delivered to the 26 S proteasome for degradation. Oxidative stress, however, arrests the function of CUL3Keap1 and thereby stabilizes Nrf2. The transcription factor is rapidly transported into the nucleus and induces the expression of phase 2 genes (all reviewed in Ref. 12).
Despite progress in understanding how Nrf2 degradation is regulated, there are still unresolved issues regarding the mechanism(s) by which Nrf2 is “sheltered” from CUL3Keap1 following oxidative stress and how the stability, nuclear accumulation, and transcriptional activity of Nrf2 are coupled. For example, it is widely held that Nrf2 is sequestered in the cytoplasm in unstressed cells but is liberated from its cytoplasmic anchor, Keap1, by oxidative stress and translocates into the nucleus to induce transcription (e.g. Refs. 13–16). In contrast, Pickett and colleagues posit that Nrf2 is a resident nuclear protein (17), whereas Keap1 is a nucleocytoplasmic shuttling protein (18) that promotes Nrf2 turnover in the nucleus (17). More recently, it has been demonstrated that, following the restoration of redox homeostasis, Keap1 enters the nucleus to bind and export Nrf2 into the cytoplasm for degradation by CUL3Keap1 (19). A common feature of these models is that Keap1 mediates the ubiquitin-dependent turnover of Nrf2. However, these models of Nrf2 repression by Keap1 differ on a key question. Does Keap1 target Nrf2 for degradation in the cytoplasm or the nucleus or both? In addition, it remains to be fully elucidated how Nrf2 is degraded following an oxidative stress, because the half-life of the transcription factor only doubles in unstressed cells versus stressed cells (8, 9). This extended, yet limited, lifespan for Nrf2 appears to be a critical regulatory mechanism as reflected by the pathological consequences associated with uncontrolled Nrf2 accumulation in Keap1 knock-out mice (20). It is thus likely that one or more additional mechanisms exist for controlling Nrf2 stability in stressed cells, and experimental evidence for such a mechanism has been reported (21, 22).
In the work presented here, we have identified the E2 UbcM2 as a novel regulator of nuclear Nrf2 stability. This regulation of Nrf2 appears to be mediated by a direct interaction between the enzyme and the transcription factor. Further, the interaction is principally mediated by a non-catalytic cysteine residue (Cys-136) within UbcM2 and by the Neh1 domain of Nrf2. The consequences of this interaction are an increased half-life and enhanced stabilization of Nrf2 in the nucleus and a corresponding increase in Nrf2 transcriptional activation. We also show that the active-site cysteine of UbcM2 contributes to regulating Nrf2 transcriptional activation. Although numerous transcription factors are modulated in a non-catalytic fashion by the small ubiquitin-like modifier (SUMO) E2 Ubc9 (23–27), this report represents the first demonstration that a ubiquitin E2 can directly engage and regulate a transcription factor utilizing a non-catalytic cysteine. Furthermore, the data support the intriguing model that Cys-136 of UbcM2 may function as a redox sensor to enhance Nrf2 stability and activity during oxidative challenge and the restoration of redox homeostasis.
EXPERIMENTAL PROCEDURES
Antibodies and Chemicals
H-300 is a rabbit polyclonal anti-Nrf2 antibody from Santa Cruz Biotechnology and was used to detect recombinant H6-S-Nrf2. Anti-HA, anti-Myc, and anti-UbcM2 have been described previously (28, 29). tert-Butylhydroquinone (tBHQ) was purchased from Sigma-Aldrich (catalog no. 112941-100G), and MG132 from Boston Biochem (catalog no. I-130).
Transfections, Immunoprecipitations, and Indirect Immunofluorescence
Human embryonic kidney (HEK293T) and HeLa cells were transfected by the calcium phosphate method (30, 31). Overexpressed Nrf2 was triple hemagglutinin-tagged (HA3) and E2s were Myc-tagged. 2–3 days post-transfection, cells were treated for 3 h with 50 μm cycloheximide to block new protein translation and then either solubilized in two times concentrated Laemmli buffer in preparation for SDS-PAGE and Western blotting or alternatively, were lysed on ice for 15 min in binding buffer (10 mm HEPES-KOH, pH 7.4, 55 mm potassium acetate, 1 mm magnesium acetate, 0.1 mm EGTA, 0.25% Tween 20, 150 mm NaCl). Lysates were clarified by centrifugation and combined with α-Myc 9E10 monoclonal antibodies and protein-G Sepharose to recover complexes. For immunofluorescence studies, 5 × 105 HeLa cells were plated on glass coverslips, transfected, and 2 days later fixed in 3.7% formaldehyde in phosphate-buffered saline (PBS). Following permeabilization with −20 °C methanol for 2 min, cells were sequentially blocked in 3% bovine serum albumin/PBS, incubated with α-Myc 9E10 (diluted 1:500), and then with an anti-mouse Alexa546 nm secondary antibody (diluted 1:2000). Counterstaining of DNA was accomplished by including 0.1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) during the secondary antibody incubation. Epifluorescence analysis was done on a Nikon TE2000U inverted fluorescence microscope equipped with a 60× water immersion objective (numerical aperture = 1.2), and images were captured with a Hamamatsu Orca AG charge-cooled CCD camera. Image processing was done with the Openlab suite of imaging software (Improvision, Inc., Lexington, MA) and Adobe Photoshop (version 8.0).
Nrf2 Half-life Experiments
6 × 105 HEK293T cells were plated in 3-cm dishes and transiently transfected by the calcium phosphate method with plasmids expressing HA3-Nrf2 and either Myc-tagged yellow fluorescent protein (YFP) or the Myc-tagged point mutation (C136F)-UbcM2. 2 days later, pulse-chase/immunoprecipitation experiments were carried out using slight modifications of the “Ultimate Mammalian Cell Pulse-Chase and Denaturing IP” methods (available on-line from the Tansey Laboratory). Cells were incubated in methionine/cysteine-free Dulbecco's modified Eagle's medium plus 10% dialyzed fetal calf serum for 30 min and then pulse-labeled with 160 μCi of Tran35Slabel (MP Bio, Inc.)/dish. After 40 min, the radioactive media was removed, and the cells were washed several times with warm PBS and then incubated in complete Dulbecco's modified Eagle's medium plus 10% fetal calf serum for 0, 15, 30, or 60 min (chase times). To prepare lysates, cells were collected in 0.5 ml of ice-cold PBS, pelleted at 500 × g for 3 min, and snap frozen in liquid N2. After collecting all samples, cell pellets were processed in parallel by thawing, resuspending in 35 μl of TS buffer (50 mm Tris-HCl (pH 7.5) plus 1% SDS), and placed in a boiling water bath for 10 min. Lysates were clarified by centrifugation at 13,200 rpm for 10 min, transferred to fresh tubes, and diluted by adding 100 μl of TNN buffer (50 mm Tris-HCl (pH 7.5) plus 150 mm NaCl plus 5 mm EDTA plus 0.5% Nonidet P-40). The lysates were subsequently pre-cleared by incubation with 20 μl of protein A Sepharose. 100 μg of pre-cleared lysate was combined with 3 μl of anti-HA 12CA5 mAb and 25 μl of protein A-Sepharose and incubated at 4 °C for 2 h. The immunoprecipitations were washed with 2 × 1 ml of ice-cold TNN buffer and solubilized in 30 μl of 2 times concentrated Laemmli buffer in preparation for SDS-PAGE. Gels were stained with Coomassie Brilliant Blue (CBB), destained, and dried onto Whatman paper, and direct exposures were made at room temperature using X-OMAT Blue film. Films were scanned, and band intensities were quantified by calculating the area under the curve using NIH ImageJ software. Half-life measurements were calculated using the formula, T½ = (t*ln2)/(ln R0 − ln Rt), where t = chase time, R0 = band intensity at t0, Rt = intensity at t. Final assignments were derived from averaging T½ measurements from three independent experiments, and statistical significance was calculated using a two-tailed Student's t test.
