

Abstract
The processing and regulated secretion of IL-1β are critical points of control of the biological activity of this important pro-inflammatory cytokine. IL-1β is produced by both monocytes and macrophages, but the rate and mechanism of release differ according to the differentiation status and the origin of these cells. We aimed to study the control of processing and release in human blood monocytes and human monocyte-derived macrophages. Toll-like receptor (TLR)-induced IL-1β production and release were investigated for dependence upon caspase-1, P2X7 receptor activation, and loss of membrane asymmetry associated with microvesicle shedding. TLR agonists induced P2X7 receptor-dependent IL-1β release in both monocytes and macrophages; however, only monocytes also showed P2X7 receptor-independent release of mature IL-1β. Furthermore, in monocytes ATP-mediated PS exposure could be activated independently of IL-1β production. Release of IL-1β from monocytes showed selectivity for specific TLR agonists and was accelerated by P2X7 receptor activation. Human monocytes released more IL-1β/cell than macrophages. These data have important implications for inflammatory diseases that involve monocyte activation and IL-1 release.
Keywords: Cytokines/Interleukins, Immunology/ Innate Immunity, Immunology/LPS, Immunology/Toll Receptors, Organisms/Human, IL-1β, P2X7, Inflammation
Introduction
The interleukin (IL)-16 family of cytokines contains important regulators of the acute inflammatory response (1) and can contribute to chronic inflammation in a number of diseases (2). Interleukin-1β is produced predominantly by activated monocytes and macrophages (3); however, mechanisms of cellular release into the extracellular environment remain the subject of much investigation. IL-1β is a leaderless protein, lacking the signal peptide that typically directs secreted proteins through the classical endoplasmic reticulum-Golgi secretory pathway (4). Human IL-1β is synthesized as a 31-kDa inactive precursor, cleaved by caspase-1 (but potentially by caspase-8 (5)), to generate a 17-kDa biologically active species.
In inflammatory cells such as the monocyte/macrophage, the process of IL-1β secretion is thought to occur via two key steps. A primary pro-inflammatory stimulus, notably bacterial LPS, induces high levels of pro-IL-1β synthesis but applied alone results in little IL-1β release (6–9). A second stimulus is required to activate caspase-1 (10), and extracellular ATP, acting on the P2X7 receptor, is widely acknowledged as having this role. The precise mechanism of P2X7 receptor-induced caspase-1 activation is not clear but is known to require K+ efflux and assembly of the NALP3 inflammasome (11, 12).
The P2X7 receptor is a cation-selective ion channel activated by extracellular ATP, but in striking contrast to other ion channels, activation also causes opening of larger pores, allowing passage of molecules up to 900 Da (13) through coupling to the Pannexin-1 hemichannel (14–16). Several reports have shown that the P2X7 receptor, and not other P2X receptors, is responsible for IL-1β release from activated myeloid cells (9, 11, 17–19). LPS-primed macrophages from transgenic mice lacking P2X7 receptors fail to release IL-1β in response to ATP, although synthesis of pro-IL-1β and caspase-1 is unaltered (20, 21).
The role of ATP/P2X receptor signaling in release of IL-1β from human monocytes has, however, been questioned. Grahames et al. (22) found no evidence of a role for ATP or the P2X7 receptor in LPS-induced IL-1β release from either THP-1 cells or primary human monocytes. In contrast a recent study demonstrated that low levels of IL-1β release were detected following chronic exposure of blood monocytes to LPS (24 h) because of endogenous ATP release (23, 24). Whether prolonged exposure of monocytes to LPS results in IL-1β secretion in the absence of a secondary stimulus or as a result of endogenous ATP release remains an important question.
Several mechanisms for trafficking leaderless IL-1β into the extracellular compartment have been proposed (8, 11, 25–27), including shedding of PS-exposed microvesicles containing IL-1β (28). Although such mechanisms are important for IL-1β release from monocytic-like cell lines such as THP-1 (22, 28–30), the mechanism of IL-1β externalization from primary human monocytes is less studied. Most studies have focused on human macrophages, but these differentiated cells produce less IL-1β than their precursor monocytes and, over a longer time course (31, 32), a phenotype particularly evident in the lung (33). Differentiated macrophages show a number of differences to blood monocytes, including altered expression of TLRs (34) and increased ability to present antigens (35), reflecting their role in linking innate to adaptive immunity.
The aims of the present study were: to determine the ATP dependence of temporal IL-1β secretion in human primary blood monocytes following TLR stimulation, to identify whether these cells undergo a loss of membrane asymmetry in response to ATP, and to test the hypothesis that the mechanisms regulating IL-1β release may be different between macrophages and monocytes. We show that human peripheral blood monocytes undergo PS exposure in response to P2X7 receptor activation, which was independent of priming stimulus, caspase-1 activation, and IL-1β secretion. We have identified that 24-h activation of monocytes with LPS or stimulation with TLR 7/8 agonists leads to P2X7 receptor-independent IL-1β secretion. This ATP-independent IL-1 release is absent in monocyte-derived macrophages. Sequential addition of ATP in both cell types resulted in a fast accentuated release response to enhance IL-1β secretion.
EXPERIMENTAL PROCEDURES
Materials
LPS (purified from Escherichia coli, serotype 0111:B4, TLR grade, Alexis) was purchased from Enzo Life Sciences Ltd. (Exeter, UK). Poly(I:C), a synthetic analogue of double-stranded RNA and TLR3 ligand (InvivoGen, San Diego, CA), was used at 25 μg/ml. CL075 (also named 3M002, purchased from InvivoGen), a thiazoloquinolone derivative that stimulates human TLRs 7 and 8, was used at 1 μg/ml unless otherwise stated. GardiquimodTM (InvivoGen), an imidazoquinoline active at the TLR7, was used at 1 or 10 μg/ml as stated. CL097 (InvivoGen), a water-soluble derivative of the imidazoquinoline compound R848 shown to induce activation of TLR7 at 0.1 μg/ml and TLR8 at 1 μg/ml, was used at each of these concentrations. For TLR2 agonists, MALP-2 was from Axxora (Nottingham, UK), and Pam2CSK4, Pam3CSK4, and FSL-1 were from InvivoGen, each used at 10 ng/ml unless otherwise stated. 3′-O-(4-benzoyl)benzyl-ATP (Bz-ATP) purchased from Sigma was added at 300 μm. The isoquinoline KN62 (calmodulin kinase II and P2X7 receptor inhibitor; Calbiochem) was used at 10 μm from a 6 mm working stock of KN62 in DMSO; hence, 0.17% (v/v) DMSO vehicle controls were applied in parallel to KN62 incubations. The selective P2X7 receptor antagonist 15d (36) (A438079 hydrochloride; Tocris Bioscience) was used at 10 μm. The caspase-1 inhibitor YVAD (Calbiochem) was incubated at 50 μm in parallel with the corresponding 0.01% (v/v) DMSO vehicle control. Fluorophores were purchased from Molecular Probes/Invitrogen or BD Biosciences, as below.
