

Abstract
The focal adhesion protein vinculin is an actin-binding protein involved in the mechanical coupling between the actin cytoskeleton and the extracellular matrix. An autoinhibitory interaction between the N-terminal head (Vh) and the C-terminal tail (Vt) of vinculin masks an actin filament side-binding domain in Vt. The binding of several proteins to Vh disrupts this intramolecular interaction and exposes the actin filament side-binding domain. Here, by combining kinetic assays and microscopy observations, we show that Vt inhibits actin polymerization by blocking the barbed ends of actin filaments. In low salt conditions, Vt nucleates actin filaments capped at their barbed ends. We determined that the interaction between vinculin and the barbed end is characterized by slow association and dissociation rate constants. This barbed end capping activity requires C-terminal amino acids of Vt that are dispensable for actin filament side binding. Like the side-binding domain, the capping domain of vinculin is masked by an autoinhibitory interaction between Vh and Vt. In contrast to the side-binding domain, the capping domain is not unmasked by the binding of a talin domain to Vh and requires the dissociation of an additional autoinhibitory interaction. Finally, we show that vinculin and the formin mDia1, which is involved in the processive elongation of actin filaments in focal adhesions, compete for actin filament barbed ends.
Keywords: Actin, Adhesion, Kinetics, Protein Self-assembly, Protein-Protein Interactions, Vinculin
Introduction
Vinculin is an actin filament (F-actin)-binding protein involved in cell-matrix adhesion (1, 2). Cells depleted of vinculin show defects in cell spreading, cell migration, cell-matrix adhesion, and focal adhesion turnover (3–6). Fluorescent speckle microscopy within focal adhesions showed that the retrograde movement of vinculin is partially coupled to actin motion, suggesting that it may tune the degree of force transmission to the substrate (7, 8). Recent studies further demonstrated that force transmission at focal adhesions requires the binding of vinculin to the actin cytoskeleton and is regulated by proteins that connect integrin to vinculin (9, 10). However, the function of vinculin in actin dynamics and its regulation are still unclear.
Vinculin is a large protein of 1066 amino acids made of a N-terminal globular head (Vh)3 comprising subdomains D1–D4, followed by a central polyproline-rich linker and a C-terminal tail (Vt) (1) (see Fig. 4A). Vt binds F-actin and cross-links actin filaments into bundles (11–13). The protein is regulated by an intramolecular interaction in which Vh interacts with Vt (14). The intramolecular interaction between Vh and Vt masks the F-actin-binding domain located in Vt (15). The binding of talin and α-actinin (16, 17) to the D1 subdomain of vinculin induces a helical bundle conversion of this subdomain, leading to the disruption of the intramolecular interaction and the exposure of the cryptic F-actin-binding domain of Vt (18–20). Interestingly, the bacterial protein IpaA from Shigella mimics the activation of vinculin by talin (21). Although it is clear that the high affinity binding of IpaA to the D1 head subdomain is sufficient to activate vinculin and induce F-actin binding (22–24), the ability of the cellular D1-binding proteins (talin and α-actinin) to fully activate vinculin is controversial (see Refs. 1 and 25 for reviews). Several lines of evidence suggest that vinculin needs a more complex mechanism of activation than the D1 conformational change. First, the high concentration of a synthetic peptide, corresponding to a vinculin binding site (VBS) of talin, required to induce the binding of vinculin to F-actin, suggests a very low affinity of the VBS peptide for the D1 head subdomain of vinculin (Kd > 100 μm) (20). Second, the disruption of the head-to-tail interaction is prerequisite rather than subsequent to the formation of vinculin-talin complexes in living cells (26). Finally, in agreement with additional activation mechanisms, the structure of the full-length vinculin and affinity measurements showed that Vt interacts not only with the D1 subdomain but also with D3 and D4 (18, 27, 28). However, the high affinity of VBS peptides for immobilized vinculin, measured by surface plasmon resonance, challenged this idea and led to the proposal that talin or α-actinin binding to D1 is sufficient to fully activate vinculin (29).
FIGURE 4.

Identification of the critical domains of vinculin involved in the barbed end capping and F-actin side binding activities. A, constructs used in this study. B, SDS-PAGE of the purified proteins used in this work. C, barbed end growth was measured in the presence of 100 pm spectrin-actin seeds (SP), 1.5 μm MgATP-G-actin (10% pyrenyl-labeled), and the indicated concentrations of vinculin full-length (residues 1–1066), ΔD1 (residues 253–1066), 812–1066, 812–1044, 812–978, and 879–1066. The rate of barbed end growth at 25% polymerization was plotted versus the concentration of each vinculin construct. D, the co-sedimentation of vinculin 812–1066 (2 μm), 812–1044 (2 μm), 812–978 (2 μm), 253–1066 (2 μm), and full-length vinculin 1–1066 (2 μm) with F-actin was measured in the presence of increasing concentrations of F-actin. The fraction of vinculin bound to F-actin was plotted versus the concentration of actin. All of the experiments were performed three times with the same results. a.u., absorbance units.