In Vitro Binding Assays
Recombinant fusion proteins were expressed and purified from BL21(star) Escherichia coli as described previously (29). Recombinant pulldown assays were done by combining equimolar amounts of glutathione S-transferase (GST) and H6-S fusion proteins with GSH-Sepharose in binding buffer for 3 h at room temperature. Reactions were supplemented with either 1 or 2 mm dithiothreitol (DTT) as noted in the text or the indicated amounts of N-ethylmaleimide (NEM). Complexes were resolved by SDS-PAGE and analyzed by Western blotting and/or CBB staining. Reciprocal pulldown assays were done as above replacing GSH-Sepharose with Ni2+-nitrilotriacetic acid resin.
Auto-ubiquitylation Assays
HEK293T cells were transiently transfected with plasmids encoding Myc-UbcM2 (wt), Myc-(C136F)-UbcM2, or Myc-(C145A)-UbcM2. 2–3 days later, the cells were lysed, and the enzymes were immunoprecipitated with α-Myc antibody and protein-G-Sepharose. One-half of each bead-bound sample was solubilized with 2 times concentrated Laemmli solubilizing buffer, and the other one-half was incubated at 37 °C for 2 h in a ubiquitin reaction mixture consisting of recombinant E1 (62.5 nm), ubiquitin (235 μm), and an ATP-regenerating system (100 mm Tris-HCl (pH 7.4), 0.4 mm MgATP, 1 mm MgCl2, 0.2 mm DTT, 2 mm creatine phosphate, 0.2% Tween 20, 108 units of creatine phosphokinase). Reactions were terminated by the addition of 2 times concentrated Laemmli solubilizing buffer, and reaction products were analyzed by SDS-PAGE and α-UbcM2 Western blotting.
Transcription Assays
1 × 105 HEK293T cells were seeded per well and co-transfected with 50 ng of a pARE-luciferase plasmid (a kind gift of Dr. Mark Hannink, University of Missouri), 1 ng of a RSV-LACZ plasmid (a kind gift of Dr. Ralf Janknecht, University of Oklahoma Health Sciences Center), and 40 ng of either pK-Myc, pK-Myc-UbcM2 (wt), pK-Myc-UbcM2 (C136F), pK-Myc-UbcM2 (C136F and C145A), pK-Myc-UbcM2 (C145A), pK-Myc-UbcM2 (C136P), or pK-Myc-UbcM2 (C136P and C145A). 2 days post-transfection, cells were treated with vehicle or 25 μm tBHQ for 16 h prior to harvesting. Cells were lysed in Passive Lysis Buffer (Promega, Madison, WI) at room temperature for 15 min. Equal volumes (25 μl) of lysate and Bright-Glo Luciferase Reagent (Promega, Inc) were combined and read in a TD-20/20 Luminometer (Turner Designs, Inc.) for 20 s. In parallel, β-galactosidase activity was measured to normalize for transfection efficiency. 10 μl of lysate was combined with 100 μl of Z-buffer (60 mm NaHPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4, adjust pH to 7.0, and add 2 mm DTT plus 1 mg/ ml ortho-nitrophenyl-β-galactoside (ONPG) fresh), incubated at 37 °C for at least 10 min, and then read in a Benchmark Microplate Reader spectrophotometer (Bio-Rad) at 405 nm.
RESULTS
Although it is clear that Nrf2 is stabilized and transcriptionally activated following an oxidative insult, open questions remain as to the mechanism(s) by which the stabilization, nuclear accumulation, and activation of the transcription factor are coupled. Our previous work indicated that the nuclear E2 UbcM2 might contribute to this process. This enzyme engages CUL3 E3 ligases in concert with their substrate adaptors (29).
To investigate a possible role for UbcM2 in oxidant-induced Nrf2 stabilization, recombinant pulldown assays were done to determine if the two proteins could directly interact. Recombinant forms of GST-UbcM2 and H6-S-Nrf2 were combined with GSH-Sepharose, and complex formation was analyzed by Western blotting. Only a minimal level of interaction was detected in the presence of 2 mm DTT (Fig. 1A, lane 1), however, the proteins interacted robustly in the presence of increasing amounts of the alkylating agent, NEM (Fig. 1A, lanes 3–6). NEM was added to mimic the thiol-reactive environment generated in cells during oxidative stress. Because NEM specifically modifies cysteines, we further determined whether complex formation required that UbcM2, Nrf2, or both proteins be alkylated. To test if alkylation of UbcM2 was necessary and sufficient to induce Nrf2 binding, bead-immobilized GST-UbcM2 was pre-treated with either buffer or NEM for 1 h and unreacted NEM was removed by washing the bead-bound fusion protein. When alkylated GST-UbcM2 was combined with DTT-treated H6-S-Nrf2, complex formation was observed (Fig. 2B, lane 4) indicating that alkylation of UbcM2 is sufficient to induce binding to Nrf2. In contrast, pretreatment of H6-S-Nrf2 with NEM was not sufficient to promote binding to non-alkylated UbcM2 (Fig. 1B, lane 3). Consistent with Fig. 1A, the addition of NEM to binding reactions promoted complex formation (Fig. 1B, lanes 2 and 5, respectively). The NEM-inducible interaction between Nrf2 and UbcM2 was also demonstrated by reciprocal pulldown assays in which H6-S-Nrf2 was immobilized on Ni2+-nitrilotriacetic acid beads and combined with a mixture of GST-UbcM2 and either DTT or NEM (Fig. 1C). Collectively, these data demonstrate that alkylation of UbcM2 is necessary and sufficient for binding to recombinant Nrf2.
FIGURE 1.

UbcM2 and Nrf2 interact directly in vitro under conditions that promote cysteine alkylation. A, recombinant pulldown assay with GST-UbcM2 and H6-S-Nrf2 in the presence of 2 mm DTT (lane 1) or increasing amounts of NEM (lanes 2–6). The top panel shows precipitated H6-S-Nrf2 detected with α-Nrf2, and the bottom panel shows immobilized GST-UbcM2 detected with CBB. 1% of the H6-S-Nrf2 input is shown in the leftmost lane of the top panel. B, similar assay to A to determine whether Nrf2, UbcM2, or both proteins require modification by NEM to induce complex formation. GST-UbcM2 immobilized on GSH-Sepharose was either pre-treated with buffer (lanes 1–3) or NEM (lanes 4–6) for 1 h, and excess NEM was washed away. For samples in which soluble H6-S-Nrf2 was pre-treated with NEM, the excess alkylating agent was quenched with DTT (lanes 3 and 6). For all other samples, H6-S-Nrf2 was added to binding reactions supplemented with either DTT (lanes 1 and 4) or NEM (lanes 2 and 5). C, same as A except H6-S-Nrf2 was immobilized on Ni2+-nitrilotriacetic acid resin and combined with GST-UbcM2 pre-treated with DTT (lanes 1 and 3) or NEM (lanes 2 and 4). GST-UbcM2 inputs are shown in the right panel. GST-UbcM2 was detected with anti-GST and H6-S-Nrf2 with α-Nrf2. D, same as A testing the requirement for an intact active-site cysteine (Cys-145) in UbcM2. All experiments were repeated three independent times.
FIGURE 2.