Isolation of Human Primary Peripheral Blood Monocytes
Peripheral venous blood was taken from healthy volunteers under written consent using a protocol approved by the South Sheffield Research Ethics Committee. Peripheral blood mononuclear cells were prepared by density centrifugation using OptiPrepTM (Axis-Shield, Kimbolton, UK). Enriched monocytes were prepared from peripheral blood mononuclear cells using the negative magnetic selection monocyte isolation kit II (Miltenyi Biotec, Bergisch Gladbach, Germany). Monocyte purity was determined by staining with anti-CD14 and analysis by flow cytometry (FACScan flow cytometer; BD Biosciences, Mountain View, CA). The data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR). Mean purity over the course of these experiments was 90.2 ± 0.74% CD14 positive (n = 32).
Monocyte-derived Macrophage (MDM) Preparation
The cells were prepared from peripheral blood mononuclear cells plated at 2 × 106 cells/ml in serum-free RPMI. After 1 h, the nonadherent cells were removed, and adherent cells were cultured in RPMI containing 10% (v/v) newborn calf serum with macrophage colony-stimulating factor (50 ng/ml) for 3 days and then cultured in RPMI with 10% (v/v) newborn calf serum for a further 11 days. Cell treatments with inflammatory modulators were performed at 37 °C (5% CO2 humidified incubator) in normal monocyte or MDM growth medium for 3 or 24 h as stated. Subsequent application of BzATP (in the absence or presence of inhibitors or DMSO vehicle control) was for 15–30 min, unless otherwise stated, at 37 °C. The supernatants were collected and separated from nonadherent cells by centrifugation.
Detection of IL-1β by ELISA
Monocytes were plated at 50,000 cells/ml (most experiments) or 200,000 cells/ml (experiments studying responses to TLR2 ligands) in a 0.5-ml volume in 24-well plates; cell-free supernatants were prepared following the incubation conditions described. IL-1β was measured using the Duo Set (DY201) ELISA from R & D Systems (Abingdon, UK), according to the manufacturer's instructions. The samples were diluted so that the optical density fell within the usable range of a log linear standard curve. The limit of detection was 19.5 pg/ml for IL-1β; where samples were below the detection limit, they were assigned the limit value.
Western Blotting
Monocytes were used at 500,000 cells/sample (in 250 μl of medium) and stimulated as described for each experiment. The cell extracts were prepared by centrifugation of supernatants to pellet suspension cells followed by resuspension in lysis buffer (phosphate-buffered saline plus 1% Triton (v/v) and protease inhibitors, Mixture III; Calbiochem). Adherent monocytes were extracted by scraping cells from multiwell plates into lysis buffer. Monocyte supernatants and cell extracts were analyzed by immunoblotting after electrophoresis on a 12% SDS (w/v) polyacrylamide gel and transfer onto polyvinylidene difluoride membrane. Equal loading of samples was verified by Ponceau S staining (Sigma) of the SDS-PAGE gel prior to transfer. Human IL-1β was detected using a monoclonal antibody (R & D Systems; Clone MAB201) at 1:1,000 dilution in phosphate-buffered saline with 0.1% (v/v) Tween 20, 5% (w/v) nonfat milk. Detection of the primary antibody was achieved by staining with an horseradish peroxidase-conjugated anti-mouse immunoglobulin at 1:2,000 dilution (New England Biolabs) and Amersham Biosciences ECLTM Western blotting detection reagents (GE Healthcare).
Detection of PS Exposure
Monocytes, prepared as described, were seeded at 1 × 106 cells/ml in 96-well Flexiwell (Dynex Technologies, Worthing, UK) plates and then treated for 3 h with buffer or LPS (1 ng/ml) in the presence of DMSO or YVAD (50 μm), followed by treatment for 20 min with buffer or BzATP (300 μm). The cells were then removed by gentle agitation in ice-cold phosphate-buffered saline (without Ca2+ and Mg2+) and centrifuged at 2000 × g for 5 min before resuspension in 96.5 ml of annexin-binding buffer (BD Biosciences). The monocytes were then stained with 3.5 μl of annexin V PE (BD Biosciences) for 20 min at room temperature in the dark before centrifugation and resuspension in CellFIXTM (BD Biosciences). The samples were acquired (10,000 total events) on a BD FACScan analyzed using FlowJo.
PS exposure on the cell surface was also measured by microscopy using annexin V-Alexa Fluor® 488 conjugate (Invitrogen), counterstained with Hoechst 33342. The cells were plated in LabTek (Fisher) 8-well chamber slides at 200,000 cells/well and incubated with buffer, LPS, or CL075 for 3 or 24 h. The permeable nuclear stain Hoechst 33342 was then added to the cells at 5 μg/ml final concentration for 15 min at 37 °C. The cells were then washed and resuspended in fresh medium. Five μl of annexin V-Alexa Fluor® 488 was added per well, along with BzATP or buffer (control) stimulus. The cells were observed using a 40× oil immersion objective (n = 1.3) on a Zeiss Meta LSM 510 (Carl Zeiss) confocal microscope for DIC digital transmission and fluorescence imaging, in a 37 °C heated chamber. The images were captured and analyzed using proprietary LSM software.
Statistics
All of the data were analyzed using the Prism 5.0 program (GraphPad, San Diego, CA). Differences between groups were analyzed by one-way ANOVA and Tukey's post-test. The data are given as the means from separate biological samples ± S.E. All of the n values represent separate biological replicates (donors).
RESULTS
Previous studies have shown that activation of the P2X7 receptor results in IL-1β release from LPS-primed THP-1 cells and blood monocytes (22), but the role of P2X7 receptor signaling in human monocyte IL-1β release is less clear. We determined whether the P2X7 receptor was required for monocyte or macrophage IL-1β release and investigated to what degree IL-1β production and processing were linked to PS flip, associated with microvesicle shedding mechanisms.
Early Release of IL-1β Release Is P2X7 Receptor-dependent
BzATP, a P2X7 receptor agonist, stimulated IL-1β secretion from LPS-primed human primary peripheral blood monocytes. IL-1β was released into the extracellular medium by monocytes primed for 3 h by 1 ng/ml LPS and then treated with BzATP for 30 min. This was completely blocked by the P2X7 receptor antagonist, KN62 (Fig. 1A).
FIGURE 1.

Fast P2X7 receptor-dependent IL-1β release in peripheral blood monocytes primed with LPS, compared with P2X7 receptor-independent IL-1β release in monocytes treated with CL075. A, monocytes were primed with buffer (control) or LPS (1 ng/ml) for 3 h in the presence of DMSO (vehicle control) or KN62, followed by a second treatment with buffer or 300 μm BzATP. Release of IL-1β was determined by ELISA (mean ± S.E., n = 5 separate donors). Significant differences for BzATP compared with buffer only controls (***, p < 0.001) or compared with KN62 (###, p < 0.001) were analyzed by one-way ANOVA and Tukey's post test. B, peripheral blood monocytes were stimulated with buffer (control), LPS (1 ng/ml), poly(I:C) (TLR3 ligand, 25 μg/ml), or CL075 (TLR7/8 ligand, 1 μg/ml) for 3 h, followed by buffer (control) or BzATP treatment for 30 min. The supernatants were collected, and IL-1β release was measured by ELISA (means ± S.E., n = 5 separate donors). Significant differences for BzATP compared with buffer only (***, p < 0.001) were analyzed by one-way ANOVA and Tukey's post test. C, stimulated monocyte supernatants (upper panel) and lysates (lower panel) were analyzed for IL-1β content by immunoblotting, where unprocessed pro-IL-1β is 31 kDa, and active processed IL-1β (along with recombinant IL-1β control, right lane) is 17 kDa. The immunoblots shown are representative of n = 3 separate donors. Con, control.