As discussed earlier, the binding of vinculin to the side of actin filaments and its regulation have been extensively studied, but little is known about the ability of vinculin to regulate actin dynamics. Recently, the study of the vinculin-binding protein IpaA, from the bacteria Shigella, showed that the complex IpaA-vinculin partially inhibits the elongation of actin filament barbed ends (24). However, whether this activity is an intrinsic property of vinculin that is unmasked by IpaA is not known.
Here we show that the Vt domain of vinculin (residues 879–1066) totally inhibits actin dynamics by blocking barbed end elongation without affecting the critical concentration of actin assembly. In low salt conditions, Vt nucleates actin filaments capped at their barbed ends. By observing single growing actin filaments by total internal reflection fluorescence (TIRF) microscopy, we determined that the interaction between vinculin and the barbed end of the actin filament is characterized by slow association and dissociation rate constants. The barbed end capping activity requires the C-terminal arm of Vt that is dispensable for actin filament side binding. Like the side-binding domain, the barbed end-capping domain of vinculin is masked by an autoinhibitory interaction between Vh and Vt. However, in contrast to the actin filament side-binding domain, the capping domain is not unmasked by the binding of the VBS1 domain of talin to the D1 subdomain and requires the dissociation of additional autoinhibitory contacts. Finally, we show that vinculin and the formin mDia1, which is involved in the processive elongation of actin filaments in focal adhesions, compete for actin filament barbed ends.
MATERIALS AND METHODS
Recombinant cDNA Constructs
Talin 482–636 (VBS1), vinculin 1–851 (Vh) and 1–258 (D1) cDNAs were constructed by PCR amplification of the full-length cDNAs and subcloning of the resulting DNA in the BamHI and EcoRI sites of pGEX-6P1 (GE Healthcare). Vinculin 879–1066 (Vt), 812–1066, 812–978, and 812–1044 cDNAs were constructed by PCR amplification of the full-length cDNA and subcloning of the resulting cDNA in the NcoI and HindIII sites of pET28a. The ΔD1-vinculin (residues 253–1066) was constructed by PCR amplification of the full-length cDNA and subcloning of the resulting cDNA first in TOPO champion (Invitrogen) and finally in the NotI and NcoI site of pET3. All of the constructs were verified by sequencing.
Protein Purification
Talin 482–636 (VBS1), vinculin 1-851, and vinculin 1–258 were expressed as glutathione S-transferase fusion proteins in Escherichia coli BL21, bound to glutathione-Sepharose (GE Healthcare), and finally eluted by PreScission protease (GE Healthcare) cleavage. His-tagged vinculin 879–1066, 812–1066, 812–978, and 812–1044 cDNAs were expressed in E. coli BL21 and purified by Ni2+-nitrilotriacetic acid (Qiagen) affinity chromatography, followed by gel filtration on a Superdex 75 column (GE Healthcare). Full-length His-tagged vinculin was expressed in E. coli BL21 and purified by Ni2+-nitrilotriacetic acid affinity chromatography, followed by a Q-Sepharose ion exchange column. His-tagged ΔD1-vinculin (residues 253–1066) mutant was expressed in E. coli BL21 and purified by Ni2+-nitrilotriacetic acid affinity chromatography, followed by a gel filtration on a Superdex 200 column (GE Healthcare).
Polymerization Assay
Actin polymerization was monitored by the increase in fluorescence of 10% pyrenyl-labeled actin. Actin polymerization was induced by the addition of 100 mm KCl, 1 mm MgCl2, and 0.2 mm EGTA to a solution of 10% pyrenyl-labeled CaATP-G-actin containing the proteins of interest. Fluorescence measurements were carried out in a Safas Xenius model FLX (Safas, Monaco) spectrophotometer. For kinetic experiments, spectrin-actin seeds were added to the reaction for barbed end elongation measurements, and gelsolin-actin (1:2) complexes were added for pointed end elongation measurements.
The affinity for barbed ends was derived as follows. The elongation rate at 25% polymerization was taken as a measure of the free barbed ends (R). The following equation was used to fit the data, in which [C0] and [V0] are the total concentration of barbed ends and vinculin, respectively, and K represents the equilibrium dissociation constant of the complex of vinculin with barbed ends. The fraction R of free barbed ends is as follows.
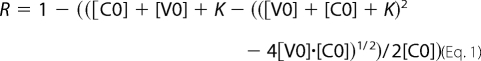 |
F-actin Co-sedimentation Assay
We performed co-sedimentation assays to measure the vinculin-VBS1 complex equilibrium dissociation constant. Increasing concentrations of VBS1 were incubated with 2 μm vinculin and 12 μm F-actin. After centrifugation at 90,000 rpm in a TL100 centrifuge (Beckman), the pellets and supernatants were separated, loaded on SDS-PAGE, and quantified using the ImageJ software. The vinculin bound fraction was plotted versus the total concentration of VBS1. The following equation was used to fit the data, in which [V0] is the concentration of vinculin, [T0] is the initial concentration of VBS1, and K is the equilibrium dissociation constant (Kd). The fraction R of vinculin bound to F-actin is as follows.