A unique cysteine shared by the class III E2s mediates recombinant Nrf2 binding. A, alignment of mammalian E2 amino acid sequences flanking the active-site cysteine in the catalytic core domain. The active-site cysteine is marked with a filled star. The two cysteine residues unique to the class III E2s (UbcM2, UbcM3, and UBE2E2) are boxed, and open stars mark the location of these residues relative to the active site. All other E2s shown have prolines at these two positions. B, same assay as done in Fig. 1 using a panel of GST-E2 fusions. Each reaction was supplemented with 1 mm NEM. The E2 class is denoted below the lane numbers. C, recombinant pulldown assay testing a series of GST-UbcM2 fusions bearing mutations at one or both candidate cysteines (136 and 173). Reactions were supplemented with either 1 mm DTT (lanes 1–5) or 1 mm NEM (lanes 6–10). D, recombinant pulldown assays testing the Nrf2-binding capacity of GST-UbcH5b mutants (lanes 3–5 and 8–10) harboring cysteines in place of prolines. The level of Nrf2 binding is being directly compared with the comparable mutants of GST-UbcM2 (lanes 1–2 and 6–7). The designations above the blots denote the genotype of the GST-E2. Reactions were supplemented with either DTT (lanes 1–5) or NEM (lanes 6–10). For B–D, co-precipitated Nrf2 was detected by blotting with α-Nrf2 (top panels), and the immobilized GST proteins were detected by CBB-staining (bottom panels). Each experiment was repeated at least three independent times.
We next tested if an intact active-site cysteine (Cys-145) was required for the NEM-inducible interaction. GST fusions of either wt or (C145A)-UbcM2 were mixed with H6-S-Nrf2, GSH-Sepharose, and either NEM or DTT and then analyzed as in Fig. 1 (A and B). The results from these experiments showed that NEM could enhance the binding of both forms of GST-UbcM2 to Nrf2 (Fig. 1D). Interestingly, (C145A)-UbcM2 exhibited slightly lower levels of Nrf2 binding as compared with wt enzyme (Fig. 1D, lanes 2 and 4, versus lanes 1 and 3) suggesting that an intact active site may contribute to complex formation.
An alignment of mammalian E2s revealed that UbcM2 possesses two cysteine residues (Cys-136 and Cys-173) in close proximity to the conserved active-site cysteine. This pair of cysteines is only present in the class III E2s (UbcM2, UbcM3, and UBE2E2), whereas nearly all other mammalian E2s harbor prolines at the corresponding positions (Fig. 2A). Consistent with a role for this pair of cysteines in the inducible interaction with Nrf2, GST fusions of each class III E2 bound H6-S-Nrf2 in the presence of NEM (Fig. 2B, lanes 4–6). In contrast, representative enzymes from each of the other classes failed to interact with the transcription factor (Fig. 2B, lanes 2, 3, and 7). Moreover, simultaneous mutation of both cysteine residues to prolines abrogated the effect of NEM on UbcM2-Nrf2 complex formation (Fig. 2C, lane 8). Single cysteine mutations revealed that Cys-136 is the primary target of the NEM-inducible binding (Fig. 2C, lanes 4, 5, 9, and 10). Mutation of this cysteine completely blocked the NEM-stimulated binding of Nrf2 by UbcM2 (Fig. 2C, lane 10).
We next analyzed whether replacing the corresponding proline (Pro-76) in UbcH5b with a cysteine could confer the capacity to bind Nrf2 in an NEM-dependent manner. UbcH5b was chosen because its catalytic core domain is 64% identical and 77% similar to the corresponding domain of UbcM2 and in particular, only differs at 7 residues in the region containing cysteines 136 and 173. Interestingly, mutation of Pro-76 in UbcH5b to a cysteine failed to confer Nrf2 binding (Fig. 2D, lanes 5 and 10). Likewise, mutation of the other conserved proline in UbcH5b (Pro-113), which corresponds to Cys-173 in UbcM2, had no effect on binding to recombinant Nrf2 (Fig. 2D, lanes 4 and 9). wt UbcH5b also did not interact (data not shown). Auto-ubiquitylation assays confirmed that the introduction of the cysteine point mutants did not result in global unfolding of recombinant UbcH5b (supplemental Fig. S1). Collectively, these studies reveal that alkylation of UbcM2 on a class III E2-specific cysteine induces binding to Nrf2. Furthermore, it appears that one or more other residues, domain, or structural context of UbcM2 contributes to the interaction as transplantation of the critical cysteine into the conserved position of UbcH5b was not sufficient to confer Nrf2 binding. This represents the first report of a direct interaction between an E2 and a transcription factor. Akin to cysteine alkylation studies with proteins such as Keap1 (11, 13, 32–35), these data position Cys-136 as a potential redox sensor.
To further validate the importance of Cys-136 in the interaction between UbcM2 with Nrf2, we generated point mutants that replaced the cysteine with a hydrophobic residue. The rationale was that the hydrophobic residue would reduce or eliminate the need to supplement the recombinant pulldown assays with NEM. Three mutations were tested (C136W, C136Y, and C136F), and each bound recombinant Nrf2 when incubated with either DTT or NEM (Fig. 3A, lanes 2–4 and 6–8). The level of Nrf2 complex formation was equivalent to or greater than that observed with the wild-type enzyme after NEM treatment (Fig. 3A, compare lane 5 with lanes 2–4 and 6–8). In addition, the NEM enhancement of Nrf2 binding by these mutants, especially the C136Y mutant (Fig. 3A, lanes 4 and 8), indicate that a second cysteine, perhaps the active site, Cys-145, contributes to complex formation.
FIGURE 3.

Biochemical characterization of Cys-136 substitution mutants. A, recombinant pulldown with wt GST-UbcM2 or the designated Cys-136 substitution mutants and H6-S-Nrf2 in the presence of 1 mm DTT (lanes 1–4) or 1 mm NEM (lanes 5–8). The top panel shows precipitated H6-S-Nrf2 detected with α-Nrf2, and the bottom panel shows immobilized GST fusion proteins detected with CBB. B, lysates from transiently transfected HEK293T cells were divided in half and processed for non-reducing (top) or reducing (bottom) SDS-PAGE and anti-Myc Western blotting. Unmodified enzyme is designated as “Myc-UbcM2” and ubiquitin-charged enzyme as “Myc-UbcM2∼Ub.” The asterisk marks a slower migrating, β-mercaptoethanol-labile form of particular UbcM2 mutants (lanes 3–5). YFP was expressed as a negative control (lane 1). The migration of molecular weight markers is indicated on the left. C, immunofluorescence analysis of the indicated Myc-UbcM2 proteins expressed in HeLa cells. As reported previously (36), mutation of the active-site cysteine, Cys-145, blocks import of the enzyme (panels c and f). Panels a–c show representative photomicrographs of anti-Myc staining and panels d–f show the corresponding DAPI images. The white bar represents 10 μm. D, auto-ubiquitylation assays were performed by immunoprecipitating the indicated Myc-UbcM2 proteins from cell lysates and combining one-half of each sample with recombinant E1, ubiquitin, and an energy-regenerating system (Ub rxn mixt). “Myc-UbcM2” denotes unmodified enzyme, “Ub-Myc-UbcM2” denotes mono-ubiquitylated enzyme, and “(Ub)n-Myc-UbcM2” denotes multiply ubiquitylated enzyme. The asterisk marks a slower migrating form of (C136F)-UbcM2. Experiments were repeated at least three independent times.
Three independent approaches were taken to rule out that introduction of a hydrophobic residue at position 136 induces global unfolding of the UbcM2 catalytic core domain. First, we expressed (C136F)-UbcM2 by transient transfection in mammalian cells and examined ubiquitin-charging of its active-site cysteine. We found that Myc-(C136F)-UbcM2 was charged at its active site with ubiquitin (Fig. 3B, lane 4), albeit less efficiently than wt enzyme (Fig. 3B, lane 2) but equivalently to (C145S)-UbcM2, a mutant that forms a β-mercaptoethanol-resistant oxyester bond with ubiquitin (Fig. 3B, lane 6, top and bottom panels). The specificity of this assay was validated using an active-site mutant that cannot be charged with ubiquitin (Fig. 3B, lane 5). In a second approach, we performed indirect immunofluorescence analysis to evaluate the cellular distribution of the mutant enzyme. Similar to wt UbcM2, the (C136F) mutant localized to the nucleus of transfected cells (Fig. 3C, panels a and b), whereas an active-site mutant failed to accumulate in the nucleus (Fig. 3C, panel c). Consistent with our previous work demonstrating that charging of the active-site cysteine with ubiquitin is required for nuclear import of UbcM2 (36), these localization data corroborate the biochemical data in Fig. 3B and support the conclusion that the introduction of a phenylalanine at residue 136 does not globally disrupt the structural integrity of the UbcM2 catalytic core domain.