The Imidazoquinoline, CL075, Activates IL-1β Synthesis and Release without a Secondary Stimulus
Following the finding that purified monocytes release IL-1β in response to brief (3 h) TLR4 ligand priming by LPS and subsequent P2X7 receptor activation and given that circulating monocytes are exposed to a variety of pathogens, we determined whether other TLR agonists prime in an analogous manner. We studied a panel of TLR agonists including CL075/3M002 (a thiazoloquinolone derivative with activity upon the TLR7/8 receptors (37–39)) for their ability to induce IL-1β secretion from peripheral blood monocytes.
Monocytes exposed to CL075 released large amounts (∼600 pg/ml) of IL-1β in the absence of any secondary stimulus (Fig. 1B). CL075 caused IL-1β release when used at concentrations above 0.3 μg/ml, reaching a plateau from 1 to 10 μg/ml (n = 4, not shown). Subsequent experiments used CL075 at 1 μg/ml. Monocytes do not express TLR3 (38), and in keeping with this the TLR3 agonist poly(I:C) did not induce IL-1β production in the absence or presence of a secondary stimulus (Fig. 1B).
CL075 stimulation induced IL-1β release in the absence of a secondary stimulus, but we hypothesized that addition of a P2X7 receptor agonist would enhance this IL-1β release. Secondary stimulation with BzATP, following 3 h of treatment with CL075, lead to an augmented level of IL-1β release (∼2,900 pg/ml; Fig. 1B).
To study the nature of the released IL-1β in more detail, supernatants and lysates from LPS- or CL075-treated monocytes were analyzed for their IL-1β content by immunoblotting (Fig. 1C). Treatment of monocytes with LPS for 3 h in the absence of a BzATP stimulus did not result in secretion of IL-1β into the supernatant, whereas CL075 treatment for 3 h resulted in the secretion of only the cleaved active 17-kDa form of IL-1β (Fig. 1C, third lane). A secondary stimulus of BzATP following either LPS or CL075 treatment resulted in release of both 31-kDa (unprocessed) and 17-kDa (active) IL-1β into the supernatant (Fig. 1C, fifth lane). In addition, an intermediate IL-1β cleavage product (∼25–28 kDa) was detected in supernatants taken from CL075/BzATP-treated monocytes. Monocyte lysates were prepared, and IL-1β cellular content was assessed by immunoblotting (Fig. 1C, lower blot). This showed 31-kDa and partial cleavage products in cells stimulated with either LPS or CL075 in both the control and ATP-stimulated cells. Increased detection of IL-1β proteins (cleaved and uncleaved) in CL075-treated versus LPS-treated cells was consistent with the increased release of IL-1β seen in response to this agonist.
TLR7/8 Agonists Activate IL-1β Release Independently of P2X7 Receptor Activity
Because previous reports for a single stimulus resulting in IL-1β release have suggested a role for endogenous ATP (22, 24), we investigated whether CL075 and other TLR7/8 agonists induced IL-1β release and whether this required activation of the P2X7 receptor pathway. Monocyte release of IL-1β mediated by CL075 (3 h) alone was not significantly altered in the presence of the P2X7 receptor antagonists, KN62 or 15d (36) (Fig. 2, A and C). However, the additional accentuated release of IL-1β induced by BzATP in CL075-treated cells was completely abolished by 15d or KN62 (Fig. 2, B and D).
FIGURE 2.

Specific TLR agonists mediate P2X7 receptor-independent IL-1β release in monocytes. A and B, IL-1β release by monocytes stimulated with the TLR7/8 agonist, CL075 for 3 h in the presence or absence of 15d (A438079) P2X7 receptor antagonist, followed by buffer (A) or BzATP (B) secondary stimulation. C and D, monocytes stimulated with CL075 for 3 h in the presence or absence of KN62 P2X7 receptor antagonist, followed by buffer (C) or BzATP (D) secondary stimulation. Release caused by CL075 alone was not significantly altered by 15d (A438079) or KN62 (NS), whereas these P2X7 receptor antagonists significantly attenuated BzATP-induced release (###, p < 0.001 for both) in CL075-primed monocytes. E and F, monocytes stimulated for 3 h with the TLR7 agonist, gardiquimod, in the absence (E) or presence (F) of BzATP did not release detectable IL-1β. G, monocytes treated for 3 h with the TLR7/8 agonist CL097 at 1 μg/ml in the absence of BzATP released significantly more IL-1β compared with buffer alone, which was not significantly altered in the presence of the P2X7 receptor antagonists 15d (A438079) or KN62. H, IL-1β secretion from monocytes stimulated for 3 h with 1 μg/ml CL097 followed by BzATP secondary stimulation (H) was significantly attenuated in the presence of 15d (A438079) or KN62. I and J, monocytes stimulated for 3 h with 0.1 μg/ml CL097 (reported to activate TLR7 in preference to TLR8 at this concentration) in the absence (I) or presence (J) of BzATP did not release detectable IL-1β. Supernatants were measured for IL-1β levels by ELISA. The data are the means ± S.E. for n = 3–5 separate donors. Significant differences (***, p < 0.001; **, p < 0.01; *, p < 0.05, NS, not significant) compared with buffer were analyzed by one-way ANOVA and Tukey's post-test.
CL075 (3M002) acts at both TLR7 and TLR8. We investigated the effect of the TLR7 agonist gardiquimod on monocyte-mediated IL-1β release. Gardiquimod treatment (10 μg/ml) for 3 h neither induced detectable IL-1β secretion (Fig. 2E) nor primed cells to release IL-1β in response to a secondary stimulus of BzATP (Fig. 2F). The TLR7/8 agonist CL097, a water-soluble derivative of the imidazoquinoline compound R848, was tested at 0.1 and 1 μg/ml because these concentrations are reported to activate TLR7 only and TLR7/8, respectively. Monocytes treated for 3 h with CL097 at 1 μg/ml released IL-1β in the absence of BzATP and in the presence of KN62 and 15d P2X7 receptor antagonists (Fig. 2G). As for CL075, augmented release occurred following secondary BzATP stimulation. This too was inhibited by 15d and KN62 (Fig. 2H). The lower dose of CL097 (0.1 μg/ml acting predominantly at TLR7) did not give any detectable release of IL-1β either in the absence or presence of subsequent BzATP (Fig. 2, I and J, respectively).
A Prolonged Single Stimulus of TLR Ligand Is Sufficient to Cause IL-1β Release Independently of P2X7 Receptor Activation
Having identified CL075 as an effective single stimulus mediating P2X7 receptor-independent IL-1β release, we investigated whether prolonged LPS stimulation without P2X7 receptor secondary stimulation could also result in IL-1β release and if so whether this was affected by subsequent ATP stimulation.
Supernatants taken from peripheral blood monocytes treated for 24 h with LPS contained IL-1β (100–150 pg/ml), which was not attenuated when KN62 or 15d was present throughout the experiment (Fig. 3A). Failure of KN62 to inhibit LPS-induced IL-1β release was not as a result of a loss of efficacy of the drug over the duration of the experiment because KN62 remained effective in blocking BzATP-mediated PS exposure after 24 h in culture conditions (data not shown, n = 3). Monocytes treated with CL075 alone over a sustained period (24 h) also released high levels of IL-1β independently of ATP (Fig. 3B). Monocytes stimulated for 24 h with the TLR7 agonist gardiquimod released IL-1β (≈100 pg/ml; Fig. 3C) in the absence and presence of 15d and KN62, as did the higher dose (1 μg/ml) of CL097 (in the order of 800 pg/ml; Fig. 3D), in contrast to 0.1 μg/ml CL097, which gave no detectable release.
FIGURE 3.
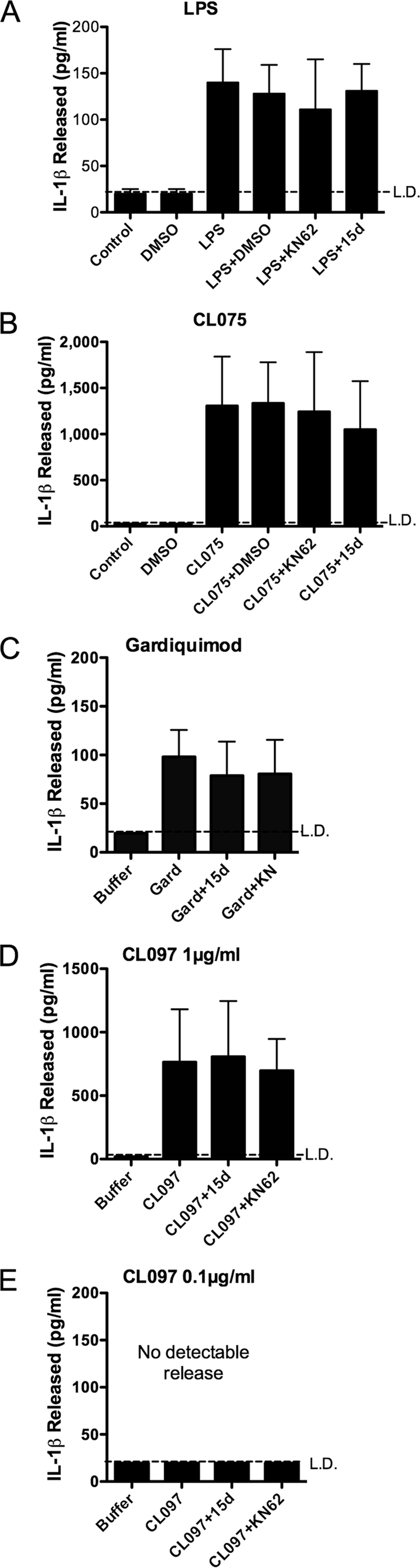
Stimulation of monocytes for 24 h with specific TLR agonists results in P2X7 receptor-independent IL-1β release. Peripheral blood monocytes treated for 24 h with 1 ng/ml LPS (A), 1 μg/ml CL075 (B), 10 μg/ml gardiquimod (C), or 1 μg/ml CL097 (D) released IL-1β as measured in supernatants by ELISA, whereas 0.1 μg/ml CL097 did not cause detectable IL-1β secretion (E). IL-1β release was not significantly inhibited by the presence of 15d (A438079) P2X7 receptor antagonist, KN62, or DMSO vehicle control. The data are the means ± S.E. for n = 3–5 separate donors.
The “One-hit” Stimulus Does Not Rely on Constitutive Activation of the IL-1β Release Mechanism
The currently accepted model for IL-1β release is a two-step control mechanism; the initial step occurs upon pathogen detection driving pro-IL-1β synthesis, whereas IL-1β processing and release requires a secondary signal, such as ATP (40). Because we were unable to inhibit IL-1β release stimulated by CL075 or prolonged LPS exposure alone using a P2X7 receptor antagonist, we considered whether the IL-1β processing and/or release pathway is constitutively active within the purified blood monocytes.
While examining TLR pathways that cause IL-1β release, we observed that activation of monocytes for 24 h with TLR2 ligands resulted in significant up-regulation of intracellular IL-1β, both in its precursor form and to a lesser extent as processed active IL-1β (Fig. 4A). However, this IL-1β was only released into the extracellular environment at very low levels compared with LPS treatment (Fig. 4B). Although monocytes stimulated with TLR2 or TLR4 ligands processed IL-1β intracellularly, the levels of intracellular IL-1β synthesis and processing by TLR2 ligands did not correlate with the released IL-1β levels. For example, of the TLR2 ligands, FSL-1 treatment resulted in the highest level of intracellular IL-1β synthesis, whereas released IL-1β remained almost undetectable, even in experiments in which monocytes were cultured at 200,000 cells/ml (4-fold higher numbers compared with other experiments investigating IL-1β release above). These data indicate that processing and release mechanisms are distinct and that the production of processed IL-1β inside the monocyte does not of necessity result in its release.
FIGURE 4.

TLR2 ligands activate IL-1β processing in the absence of release. A, whole cell lysates from monocytes treated for 24 h with LPS (1 ng/ml) or TLR2 ligands Pam3CSK4 (10 ng/ml), Pam2CSK4 (10 ng/ml), MALP-2 (10 ng/ml), or FSL-1 (10 ng/ml) were subjected to IL-1β immunoblotting. The 17-kDa bands, corresponding to active IL-1β, were analyzed by densitometry using NIH Image (v1.62f) (normalized to actin loading controls) where the data are the means ± S.E. for n = 4 separate donors. Below is a representative immunoblot for treatments corresponding to these ratios. B, IL-1β release measured by ELISA in supernatants from monocytes treated for 24 h with increasing concentrations of LPS or TLR2 ligands Pam3CSK4, MALP-2, Pam2CSK4, and FSL-1 (the data are the means ± S.E. for n = 3 separate donors).
Both P2X7 Receptor-dependent and -independent IL-1β Release Requires Caspase-1
Given recent evidence that caspase-8 can be involved in processing of IL-1β induced by TLR agonists (5), we tested the caspase-1 dependence of the newly identified P2X7 receptor-independent IL-1β release pathway in human monocytes. The caspase-1 inhibitor YVAD significantly attenuated IL-1β release for all stimulation conditions: 3 h CL075 (with or without BzATP; Fig. 5, B and C) or 3 h LPS (with or without BzATP; Fig. 5A) or sustained incubation with LPS (Fig. 5D) or CL075 (Fig. 5E) alone. The caspase-1 inhibitor was effective in reducing relative levels of secreted 17-kDa IL-1β, where supernatants were analyzed by immunoblotting (Fig. 5F). Treatment for 3 h with CL075 resulted in release of predominantly 17-kDa IL-1β, compared with mostly unprocessed 31-kDa IL-1β in the presence of YVAD (Fig. 5F, second and third lanes). A secondary stimulus of BzATP following CL075 gave rise to both 31- and 17-kDa IL-1β and an intermediate cleavage product, as above, compared with in the presence of YVAD where this level of processing was reduced, with the major product at 31 kDa (Fig. 5F, fourth and fifth lanes). These data indicate that both ATP-dependent and -independent pathways of IL-1β processing and release rely on caspase-1 activation.
FIGURE 5.