In a third approach, we tested whether the phenylalanine substitution permitted transfer of ubiquitin from the active-site cysteine to an acceptor lysine. The Myc-UbcM2 proteins were immunoprecipitated from transfected cells and incubated with recombinant E1, ubiquitin, and an energy-regenerating system. Auto-ubiquitylation of the precipitated enzymes was then assessed by Western blotting. Both wt and the (C136F) mutant underwent extensive auto-ubiquitylation (Fig. 3D, lanes 1 and 5, respectively), whereas a catalytically dead mutant did not (Fig. 3D, lane 3). The difference in auto-ubiquitylation patterns between wt and the (C136F) mutant appears to relate to the slower migrating species observed with the mutant (Fig. 3B, lane 4, asterisk, and Fig. 3D, lanes 5 and 6, asterisks). Nevertheless, these results demonstrate that the phenylalanine mutation does not compromise the capacity of the mutant enzyme to transfer ubiquitin from its active site to an acceptor lysine. This result is intriguing considering that the phenylalanine is adjacent to the conserved asparagine residue required for E2-mediated isopeptide bond formation (37).
To analyze the impact of Cys-136 on the capacity of UbcM2 to regulate Nrf2 stability in cells, we expressed increasing amounts of either Myc-tagged wt or (C136F)-UbcM2 with HA3-tagged Nrf2 in HEK293T cells. 3 h prior to harvesting, cells were treated with cycloheximide to block new protein synthesis and the levels of HA3-Nrf2 were determined by Western blotting. We used the proteasome inhibitor, MG132, as a positive control to stabilize Nrf2 (Fig. 4A, lane 1) (38). (C136F)-UbcM2 stabilized HA3-Nrf2 in a dose-dependent manner (Fig. 4A, lanes 7–9). In contrast, wt enzyme showed a minimal capacity to stabilize the transcription factor when expressed at levels comparable to the mutant (Fig. 4A, lanes 4–6) but reproducibly induced a modest stabilization when expressed severalfold above mutant levels (supplemental Fig. S2). The need for such high levels of wt UbcM2 overexpression to elicit a modest stabilization of Nrf2 likely reflects that only a small fraction of the wt enzyme is alkylated or oxidized at C136. Similar stabilization capacities for wt and (C136F)-UbcM2 were obtained in HeLa cells (data not shown).
FIGURE 4.

(C136F)-UbcM2 increases the half-life of Nrf2 in the nucleus. A, co-transfection assay demonstrating dose-dependent stabilization of HA3-Nrf2 by (C136F)-UbcM2. MG132 treatment (lane 1) is a positive control for stabilization of the transcription factor and DMSO (lane 2) is the vehicle control for MG132. A plasmid expressing YFP was used to normalize the amount of transfected DNA (lanes 1–3). wt UbcM2 is expressed in lanes 4–6 and (C136F)-UbcM2 in lanes 7–9. HA3-Nrf2 was detected by α-HA (top panel) and Myc-UbcM2 proteins by α-UbcM2 (bottom panel). B, representative pulse-chase autoradiographic data used to determine the half-life of HA3-Nrf2 when co-expressed with either YFP (lanes 1–4) or (C136F)-UbcM2 (lanes 5–8). Chase times are indicated above the autoradiograph. The graph was obtained by averaging half-life measurements from three independent experiments. Error bars represent standard deviation, and the asterisk denotes statistical significance determined by two-tailed Student's t test (p < 0.0002). C, immunofluorescence analysis of the indicated Myc-UbcM2 proteins expressed in HeLa cells. Fusion of an NES relocalizes both (C136F)-UbcM2 and wt enzyme to the cytoplasm (compare panels a to b and c to d). Panels a–d are anti-Myc labeling and panels e–h are the corresponding DAPI images. The white bar represents 10 μm. D, same assay as A in HeLa cells testing how relocalizing (C136F)-UbcM2 to the cytoplasm influences Nrf2 stabilization. Experiments were performed at least three independent times.
To begin dissecting the mechanism of Nrf2 stabilization by (C136F)-UbcM2, we carried out pulse-chase experiments to determine whether the mutant enzyme increased the half-life of the transcription factor. HEK293T cells co-expressing HA3-Nrf2 and either Myc-YFP or Myc-(C136F)-UbcM2 were pulsed with 35S-labeled methionine and cysteine and then chased for 0, 15, 30, or 60 min. The 35S-labeled HA3-Nrf2 was then immunoprecipitated from 100 μg of lysate at each time point and resolved by SDS-PAGE, and half-life measurements were made as described under “Experimental Procedures.” In cells expressing Myc-YFP, the half-life of HA3-Nrf2 was determined to be 25.7 ± 4.6 min, in reasonable agreement with published reports for the half-life of endogenous Nrf2 (e.g. Ref. 10). In cells co-expressing Myc-(C136F)-UbcM2 and HA3-Nrf2, the half-life increased nearly 3-fold to 70.2 ± 26.0 min (Fig. 4B). These data show that the mutant enzyme increases the stability of Nrf2.
To differentiate between whether this effect of Myc-(C136F)-UbcM2 on HA3-Nrf2 half-life was occurring through CUL3Keap1 or was downstream of CUL3Keap1, we examined if Nrf2 stabilization by the mutant enzyme was a cytoplasmic or a nuclear event. The rationale for this is that CUL3Keap1-mediated degradation of Nrf2 takes place in the cytoplasm (14, 35, 39). To do this, we mis-targeted (C136F)-UbcM2 to the cytoplasm by appending the nuclear export signal (NES) from the cAMP-dependent protein kinase inhibitor (40) to the N terminus of the mutant. The fused NES successfully relocalized the mutant to the cytoplasm (Fig. 4C, panel b) and reduced the mobility of the protein in SDS-PAGE (Fig. 4D, lower panel, lane 3). Importantly, this mis-targeting abrogated the Nrf2-stabilizing capacity of (C136F)-UbcM2 (Fig. 4D, upper panel, compare lanes 2 and 3). In comparison, comparable levels of wt UbcM2, with or without the fused NES, failed to stabilize Nrf2 (Fig. 4D, upper panel, lanes 4 and 5, respectively). We interpret these data to indicate that Nrf2 stabilization by (C136F)-UbcM2 takes place in the nucleus and is therefore downstream of CUL3Keap1.
Biochemical data consistent with this interpretation came from a series of co-precipitation experiments. We first demonstrated that (C136F)-UbcM2 could recover HA3-Nrf2 from co-transfected cell lysates (Fig. 5A, lane 2). The specificity of this interaction was established by the failure of (C136P)-UbcM2 (Fig. 5A, lane 3) or a different E2, UbcH10 (Fig. 5A, lane 4), to efficiently recover HA3-Nrf2 from lysates. Using this assay, we tested whether (C136F)-UbcM2 could bind to mutants of Nrf2 that localize to the nucleus, because they are deficient in binding to Keap1 (e.g. Refs. 20, 41). Keap1 functions both to retain Nrf2 in the cytoplasm and to mediate the ubiquitin-dependent degradation of the transcription factor (14, 35, 39). The domain required for the Keap1-Nrf2 interaction is well characterized and, using this information, two different Nrf2 mutants were generated that fail to stably interact with Keap1. (ΔNeh2)-Nrf2 lacks the N-terminal Neh2 domain and (ΔETGE)-Nrf2 lacks a 4-residue motif within Neh2 required for stable binding to Keap1 (14, 22, 42). Co-immunoprecipitation assays confirmed that FLAG-Keap1 co-precipitated wt HA3-Nrf2 but did not efficiently bind either mutant (Fig. 5B, lanes 4–6). In contrast, (C136F)-UbcM2 co-precipitated the wt and mutant Nrf2 proteins comparably (Fig. 5C, lanes 7–9). Because the cytoplasmic tethering of Nrf2 requires Keap1 binding (14, 20, 39), these data are consistent with UbcM2 engaging a population of Nrf2 that is not retained in the cytoplasm by Keap1. These data also indicate that, similar to the recombinant pulldown assays (Fig. 1), UbcM2 and Nrf2 complex formation does not require Keap1 to bridge the interaction.
FIGURE 5.

(C136F)-UbcM2 binds to the Neh1 domain of Nrf2. A, co-immunoprecipitation assay demonstrating that (C136F)-UbcM2 can precipitate HA3-Nrf2 from transfected cell lysates. (C136P)-UbcM2 (lane 3) and wt UbcH10 (lane 4) were included as specificity controls. Top and middle panels are α-HA blots to detect co-precipitated and input HA3-Nrf2 levels, respectively. The bottom panel is an α-Myc blot to detect Myc-E2s. B, co-immunoprecipitation assay demonstrating that FLAG-Keap1 can stably bind wt HA3-Nrf2 (lane 4) but does not efficiently interact with mutants of the transcription factor lacking either the ETGE residues (ΔETGE, lane 5) or the entire Neh2 domain (ΔNeh2, lane 6). The amount of each Nrf2 protein present in the input lysates is shown in lanes 1–3. The top blots were probed with α-HA, and the bottom blot was probed with anti-FLAG. C, same assay as A and B demonstrating the interaction between Myc-(C136F)-UbcM2 and the different forms of HA3-Nrf2. The α-Myc antibody was omitted from the control IPs (lanes 4–6) to determine the level of nonspecific binding of the HA3-Nrf2 proteins to protein A-Sepharose. Binding of the HA3-Nrf2 proteins to Myc-tagged (C136F)-UbcM2 is shown in lanes 7–9. D, same assay as C testing Myc-(C136F)-UbcM2 binding to a panel of N- and C-terminal truncation mutants of HA3-Nrf2. The level of nonspecific binding of the HA3-Nrf2 proteins is shown in lanes 8–14. The Δ symbol indicates which Neh domain(s) has(have) been deleted. E, pulldown assay to assess the binding of Myc-(C136F)-UbcM2 to the indicated domains of Nrf2. Control immunoprecipitations (lanes 6–10) lacked the α-Myc antibody. The top panels represent anti-HA blots, and the bottom panels represent anti-UbcM2 blots. The asterisk in the top input panel marks the nonspecific anti-HA band. For A–D, the migration of molecular weight markers is indicated to the left of each blot and for E, on the right side. Each experiment was repeated three independent times.
Using this co-precipitation approach, we mapped the domain(s) of Nrf2 bound by (C136F)-UbcM2. The transcription factor contains six highly conserved Neh domains, Neh1 through -6. Neh1 harbors the DNA binding domain and the Cap “n” collar basic leucine zipper domain that heterodimerizes with small Maf proteins on the promoters of target genes. As mentioned above, Neh2 mediates the interaction with Keap1. Neh3, Neh4, and Neh5 regulate the transactivation capacity of Nrf2 and Neh6 reportedly functions as a nuclear degron (22). Myc-(C136F)-UbcM2 co-precipitated full-length Nrf2 as well as mutants lacking Neh2 and Neh3 (Fig. 5D, top panel, lanes 1, 2, and 5, respectively). The enzyme showed a weaker but specific interaction with truncation mutants lacking Neh 2/4 and Neh 2/4/5 (Fig. 5D, lanes 3 and 4, respectively) suggesting that Neh4 may contribute to the Nrf2-UbcM2 interaction. Truncation mutants lacking Neh1 and Neh6 showed markedly impaired binding to Myc-(C136F)-UbcM2 (Fig. 5D, lanes 6 and 7). Control precipitations with FLAG-tagged Keap1 demonstrated that the various C-terminally deleted Nrf2 fragments, which did not bind Myc-(C136F)-UbcM2, were functional for co-precipitation by Keap1 (supplemental Fig. S3, lanes 9 and 10). In complementary pulldown assays, we observed that an HA3-Nrf2 fragment encompassing Neh1 and Neh6 was efficiently co-precipitated by Myc-(C136F)-UbcM2 and further, that a fragment representing Neh1 alone was bound (Fig. 5E, lanes 13 and 14), whereas Neh6 in isolation was not (Fig. 5E, lane 15). Interestingly, a fragment encompassing Neh4/5 also failed to be co-precipitated by Myc-(C136F)-UbcM2 (Fig. 5E, lane 12) consistent with the idea that Neh4 is not the primary mediator of complex formation. We interpret these data to indicate that Neh1 is the primary domain mediating the interaction between UbcM2 and Nrf2 and that Neh4 may contribute to stabilizing the complex.
We next tested if an intact active-site cysteine was required for (C136F)-UbcM2 to stabilize Nrf2 in vivo. Mutation of the active site to an alanine (C145A) in the context of (C136F)-UbcM2 caused a dramatic decrease in HA3-Nrf2 stabilization (Fig. 6A, upper panel, compare lanes 2 and 4). However, the levels of the transcription factor were consistently higher than those observed with a mutant harboring only an active-site mutation (Fig. 6A, upper panel, compare lanes 3 and 4). Two possible explanations for this finding are that either: (i) the catalytic activity of (C136F)-UbcM2 contributes to Nrf2 stabilization or (ii) mutation of the active site blocks the nuclear import of (C136F)-UbcM2, a requirement for stabilizing the transcription factor (Fig. 4C). This nuclear accumulation hypothesis stems from our previous report that ubiquitin charging of the active site is required for UbcM2 nuclear import (36) and by the demonstration that Myc-(C136F,C145A)-UbcM2 is mislocalized to the cytoplasm (Fig. 6B, panel c). To validate this explanation, we attempted to rescue the mislocalization of Myc-(C136F,C145A)-UbcM2 by fusing the nuclear localization signal (NLS) from the SV40 large T antigen to it. Of note, this NLS utilizes the importin α/β heterodimer to enter the nucleus (43, 44), whereas wt UbcM2 accesses the nucleus via the importin-11 pathway (45). Similar to the NES fusions in Fig. 4D, addition of the NLS retarded the migration of the fusion protein in SDS-PAGE (Fig. 6A, bottom blot, lane 5). As predicted, fusion of the NLS rescued the nuclear localization of the double mutant (Fig. 6B, panel d), but it did not rescue the ability to stabilize HA3-Nrf2 (Fig. 6A, upper blot, lane 5). Together, we interpret these results to indicate that the catalytic activity of (C136F)-UbcM2 contributes to Nrf2 stabilization in the nucleus.
FIGURE 6.