Caspase-1 dependence of IL-1β release. A, peripheral blood monocytes were primed with buffer (control) or LPS (1 ng/ml) for 3 h in the presence or absence of YVAD caspase-1 inhibitor or DMSO vehicle control, with or without a secondary stimulus of BzATP for 30 min. B and C, monocytes stimulated with CL075 for 3 h in the presence of buffer, DMSO (vehicle control), or YVAD followed by buffer (B) or BzATP secondary stimulation (C). D, monocytes were treated for 24 h with buffer (control) or LPS (1 ng/ml) in the presence of DMSO (vehicle control) or caspase-1 inhibitor YVAD. E, monocytes were treated for 24 h with buffer (control) or CL075 in the presence of DMSO (vehicle control) or caspase-1 inhibitor YVAD. The supernatants were measured for IL-1β levels by ELISA (means ± S.E., n = 5 separate donors). Significant differences for BzATP compared with buffer-treated controls (***, p < 0.001; **, p < 0.01; *, p > 0.05) or compared with YVAD (##, p < 0.01) were analyzed by one-way ANOVA and Tukey's post test. F, stimulated monocyte supernatants were analyzed for IL-1β content by immunoblotting, where unprocessed pro-IL-1β is 31 kDa, and active processed IL-1β is 17 kDa. The cells were treated for 3 h with buffer (Con, control), CL075, or CL075+YVAD in the absence or presence of 30 min of treatment with BzATP. A representative immunoblot is shown of four separate donors tested.
P2X7 Receptor-induced PS Flip Is Dissociated from IL-1β Release
In differentiated THP-1 monocytic cells, phosphatidylserine exposure and microvesicle shedding has been identified as a mechanism for packaging IL-1β and trafficking to the extracellular environment (28). We aimed to determine whether purified primary monocytes undergo PS flip and whether this is coupled to IL-1β release mechanisms. Annexin V binding was used to detect PS exposure both by fluorescence microscopy, using annexin V-Alexa Fluor 488 (not shown), and by flow cytometry using annexin V PE.
Fig. 6A shows that peripheral blood monocytes primed for 3 h by LPS were positively labeled for PS by annexin V PE upon stimulation with BzATP. The vesicles were observed separating from the plasma membrane in real time and using time lapse imaging (not shown). The PS exposure was quantified by flow cytometry for an ungated population of purified monocytes, shown by representative histograms (Fig. 6).
FIGURE 6.

PS exposure is dependent upon P2X7 receptor activation but independent of IL-1β release. PS exposure was assessed by flow cytometry for monocytes labeled with annexin V-PE. For each pair of micrographs displayed, the corresponding flow cytometric data for the same incubation conditions are shown below. A, PS staining caused by BzATP. Monocytes were primed with buffer (control, panel (i)) or LPS (1 ng/ml, panel (ii)), for 3 h and then stimulated with buffer or BzATP for 20 min. B, PS exposure inhibition by a P2X7 receptor antagonist. Monocytes were primed with buffer (control, panel (i)) or LPS (1 ng/ml, panel (ii)) for 3 h in the presence of DMSO (vehicle control) or KN62 and then stimulated with buffer or BzATP for 20 min. C, the effect of caspase-1 inhibition on PS exposure. Monocytes were primed with buffer (control, panel (i)) or LPS (1 ng/ml, panel (ii)) for 3 h in the presence of DMSO (vehicle control) or YVAD caspase-1 inhibitor and then stimulated with buffer or BzATP for 20 min. D, the effect of CL075 on PS exposure at 3 h in the presence of DMSO (vehicle control, panel (i)) or KN62 panel (ii). Monocytes treated with CL075 were subsequently stimulated with buffer or BzATP for 20 min in the absence or presence of KN62. The data shown are histograms taken from the same donor, which are typical of n = 3–4 separate donors. Further representative flow cytometric data for corresponding monocyte treatments are shown in supplemental Figs. S1 and S2.
Release of IL-1β, as measured by ELISA, only occurred for monocytes primed with LPS followed by BzATP activation (Fig. 1A), whereas nonprimed cells underwent PS exposure and vesicle shedding upon the addition of BzATP in the absence of IL-1β synthesis and release (Fig. 6A). ATP-mediated PS flip was completely inhibited by the P2X7 receptor antagonist KN62, in both buffer- and LPS-primed monocytes (Fig. 6B). Conversely, the caspase-1 inhibitor YVAD was ineffective at inhibiting these processes (Fig. 6C), clearly indicating that, under the conditions studied, PS exposure is dissociated from IL-1β processing and release.
Monocyte treatment with CL075 for 3 h (without P2X7 receptor secondary stimulation) resulted in a significant release of IL-1β at a similar, if not higher, amount as that released from cells stimulated with LPS + BzATP at the same time point. CL075 treatment alone did not, however, result in any detectable PS exposure (Fig. 6D, panel (i)), although cells retained the ability to undergo PS flip, because the addition of BzATP caused subsequent annexin V binding (Fig. 6D, panel (ii)). We also were unable to detect PS exposure for monocytes exhibiting P2X7 receptor-independent IL-1β release resulting from 24 h of LPS treatment (not shown). Monocyte annexin V-PE binding was quantified by flow cytometry; the mean data from 3 or 4 separate donors was consistent with the typical responses shown in Fig. 6 (supplemental Fig. S1).
Macrophages are absolutely dependent on ATP for IL-1β release; previously, work performed predominantly in macrophages had not reported any P2X7 receptor-independent production of IL-1β or the potential dissociation between IL-1β production and release and microvesicle formation. We therefore investigated the mechanism of IL-1β secretion evoked by TLR ligands and P2X7 receptor engagement in MDMs. IL-1β release from MDMs was detected in response to both 3- and 24-h pretreatment with either LPS or CL075 only when followed by a secondary stimulus of BzATP (Fig. 7, A and C). No IL-1β release occurred in the absence of BzATP, in contrast to the P2X7 receptor-independent CL075/slow LPS response in peripheral monocytes described above. Release of IL-1β from MDM following either CL075 or LPS priming followed by BzATP was studied, in the absence or presence of the P2X7 receptor antagonists KN62 or 15d (Fig. 7, B and D, respectively). Both of these antagonists significantly attenuated the BzATP-induced IL-1β secretion. Similar to the monocytic response, BzATP caused PS exposure at the cell surface in MDMs, as shown by confocal microscopy of macrophages labeled with annexin V Alexa Fluor® 488 (Fig. 7E). The ATP-mediated PS flip in MDMs also occurred independently of a primary LPS stimulus (Fig. 7E, panels (ii) and (iv)).
FIGURE 7.

ATP dependence of IL-1β release from MDMs. A–D, IL-1β was measured by ELISA from supernatants of MDMs. The cells were treated with 0.1, 1, or 10 ng/ml LPS or 0.3 and 1 μg/ml CL075 for 3 h (A) or 24 h (C), followed by a 30-min stimulus with buffer or BzATP. Release of IL-1β was only detected upon a secondary stimulus of BzATP (filled bars) compared with no release in the absence of BzATP (open bars). The addition of the P2X7 receptor antagonists KN62 or 15d (A438079) significantly attenuated BzATP induced IL-1β release from MDM for pretreatment over 3 h with CL075 (B) or 24 h with 1 ng/ml LPS (D). Significant differences (***, p < 0.001; *, p < 0.05) for comparisons indicated analyzed by one-way ANOVA and Tukey's post test. E, MDMs were primed with buffer (control) or LPS (1 ng/ml) for 3 h, stained with Hoechst 33342 and annexin V, Alexa Fluor® 488, and then stimulated with BzATP for 20 min and imaged by confocal microscopy. The images are typical of those obtained for n = 3 separate donors.