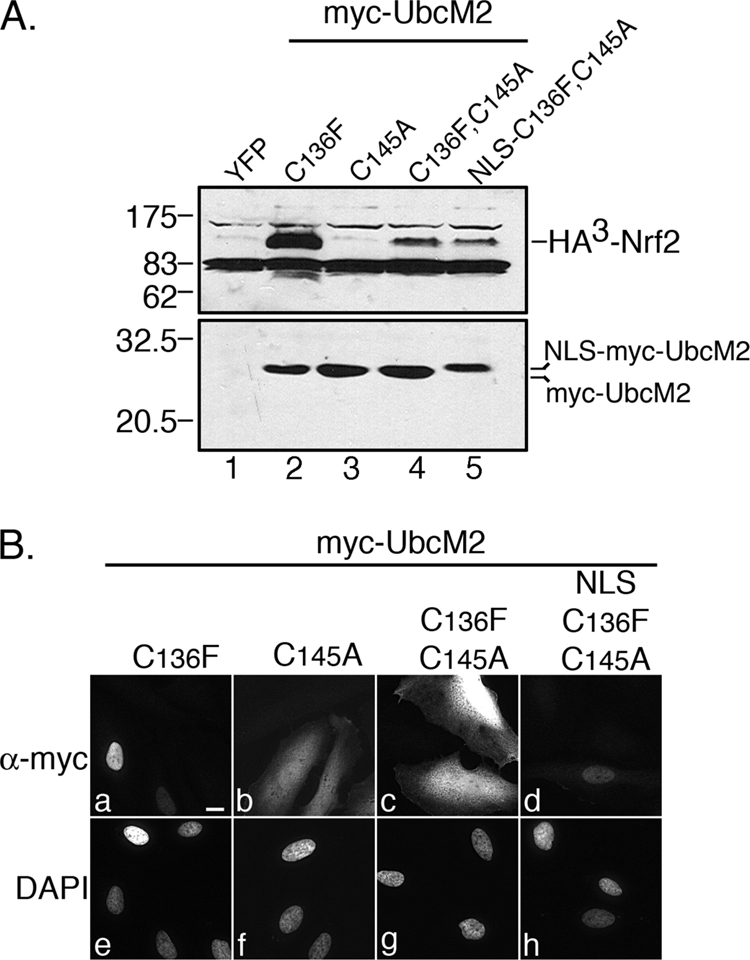
The active-site cysteine of (C136F)-UbcM2 contributes to Nrf2 stabilization. A, co-transfection assay in HeLa cells demonstrating a dramatic reduction of HA3-Nrf2 stabilization by the double mutant, (C136F,C145A)-UbcM2 (lane 4), as compared with (C136F)-UbcM2 (lane 2). Fusion of the SV40 large T antigen NLS to (C136F,C145A)-UbcM2 does not rescue the capacity to stabilize Nrf2 (lane 5). The single active-site mutant is deficient in Nrf2 stabilization (lane 3). The top panel is an α-HA blot, and the bottom panel is an α-UbcM2 blot. The migration of molecular weight markers is indicated to the left of each blot. B, immunofluorescence analysis of the indicated Myc-UbcM2 proteins expressed in HeLa cells. Fusion of the NLS relocalizes (C136F,C145A)-UbcM2 to the nucleus (compare panels c and d). Panels a–d are anti-Myc labeling and panels e–h are the corresponding DAPI images. The white bar represents 10 μm. Each experiment was repeated three independent times.
A luciferase reporter assay was used to determine if the effect of (C136F)-UbcM2 on Nrf2 stabilization resulted in an increase in transcriptional activity. Cells were transfected with plasmids encoding 1) firefly luciferase under control of the Nrf2-driven, antioxidant response element (ARE) (35), 2) β-galactosidase driven by the Rous sarcoma virus (RSV) promoter to normalize for transfection efficiency, and 3) the various Myc-tagged UbcM2 constructs. The RSV-β-galactosidase plasmid enabled us to normalize for transfection efficiency and to rule out that the Myc-UbcM2 proteins indiscriminately up-regulate transcription as the levels of β-galactosidase activity were comparable in all samples. 2 days post-transfection, cells were treated with vehicle or tBHQ, lysed, and luciferase activity assayed. The effects of the various Myc-tagged UbcM2 proteins were compared with cells transfected with the empty “Myc” plasmid (Fig. 7, light gray bars). These experiments revealed that, in the absence of an exogenously added pro-oxidant, (C136F)-UbcM2 enhanced the transcriptional activity of endogenous Nrf2 ∼6-fold, whereas (C136P)-UbcM2 had no effect. The double mutant, (C136F,C145A)-UbcM2, showed a reduced capacity to induce Nrf2 activity compared with (C136F)-UbcM2, consistent with a contribution from the active-site cysteine (Fig. 6). Unexpectedly, the isolated active-site mutant, (C145A)-UbcM2, enhanced Nrf2-mediated transcription in the absence of pro-oxidant addition. This result was surprising considering this mutant did not increase Nrf2 steady-state levels in our co-transfection assays (Fig. 6A). However, such a disconnect between Nrf2 stabilization and activation has been reported for particular chemopreventive agents (e.g. indole-3-carbinol) (46). The effect of this active-site mutant on Nrf2-driven transcription was attenuated when coupled to mutation of Cys-136 to a proline (C136P,C145A) thus further establishing cooperation between these two cysteine residues in regulating Nrf2.
FIGURE 7.

Cys-136 and Cys-145 of UbcM2 contribute to the transcriptional activation of endogenous Nrf2. Transcriptional reporter assays comparing ARE-driven luciferase expression in cells expressing empty vector (Myc), Myc-UbcM2 (wt), Myc-(C136F)-UbcM2, Myc-(C136F,C145A)-UbcM2, Myc-(C145A)-UbcM2, Myc-(C136P)-UbcM2, and Myc-(C136P,C145A). Samples were treated for 16 h with vehicle (ETOH, light gray bars) or oxidant (25 μm tBHQ, dark gray bars). The ARE corresponds to the promoter element present in the GST Ya subunit gene, an Nrf2 target gene. All cells were co-transfected with an RSV-driven Lac Z plasmid to measure β-galactosidase activity as a means of normalizing samples for transfection efficiency. The graph represents averaged data obtained from at least three independent experiments in which each sample was done in triplicate. Error bars represent standard deviation. Relative to the Myc control, all UbcM2-expressing samples displayed statistically significance differences in Nrf2 transcriptional activity (ethanol-treated samples; p ≤ 0.05; tBHQ-treated samples; p ≤ 0.001). “$” denotes statistically significance difference between Myc-UbcM2 (wt) and Myc-(C136P,C145A)-UbcM2 (tBHQ-treated samples; p < 0.025). Statistical significance was calculated using a one-way analysis or variance with post-hoc test.
In parallel, we also measured the effects of the various UbcM2 proteins on Nrf2 transcriptional activity in cells exposed to the pro-oxidant, tBHQ (Fig. 7, black bars). In all samples, tBHQ induced an additional 2- to 5-fold increase in endogenous Nrf2 activity over that observed without the pro-oxidant (Fig. 7, dark gray bars). We attribute this response to the capacity of tBHQ to stabilize and activate Nrf2 by dissociating cytoplasmic CUL3Keap1 (11). The additive effects of tBHQ and the UbcM2 mutants are consistent with UbcM2 influencing endogenous Nrf2 activation at a step distinct from CUL3Keap1 regulation. These experiments also revealed that the double mutant (C136P,C145A) showed a modest but statistically significant attenuation in Nrf2 activation as compared with wt enzyme in response to tBHQ. These data support a role for Cys-136 and Cys-145 of UbcM2 jointly contributing to Nrf2 modulation in response to an oxidative insult.
DISCUSSION
The stability and activity of the master antioxidant transcription factor, Nrf2, is regulated by cellular redox status (reviewed in Ref. 47). Nrf2 levels are suppressed in the absence of oxidative stress by the coordinated action of the UPS E3 ligase, CUL3Keap1, and the 26 S proteasome (10, 11, 48). In response to oxidative stress, CUL3Keap1 activity is disrupted (e.g. Ref. 11). This results in Nrf2 stabilization, followed by nuclear import of the transcription factor, and the induction of phase 2 genes. In this report, we demonstrate that the highly conserved metazoan E2 UbcM2 has the capacity to regulate Nrf2 function in a novel way. Unexpectedly, this function of UbcM2 appears to be independent of CUL3Keap1 (Figs. 4D, 5, and 7). Furthermore, we show that the capacity of UbcM2 to stabilize the transcription factor is primarily mediated by a non-catalytic cysteine in the enzyme, takes place in the nucleus, requires an intact active site, and is mediated through the Neh1 domain of Nrf2. This work is significant, because it demonstrates that E2s of the UPS can be harnessed to directly modulate transcription factor function. In addition, these studies identify a potential redox sensor function for a unique cysteine residue shared by the class III E2s: UbcM2, UbcM3, and UBE2E2.