DISCUSSION
We have previously shown that IL-1β production from monocytes is crucial to the establishment of inflammatory responses (41). We therefore investigated the mechanisms underpinning TLR-dependent IL-1β release from primary human monocytes. We determined that these cells have multiple regulators of IL-1β release, in which a TLR-specific response can be rapidly amplified by ATP, a product of tissue damage, potentially allowing for the rapid induction of inflammation at sites of pathogen invasion or tissue damage.
The data presented here show that LPS-primed human primary blood monocytes release IL-1β in response to P2X7 receptor activation, in keeping with previous findings (12, 22). Importantly this study also demonstrates that purified human primary peripheral blood monocytes release IL-1β independently of P2X7 receptor activity and in response to a one-hit stimulus. This was most clearly seen when monocytes were stimulated with the imidazoquinoline CL075, which even in the absence of ATP caused high level and rapid (3 h) IL-1β release. Our work is supported by the recent findings by Hurst et al. (42), who describe TLR7/8 agonist induction of IL-1β secretion. Our data provide additional mechanistic insight, showing that this pathway does not rely on autocrine ATP release acting on the P2X7 receptor, because the antagonists KN62 and 15d did not inhibit IL-1β secretion. The failure of KN62 to inhibit this response was not because the drug lost efficacy, and to confirm this again we have also taken supernatants from cells treated with KN62 for 24 h and shown that these supernatants still block the actions of BzATP on fresh cells (data not shown). We also treated monocytes with apyrase to degrade any endogenous ATP production. However, high levels of IL-1β were released, for both 3- and 24-h incubations including treatments with apyrase alone, indicating the presence of contaminants in the apyrase preparation that lead to inflammatory activation, whose effects were also likely ATP-independent.
The imidazoquinoline CL075 is reported to act at TLRs 7 and 8, with higher efficacy at TLR8. To assess the relative contribution of TLR7 or TLR8 in mediating ATP-independent IL-1β release from monocytes, we tested gardiquimod and CL097 ligands. Gardiquimod, when applied at supramaximal concentrations required to activate TLR7, was ineffective at mediating any early (3 h) ATP-independent IL-1β release. Similarly, CL097, applied at 0.1 μg/ml to selectively activate TLR7, had no effect on this release. However, when CL097 was applied at 1 μg/ml, a concentration reported to activate TLR8, a rapid ATP-independent IL-1β release was detected, similar to that observed for CL075. These data support a role for TLR8 in the early ATP-independent IL-1β secretion response in human blood monocytes. These data are consistent with observations showing that TLR8 engagement is more effective than TLR7 activation in inducing proinflammatory cytokine production from human monocytes (37).
Our work aimed to address whether IL-1β release from monocytes, following prolonged (24 h) LPS stimulation, is dependent on ATP. Our findings fit with those of Grahames et al. (22) whereby 24-h LPS-induced IL-1β release was independent of P2X7 receptor activation. These data are, however, at odds with recent work showing that sustained LPS causes IL-1β secretion as a result of autocrine ATP release (24). This apparent disparity may be reconciled by considering the differences in monocyte isolation methods; we have used negative magnetic selection, depleting CD16+ “inflammatory” monocyte subsets (43) and retaining nonadherent cells, whereas Piccini et al. (24) isolated peripheral blood mononuclear cells using buffy coats and selected monocytes by adherence. In addition we have used a lower concentration of LPS (1 ng/ml in this study, compared with 1 μg/ml); TLR4 agonist dose is reported to recruit different signaling proteins (55), which may account for some discrepancy over the ATP dependence and time course of IL-1β secretion. It will be of future interest to understand the differential roles that subsets of monocytes and relative LPS exposure play in inflammation.
We found that TLR2 ligands were able to activate the synthesis and intracellular processing of IL-1β without significant IL-1β release. These agonists were effective inducers of CXCL8 generation in monocytes (34). LPS and CL075 caused IL-1β processing and release, but our panel of TLR2 agonists caused IL-1β processing only. Thus, although our data would be consistent with constitutive or agonist-induced activation of the inflammasome in monocytes (23, 24, 44), it is not consistent with constitutive activation of the IL-1β release machinery or an absolute link between monocyte activation and IL-1β release.
Caspase-1 inhibition significantly attenuated all pathways resulting in IL-1β release from our preparations of blood monocytes. Thus, although a role for caspase-8 in the processing of IL-1β has recently been described (5), in monocytes the dominant processing is likely via caspase-1. Although it is clear from our data and that of others (24) that IL-1β secretion can be amplified by ATP acting via P2X7 receptors, our results, and those of Grahames et al. (22), now strongly suggest that in monocytes under certain conditions, these pathways are not absolutely required.
The addition of ATP to purified monocytes caused an additional and much more rapid release of IL-1β in a P2X7 receptor-dependent fashion. This amplification of IL-1β release was seen when ATP was added to cells activated with either LPS or CL075. Although cells stimulated with CL075 alone preferentially released the cleaved, 17-kDa active form of IL-1β, amplification of release by the addition of ATP caused both the pro-IL-1β and active cleaved IL-1β species to be released. These data further reinforce the difference between the two release mechanisms, because the ATP/microvesicle pathway results in a less discriminate packaging of IL-1β isoforms. The ATP-driven, accelerated release of IL-1β may reflect a rapid response mechanism to cell damage or platelet degranulation, on top of a sustained release caused by the presence of circulating viral or bacterial pathogens.
The loss of membrane symmetry and microvesicle shedding, as a mechanism of IL-1β secretion, has been previously reported in differentiated THP-1 monocytic cell lines (28) and in a mouse macrophage RAW264.7 cell line (45). We found that purified primary human monocytes also undergo PS flip in response to ATP, which was specifically inhibited by P2X7 receptor antagonists. Our data show that PS exposure occurred both in the absence and in the presence of TLR priming or caspase-1 activity. Hence the loss of membrane asymmetry linked to microvesicle shedding is not selective for IL-1β release and/or processing but is a response to P2X7 receptor activation that results in packaging of cellular content, whether or not the cell has been primed to up-regulate IL-1β, in line with related findings in other cell types (27, 46). IL-1β secretion mediated by both sustained LPS, or CL075 alone, was not accompanied by PS exposure.
Our new results showing the separation of IL-1β release mechanisms from ATP dependence reinforce the importance of differences between observations from different groups in the field. We explored why our data pointed to a more complex regulation of IL-1β release than has historically been appreciated. We therefore compared the mechanisms regulating IL-1β release in MDMs. We found that, in keeping with the prevailing literature, MDMs were unable to release IL-1β in response to CL075 or LPS in the absence of the ATP secondary stimulus acting at the P2X7 receptor, in contrast to peripheral monocytes. Thus, we reveal that monocytes possess the ability to release IL-1β in the absence of P2X7 receptor activation, which is not seen in macrophages.
These data suggest the existence of a release mechanism of IL-1β that has two points of regulation: an initial phase of IL-1β release rapidly amplified by ATP signaling potentially providing monocytes with an ability to regulate inflammation in a graded manner, in which contact with pathogens would initiate IL-1β production, and an accentuated release to rapidly amplify local effects, perhaps reflecting tissue damage or platelet activation (both good sources of ATP (47, 48)). The phenotypes of responses between donors to TLR agonists (with or without BzATP) for both monocytes and MDM were robust in all experiments.