Multiple independent lines of evidence support the conclusion that the stabilization of Nrf2 by (C136F)-UbcM2 is a nuclear event downstream of CUL3Keap1 function. First, (C136F)-UbcM2 localizes to the nucleus (Figs. 3C and 4C) and mis-targeting of the enzyme to the cytoplasm abrogates Nrf2 stabilization (Fig. 4D). This is opposed to the degradation of Nrf2 by CUL3Keap1, which occurs in the cytoplasm (e.g. Refs. 14, 35, 39). Second, (C136F)-UbcM2 can bind directly to recombinant Nrf2 (Fig. 3A) and can interact with Nrf2 mutants that are deficient in Keap1 binding (Fig. 5). In fact, (C136F)-UbcM2 binding is mediated by the Neh1 domain, whereas Keap1 binding utilizes the Neh2 domain of the transcription factor. Third, activation of endogenous Nrf2 activity by (C136F)-UbcM2 can be further enhanced by an oxidant that is known to disrupt CUL3Keap1 function (Fig. 7) indicating that the mutant enzyme operates at a step distinct from CUL3Keap1. To date, we do not know exactly how UbcM2 promotes nuclear Nrf2 stabilization and activation. An attractive idea is that UbcM2 enhances the interaction of Neh1 with the ARE promoter regions of phase two genes thereby limiting the nuclear export and subsequent recruitment of Nrf2 to cytoplasmic CUL3Keap1 for degradation (19).
Our data also support the possibility that Cys-136 of UbcM2 (and likely the corresponding cysteines in UbcM3 and UBE2E2) functions as a redox sensor. In vitro, mutation of this cysteine to the conserved proline present in all other E2s, blocks the NEM-inducible interaction between UbcM2 and Nrf2 (Fig. 2C). Likewise, replacement of the cysteine with a bulky residue (e.g. Phe, Trp, or Tyr) yielded mutant enzymes that no longer required NEM modification for Nrf2 binding (Fig. 3A). One of these mutants, (C136F)-UbcM2, was further studied in cells and found to stabilize Nrf2 in a dose-dependent manner (Fig. 4A) by increasing the half-life of the transcription factor (Fig. 4B). These data are consistent with a model in which Cys-136 becomes modified in cells exposed to oxidative stress, and this leads to a nuclear complex between UbcM2 and Nrf2. This complex sequesters Nrf2 from nuclear export and degradation until redox homeostasis can be restored. In this model, the protective role of UbcM2 would be temporally limited to the window of time necessary for the restoration of redox homeostasis. Validation of this model will ultimately require mass spectrometric confirmation that Cys-136 is modified in cells exposed to oxidant. This modification may consist of an alkylation or a different post-translational modification (e.g. glutathionylation). A similar strategy was necessary to identify the putative redox-sensing cysteines in Keap1 (e.g. Ref. 34) following a series of mutagenesis and in vitro studies to identify candidate cysteines (13, 32, 33). Alkylation of these cysteines, in particular Cys-151, integrates cellular exposure to reactive electrophiles with biochemical changes in CUL3Keap1 and culminates in the induction of cytoprotective Nrf2 target genes (11, 35, 49). The intriguing possibility that Cys-136 of UbcM2 represents a second, complementary redox sensor in the Nrf2 regulatory circuit is attractive in light of the numerous dietary and chemotherapeutic agents that target Cys-151 of Keap1 and are currently being pursued for clinical applications. Curiously, both sensors promote Nrf2 stability, but do so in opposite fashions. Whereas modification of Cys-151 disrupts complex formation between Keap1 and CUL3 (11), our data indicate that modification of Cys-136 promotes complex formation between UbcM2 and Nrf2.
In addition to a role for the non-catalytic cysteine, our studies have also revealed a secondary requirement for an intact active-site cysteine. The first evidence came from recombinant pulldown assays in which (C145A)-UbcM2 showed a modestly reduced capacity to precipitate Nrf2 as compared with wt enzyme (Fig. 1D). In vivo, the introduction of an active-site mutation into (C136F)-UbcM2 produced a version of the enzyme that no longer accumulated in the nucleus (Fig. 6B), no longer stabilized Nrf2 (Fig. 6A), and failed to activate endogenous Nrf2 as well as the catalytically active (C136F)-UbcM2 (Fig. 7). Further, rescuing the nuclear localization of the double mutant by appending a heterologous NLS was insufficient to rescue Nrf2 stabilization (Fig. 6). Additional support for a joint contribution from Cys-136 and Cys-145 came from endogenous Nrf2 transcription reporter assays in which the double mutant (C136P,C145A) displayed a modestly attenuated capacity (as compared with wt enzyme) to activate Nrf2 in response to oxidative stress (Fig. 7). Collectively, these data imply that the mechanism of Nrf2 stabilization and activation by UbcM2 involves modification of Cys-136, modification of the active-site cysteine (e.g. ubiquitin charging) and/or transfer of ubiquitin from the enzyme to Nrf2 or an additional factor.
In summary, we have identified UbcM2 as a novel component of the Nrf2 regulatory pathway. The role of UbcM2 in stabilizing and activating Nrf2 requires both a non-catalytic cysteine of the enzyme that may function as a putative redox sensor as well as an intact active site. This work expands the repertoire of functions and mechanisms by which UPS E2s can contribute to critical cellular functions. It also offers an explanation for why the class III E2s harbor a unique cysteine residue in place of a proline in the highly conserved His-Pro-Asn triad required for E2-mediated isopeptide bond formation (37).
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant P20RR024215 from the National Center for Research Resources (title: Mentoring Diabetes Research in Oklahoma). Additional funding (to S. M. P.) in support of this work was kindly provided by the American Health Assistance Foundation (Grant M2009095), The Oklahoma Center for the Advancement of Science and Technology (Grant HR08-076), and a Karl Kirchgessner Foundation Vision Research Grant.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- Nrf2
- nuclear factor E2-related factor 2
- UPS
- ubiquitin proteolytic system
- E2
- ubiquitin-conjugating enzyme
- E3
- ubiquitin-protein isopeptide ligase
- ARE
- antioxidant response element
- CUL3
- cullin 3
- RING
- really interesting new gene
- tBHQ
- tert-butylhydroquinone
- HEK
- human embryonic kidney
- HA3
- triple hemagglutinin
- PBS
- phosphate-buffered saline
- DAPI
- 4′,6-diamidino-2-phenylindole
- YFP
- yellow fluorescent protein
- CBB
- Coomassie Brilliant Blue
- GST
- glutathione S-transferase
- DTT
- dithiothreitol
- NEM
- N-ethylmaleimide
- Neh
- Nrf2 ECH homology domain
- wt
- wild type
- NES
- nuclear export signal
- NLS
- nuclear localization signal
- RSV
- Rous sarcoma virus.