The one-hit stimulus of either CL075 or LPS results in ATP-independent IL-1β secretion, without the loss of membrane asymmetry, whereas a subsequent hit with ATP drives a rapid amplification of IL-1β release via PS exposure and microvesicle shedding (28). This supports a model for the one-hit pathway engaging alternative extracellular trafficking to the microvesicle shedding, which may involve secretory lysosomes (4, 49) or phagosomal destablization (50). Many models for IL-1β release have been reported, including microvesicle shedding, endolysosomal pathways, exosomal transport (27, 51), and phagosomal destablization. Hence the question of how IL-1β exits the cell remains enigmatic. Our data support the concept that, in purified primary human monocytes, IL-1β may be released by engaging multiple distinct secretory mechanisms, according to which receptors are activated.
Recently, a TLR-mediated biphasic response, dependent on ROS production followed by a delayed antioxidative response, has been shown to control IL-1β processing and release in human monocytes (52). The role of ROS and antioxidative responses in the CL075 P2X7-independent IL-1β release mechanism, compared with P2X7-dependent release, will provide an interesting future investigation, beyond the scope of this study.
An important role for the P2X7 receptor in chronic inflammatory disease models has been demonstrated where P2X7 receptor antagonists are effective in animal models of pain and inflammation (53), and P2X7 receptor knock-out mice show markedly attenuated collagen-induced arthritis (20). These chronic inflammatory conditions are likely to involve tissue macrophages, which are absolutely dependent on P2X7 receptor activity for IL-1β secretion. Conversely, some IL-1-driven inflammatory disease models refute a role for the P2X7 receptor; IL-1-mediated neointima formation is unaltered in P2X7 receptor-deficient mice (54). Our data showing that monocytes undergo P2X7 receptor-independent IL-1β release may help to explain these findings and may indicate where disease processes are driven predominantly by circulating monocytes or the tissue macrophage.
The differences between monocytic and macrophage IL-1β release mechanisms may well underlie differences in their physiological roles. Activation of monocytes generates IL-1β production, which occurs over a few hours, and will induce inflammation in various tissue models of disease (41). Monocytes are rapidly recruited to inflammatory sites and as circulating cells may also contact sites of acute vascular damage where platelet activation has occurred. Where tissue damage or platelet activation has released ATP, a primed monocyte is capable of initiating a rapid amplification of inflammation via quickly accentuated IL-1β release. In contrast, in macrophage populations such as the alveolar macrophage, IL-1β production occurs at reduced levels and over slower time courses (31, 32). Such down-regulation of IL-1β production may facilitate a scavenging role for these cells, allowing them to survey and patrol epithelial surfaces, engage with limited stimuli that have not yet established substantial tissue damage (e.g. small numbers of inhaled bacteria), and resolve small stimuli without rapid induction of a major inflammatory response. However, once an initial threshold of activation is overcome and monocyte recruitment is initiated from inflamed tissue or a damaged microvasculature, rapid IL-1β production and release will serve to initiate effective local inflammation.
This study has provided important insight into the mechanisms leading to monocyte/macrophage inflammatory responses upon host-pathogen interaction. The differences in P2X7 receptor activation requirement for IL-1β release between monocytes and MDMs may also help reconcile some differences between the monocyte/macrophage cell-based models, whether derived from primary sources or from cell lines. In addition, these findings offer important implications for inflammatory disease processes, which may have pathogenic mechanisms based predominantly in either circulating monocytes or the tissue macrophage.
Supplementary Material
Acknowledgments
We are grateful to Elizabeth Prestwich, Nazia Chaudhuri, and Sadia Anwar for assistance in monocyte preparations and to Clare Druce for protocol optimization.
This work was supported by British Heart Foundation Grants FS/06/004 (to J. R. W.) and FS/05/081/19423 (to P. W. W.) and funds from the Medical Research Council (to L. C. P.) and Research Councils UK (to H. L. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- IL
- interleukin
- TLR
- Toll-like receptor
- LPS
- lipopolysaccharide
- PS
- phosphatidylserine
- DMSO
- dimethyl sulfoxide
- MDM
- monocyte-derived macrophage
- ELISA
- enzyme-linked immunosorbent assay
- ANOVA
- analysis of variance.
REFERENCES
- 1.Dinarello C. A. (2009) Annu. Rev. Immunol. 27, 519–550 [DOI] [PubMed] [Google Scholar]
- 2.Dinarello C. A. (1996) Blood 87, 2095–2147 [PubMed] [Google Scholar]
- 3.Dinarello C. A. (1998) Int. Rev. Immunol. 16, 457–499 [DOI] [PubMed] [Google Scholar]
- 4.Zocchi M. R., Rubartelli A. (2001) in Frontiers of Life (Baltimore D., Dulbecco R., Jacob F., Levi-Montalcini R. eds) pp. 477–488, Academic Press, Orlando, FL [Google Scholar]
- 5.Maelfait J., Vercammen E., Janssens S., Schotte P., Haegman M., Magez S., Beyaert R. (2008) J. Exp. Med. 205, 1967–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrari D., Chiozzi P., Falzoni S., Hanau S., Di Virgilio F. (1997) J. Exp. Med. 185, 579–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogquist K. A., Nett M. A., Unanue E. R., Chaplin D. D. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 8485–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perregaux D., Gabel C. A. (1994) J. Biol. Chem. 269, 15195–15203 [PubMed] [Google Scholar]
- 9.Sanz J. M., Di Virgilio F. (2000) J. Immunol. 164, 4893–4898 [DOI] [PubMed] [Google Scholar]
- 10.Martinon F., Gaide O., Pétrilli V., Mayor A., Tschopp J. (2007) Semin. Immunopathol. 29, 213–229 [DOI] [PubMed] [Google Scholar]
- 11.Ferrari D., Pizzirani C., Adinolfi E., Lemoli R. M., Curti A., Idzko M., Panther E., Di Virgilio F. (2006) J. Immunol. 176, 3877–3883 [DOI] [PubMed] [Google Scholar]
- 12.Mariathasan S., Weiss D. S., Newton K., McBride J., O'Rourke K., Roose-Girma M., Lee W. P., Weinrauch Y., Monack D. M., Dixit V. M. (2006) Nature 440, 228–232 [DOI] [PubMed] [Google Scholar]
- 13.Virginio C., MacKenzie A., North R. A., Surprenant A. (1999) J. Physiol. 519, 335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pelegrin P., Surprenant A. (2006) EMBO J. 25, 5071–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelegrin P., Barroso-Gutierrez C., Surprenant A. (2008) J. Immunol. 180, 7147–7157 [DOI] [PubMed] [Google Scholar]
- 16.Pelegrin P., Surprenant A. (2007) J. Biol. Chem. 282, 2386–2394 [DOI] [PubMed] [Google Scholar]
- 17.Griffiths R. J., Stam E. J., Downs J. T., Otterness I. G. (1995) J. Immunol. 154, 2821–2828 [PubMed] [Google Scholar]
- 18.Le Feuvre R., Brough D., Rothwell N. (2002) Eur. J. Pharmacol. 447, 261–269 [DOI] [PubMed] [Google Scholar]
- 19.Mehta V. B., Hart J., Wewers M. D. (2001) J. Biol. Chem. 276, 3820–3826 [DOI] [PubMed] [Google Scholar]
- 20.Labasi J. M., Petrushova N., Donovan C., McCurdy S., Lira P., Payette M. M., Brissette W., Wicks J. R., Audoly L., Gabel C. A. (2002) J. Immunol. 168, 6436–6445 [DOI] [PubMed] [Google Scholar]
- 21.Solle M., Labasi J., Perregaux D. G., Stam E., Petrushova N., Koller B. H., Griffiths R. J., Gabel C. A. (2001) J. Biol. Chem. 276, 125–132 [DOI] [PubMed] [Google Scholar]
- 22.Grahames C. B., Michel A. D., Chessell I. P., Humphrey P. P. (1999) Br. J. Pharmacol. 127, 1915–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Netea M. G., Nold-Petry C. A., Nold M. F., Joosten L. A., Opitz B., van der Meer J. H., van de Veerdonk F. L., Ferwerda G., Heinhuis B., Devesa I., Funk C. J., Mason R. J., Kullberg B. J., Rubartelli A., van der Meer J. W., Dinarello C. A. (2009) Blood 113, 2324–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piccini A., Carta S., Tassi S., Lasiglié D., Fossati G., Rubartelli A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8067–8072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brough D., Rothwell N. J. (2007) J. Cell Sci. 120, 772–781 [DOI] [PubMed] [Google Scholar]
- 26.Kahlenberg J. M., Dubyak G. R. (2004) Am. J. Physiol. Cell Physiol. 286, C1100–C1108 [DOI] [PubMed] [Google Scholar]
- 27.Qu Y., Franchi L., Nunez G., Dubyak G. R. (2007) J. Immunol. 179, 1913–1925 [DOI] [PubMed] [Google Scholar]
- 28.MacKenzie A., Wilson H. L., Kiss-Toth E., Dower S. K., North R. A., Surprenant A. (2001) Immunity 15, 825–835 [DOI] [PubMed] [Google Scholar]
- 29.Moore S. F., MacKenzie A. B. (2007) Cell Signal. 19, 855–866 [DOI] [PubMed] [Google Scholar]
- 30.Schilling W. P., Wasylyna T., Dubyak G. R., Humphreys B. D., Sinkins W. G. (1999) Am. J. Physiol. 277, C766–C776 [DOI] [PubMed] [Google Scholar]
- 31.Chin J., Kostura M. J. (1993) J. Immunol. 151, 5574–5585 [PubMed] [Google Scholar]
- 32.Perregaux D. G., Laliberte R. E., Gabel C. A. (1996) J. Biol. Chem. 271, 29830–29838 [DOI] [PubMed] [Google Scholar]
- 33.Herzyk D. J., Allen J. N., Marsh C. B., Wewers M. D. (1992) J. Immunol. 149, 3052–3058 [PubMed] [Google Scholar]
- 34.West P. W., Parker L. C., Ward J. R., Sabroe I. (2008) Immunology 125, 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laupéze B., Fardel O., Onno M., Bertho N., Drénou B., Fauchet R., Amiot L. (1999) Hum. Immunol. 60, 591–597 [DOI] [PubMed] [Google Scholar]
- 36.Nelson D. W., Gregg R. J., Kort M. E., Perez-Medrano A., Voight E. A., Wang Y., Grayson G., Namovic M. T., Donnelly-Roberts D. L., Niforatos W., Honore P., Jarvis M. F., Faltynek C. R., Carroll W. A. (2006) J. Med. Chem. 49, 3659–3666 [DOI] [PubMed] [Google Scholar]
- 37.Gorden K. B., Gorski K. S., Gibson S. J., Kedl R. M., Kieper W. C., Qiu X., Tomai M. A., Alkan S. S., Vasilakos J. P. (2005) J. Immunol. 174, 1259–1268 [DOI] [PubMed] [Google Scholar]
- 38.Hornung V., Rothenfusser S., Britsch S., Krug A., Jahrsdörfer B., Giese T., Endres S., Hartmann G. (2002) J. Immunol. 168, 4531–4537 [DOI] [PubMed] [Google Scholar]
- 39.Stockfleth E., Trefzer U., Garcia-Bartels C., Wegner T., Schmook T., Sterry W. (2003) Br. J. Dermatol. 149, (Suppl. 66) 53–56 [DOI] [PubMed] [Google Scholar]
- 40.Di Virgilio F. (2007) Trends Pharmacol. Sci. 28, 465–472 [DOI] [PubMed] [Google Scholar]
- 41.Morris G. E., Whyte M. K., Martin G. F., Jose P. J., Dower S. K., Sabroe I. (2005) Am. J. Respir. Crit. Care Med. 171, 814–822 [DOI] [PubMed] [Google Scholar]
- 42.Hurst J., Prinz N., Lorenz M., Bauer S., Chapman J., Lackner K. J., von Landenberg P. (2009) Immunobiology 214, 683–691 [DOI] [PubMed] [Google Scholar]
- 43.Simpson R. J., McFarlin B. K., McSporran C., Spielmann G., ó Hartaigh B., Guy K. (2009) Brain Behav. Immun. 23, 232–239 [DOI] [PubMed] [Google Scholar]
- 44.Kleinnijenhuis J., Joosten L. A., van de Veerdonk F. L., Savage N., van Crevel R., Kullberg B. J., van der Ven A., Ottenhoff T. H., Dinarello C. A., van der Meer J. W., Netea M. G. (2009) Eur. J. Immunol. 39, 1914–1922 [DOI] [PubMed] [Google Scholar]
- 45.Wilson H. L., Francis S. E., Dower S. K., Crossman D. C. (2004) J. Immunol. 173, 1202–1208 [DOI] [PubMed] [Google Scholar]
- 46.Keller M., Rüegg A., Werner S., Beer H. D. (2008) Cell 132, 818–831 [DOI] [PubMed] [Google Scholar]
- 47.Di Virgilio F., Solini A. (2002) Br. J. Pharmacol. 135, 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knöfler R., Weissbach G., Kuhlisch E. (1997) Am. J. Hematol. 56, 259–265 [DOI] [PubMed] [Google Scholar]
- 49.Carta S., Tassi S., Semino C., Fossati G., Mascagni P., Dinarello C. A., Rubartelli A. (2006) Blood 108, 1618–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qu Y., Ramachandra L., Mohr S., Franchi L., Harding C. V., Nunez G., Dubyak G. R. (2009) J. Immunol. 182, 5052–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tassi S., Carta S., Vené R., Delfino L., Ciriolo M. R., Rubartelli A. (2009) J. Immunol. 183, 1456–1462 [DOI] [PubMed] [Google Scholar]
- 53.Broom D. C., Matson D. J., Bradshaw E., Buck M. E., Meade R., Coombs S., Matchett M., Ford K. K., Yu W., Yuan J., Sun S. H., Ochoa R., Krause J. E., Wustrow D. J., Cortright D. N. (2008) J. Pharmacol. Exp. Ther. 327, 620–633 [DOI] [PubMed] [Google Scholar]
- 54.Chamberlain J., Evans D., King A., Dewberry R., Dower S., Crossman D., Francis S. (2006) Am. J. Pathol. 168, 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perera P. Y., Mayadas T. N., Takeuchi O., Akira S., Zaks-Zilberman M., Goyert S. M., Vogel S. N. (2001) J. Immunol. 166, 574–581 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.