REFERENCES
- 1.Itoh K., Igarashi K., Hayashi N., Nishizawa M., Yamamoto M. (1995) Mol. Cell. Biol. 15, 4184–4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marini M. G., Chan K., Casula L., Kan Y. W., Cao A., Moi P. (1997) J. Biol. Chem. 272, 16490–16497 [DOI] [PubMed] [Google Scholar]
- 3.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. (1997) Biochem. Biophys. Res. Commun. 236, 313–322 [DOI] [PubMed] [Google Scholar]
- 4.Enomoto A., Itoh K., Nagayoshi E., Haruta J., Kimura T., O'Connor T., Harada T., Yamamoto M. (2001) Toxicol. Sci. 59, 169–177 [DOI] [PubMed] [Google Scholar]
- 5.Chan K., Kan Y. W. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 12731–12736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos-Gomez M., Kwak M. K., Dolan P. M., Itoh K., Yamamoto M., Talalay P., Kensler T. W. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3410–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMahon M., Itoh K., Yamamoto M., Chanas S. A., Henderson C. J., McLellan L. I., Wolf C. R., Cavin C., Hayes J. D. (2001) Cancer Res. 61, 3299–3307 [PubMed] [Google Scholar]
- 8.Nguyen T., Sherratt P. J., Huang H. C., Yang C. S., Pickett C. B. (2003) J. Biol. Chem. 278, 4536–4541 [DOI] [PubMed] [Google Scholar]
- 9.Stewart D., Killeen E., Naquin R., Alam S., Alam J. (2003) J. Biol. Chem. 278, 2396–2402 [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi A., Kang M. I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., Yamamoto M. (2004) Mol. Cell. Biol. 24, 7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang D. D., Lo S. C., Cross J. V., Templeton D. J., Hannink M. (2004) Mol. Cell. Biol. 24, 10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W., Kong A. N. (2009) Mol. Carcinog. 48, 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinkova-Kostova A. T., Holtzclaw W. D., Cole R. N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11908–11913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J. D., Yamamoto M. (1999) Genes Dev. 13, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wakabayashi N., Dinkova-Kostova A. T., Holtzclaw W. D., Kang M. I., Kobayashi A., Yamamoto M., Kensler T. W., Talalay P. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2040–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watai Y., Kobayashi A., Nagase H., Mizukami M., McEvoy J., Singer J. D., Itoh K., Yamamoto M. (2007) Genes Cells 12, 1163–1178 [DOI] [PubMed] [Google Scholar]
- 17.Nguyen T., Sherratt P. J., Nioi P., Yang C. S., Pickett C. B. (2005) J. Biol. Chem. 280, 32485–32492 [DOI] [PubMed] [Google Scholar]
- 18.Karapetian R. N., Evstafieva A. G., Abaeva I. S., Chichkova N. V., Filonov G. S., Rubtsov Y. P., Sukhacheva E. A., Melnikov S. V., Schneider U., Wanker E. E., Vartapetian A. B. (2005) Mol. Cell. Biol. 25, 1089–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Z., Zhang S., Chan J. Y., Zhang D. D. (2007) Mol. Cell. Biol. 27, 6334–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wakabayashi N., Itoh K., Wakabayashi J., Motohashi H., Noda S., Takahashi S., Imakado S., Kotsuji T., Otsuka F., Roop D. R., Harada T., Engel J. D., Yamamoto M. (2003) Nat. Genet. 35, 238–245 [DOI] [PubMed] [Google Scholar]
- 21.McMahon M., Itoh K., Yamamoto M., Hayes J. D. (2003) J. Biol. Chem. 278, 21592–21600 [DOI] [PubMed] [Google Scholar]
- 22.McMahon M., Thomas N., Itoh K., Yamamoto M., Hayes J. D. (2004) J. Biol. Chem. 279, 31556–31567 [DOI] [PubMed] [Google Scholar]
- 23.Guo Y., Yang M. C., Weissler J. C., Yang Y. S. (2008) Biochem. Biophys. Res. Commun. 374, 570–575 [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi S., Shibata H., Kurihara I., Yokota K., Suda N., Saito I., Saruta T. (2004) J. Mol. Endocrinol. 32, 69–86 [DOI] [PubMed] [Google Scholar]
- 25.Pan X., Li H., Zhang P., Jin B., Man J., Tian L., Su G., Zhao J., Li W., Liu H., Gong W., Zhou T., Zhang X. (2006) Biochem. Biophys. Res. Commun. 344, 727–734 [DOI] [PubMed] [Google Scholar]
- 26.Poukka H., Aarnisalo P., Karvonen U., Palvimo J. J., Jänne O. A. (1999) J. Biol. Chem. 274, 19441–19446 [DOI] [PubMed] [Google Scholar]
- 27.Yokota K., Shibata H., Kurihara I., Kobayashi S., Suda N., Murai-Takeda A., Saito I., Kitagawa H., Kato S., Saruta T., Itoh H. (2007) J. Biol. Chem. 282, 1998–2010 [DOI] [PubMed] [Google Scholar]
- 28.Plafker K. S., Farjo K. M., Wiechmann A. F., Plafker S. M. (2008) Invest. Ophthalmol. Vis. Sci. 49, 5611–5618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plafker K. S., Singer J. D., Plafker S. M. (2009) Biochemistry 48, 3527–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen C., Okayama H. (1987) Mol. Cell. Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C. A., Okayama H. (1988) BioTechniques 6, 632–638 [PubMed] [Google Scholar]
- 32.Hong F., Freeman M. L., Liebler D. C. (2005) Chem. Res. Toxicol. 18, 1917–1926 [DOI] [PubMed] [Google Scholar]
- 33.Hong F., Sekhar K. R., Freeman M. L., Liebler D. C. (2005) J. Biol. Chem. 280, 31768–31775 [DOI] [PubMed] [Google Scholar]
- 34.Rachakonda G., Xiong Y., Sekhar K. R., Stamer S. L., Liebler D. C., Freeman M. L. (2008) Chem. Res. Toxicol. 21, 705–710 [DOI] [PubMed] [Google Scholar]
- 35.Zhang D. D., Hannink M. (2003) Mol. Cell. Biol. 23, 8137–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plafker S. M., Plafker K. S., Weissman A. M., Macara I. G. (2004) J. Cell Biol. 167, 649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu P. Y., Hanlon M., Eddins M., Tsui C., Rogers R. S., Jensen J. P., Matunis M. J., Weissman A. M., Wolberger C., Pickart C. M. (2003) EMBO J. 22, 5241–5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekhar K. R., Yan X. X., Freeman M. L. (2002) Oncogene 21, 6829–6834 [DOI] [PubMed] [Google Scholar]
- 39.Itoh K., Wakabayashi N., Katoh Y., Ishii T., O'Connor T., Yamamoto M. (2003) Genes Cells 8, 379–391 [DOI] [PubMed] [Google Scholar]
- 40.Wen W., Meinkoth J. L., Tsien R. Y., Taylor S. S. (1995) Cell 82, 463–473 [DOI] [PubMed] [Google Scholar]
- 41.Shibata T., Ohta T., Tong K. I., Kokubu A., Odogawa R., Tsuta K., Asamura H., Yamamoto M., Hirohashi S. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 13568–13573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi M., Itoh K., Suzuki T., Osanai H., Nishikawa K., Katoh Y., Takagi Y., Yamamoto M. (2002) Genes Cells 7, 807–820 [DOI] [PubMed] [Google Scholar]
- 43.Adam S. A., Gerace L. (1991) Cell 66, 837–847 [DOI] [PubMed] [Google Scholar]
- 44.Radu A., Blobel G., Moore M. S. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 1769–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plafker S. M., Macara I. G. (2000) EMBO J. 19, 5502–5513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeong W. S., Keum Y. S., Chen C., Jain M. R., Shen G., Kim J. H., Li W., Kong A. N. (2005) J. Biochem. Mol. Biol. 38, 167–176 [DOI] [PubMed] [Google Scholar]
- 47.Nguyen T., Sherratt P. J., Pickett C. B. (2003) Annu. Rev. Pharmacol. Toxicol. 43, 233–260 [DOI] [PubMed] [Google Scholar]
- 48.Furukawa M., Xiong Y. (2005) Mol. Cell. Biol. 25, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi A., Kang M. I., Watai Y., Tong K. I., Shibata T., Uchida K., Yamamoto M. (2006) Mol. Cell. Biol. 26, 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.