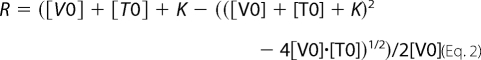 |
Fluorescence Anisotropy Measurement
Fluorescence anisotropy measurements were carried out at 20 °C in a Safas Xenius model FLX (Safas, Monaco) equipped with a device for automatic anisotropy measurements. Each data point reflects the average of at least 25 anisotropy measurements. The VBS1-bound fraction was plotted versus the total concentration of vinculin. An equation similar to the one used for co-sedimentation measurement was used to fit the data.
Labeling of the single cysteine of VBS1 with IAEDANS was performed by adding a 10-fold excess IAEDANS to VBS1 in 20 mm Hepes, pH 6.8, 100 mm KC1, for 1 h at 4 °C. The reaction was stopped by the addition of 5 mm DTT before application of the sample to a PD10 desalting column (GE Healthcare) to remove the unreacted dye.
Observation and Measurement of Single Actin Filament Elongation by TIRF Microscopy
Our protocol is a modification of the protocol used to study the heterodimeric capping protein (CP) (30). Flow cells containing 40–60 μl of fluid were first prepared by adapting a coverslip on a slide with double face adhesive spacers. In order to capture the filaments on the coverslip, inactivated NEM-myosin (100 μl at 0.1 mg/ml) in buffer T1 (5 mm Tris, pH 7.8, 0.2 mm ATP, 1 mm MgCl2, 0.1 mm CaCl2, 10 mm DTT, 500 mm KCl) was loaded in the flow chamber. After a 1-min incubation time at room temperature, the flow chamber was washed with 100 μl of buffer T1, 100 μl of buffer T1 containing 1% bovine serum albumin, and twice 100 μl of buffer T2 (5 mm Tris, pH 7.8, 0.2 mm ATP, 1 mm MgCl2, 0.1 mm CaCl2, 10 mm DTT, 100 mm KCl). We loaded MgATP-G-actin (0.6 μm), 10% Alexa488-labeled, and the proteins of interest in buffer T2 containing 0.2% methyl-cellulose and 0.2 mm 1,4-diazabicyclo(2,2,2)-octane. Finally, we sealed the flow chamber with VALAP (a mixture of vaseline, lanolin, and paraffin) and observed the reaction on an Olympus AX71 inverted microscope equipped with a ×60 (numerical aperture 1.45) objective (Olympus) and a Blues 473-nm laser (Cobolt). The time lapse videos were acquired by Metamorph and subsequently analyzed by the ImageJ software.
Measurement of the Association and Dissociation Rate Constants from TIRF Microscopy
We first verified that, in our experimental conditions, 0.6 μm MgATP-actin containing 10% Alexa488-labeled actin assembled at the barbed end with the same association rate constant as previously measured in solution experiments. At 0.6 μm actin, pointed end growth is negligible. By measuring the slopes of the kymographs from movies, we determined a k+ of 9.5 μm−1 s−1, which is in good agreement with the known values (k+ = 10–12.9 μm−1 s−1) (31–33). In the presence of Vt, actin filaments displayed a binary mode of growth with phases of continuous growth followed by pauses during which the elongation is totally arrested. We assumed that the times at which the pauses ended represented the dissociation times of vinculin from actin filament barbed ends and that the times at which the pauses started represented the association times. From time lapse videos, we generated kymographs of growing actin filaments by using the ImageJ software. We measured manually the time intervals corresponding to periods of growth and arrest. A total of 200–300 filaments were analyzed for each concentration of Vt. The exponential fit of the time distributions gave values for the association and dissociation rates. The association and dissociation rates were plotted versus the concentration of Vt. The slope of the association rate plot gave the association rate constant. Both the y axis intercept of the association rate plot and the average value of the dissociation rate plot were used to obtain the value of the dissociation rate constant. As a control, we varied the frequency of acquisition of the time lapse videos from 4 to 20 s and verified that the observed dissociation and association rates were not significantly affected. We also verified that the capping activity of vinculin is also observed when the filaments are free in solution, in a flow chamber that is not coated with NEM-myosin.
Cleavage of Vinculin by V8 Protease
Vinculin was incubated during 1 h at 37 °C in the presence of V8 protease. V8 protease and His-tagged vinculin were then separated by Ni2+-nitrilotriacetic acid (Qiagen) chromatography. After elution by imidazole (250 mm), vinculin was desalted using a PD10 column (GE Healthcare). As a control for polymerization assays, a sample of vinculin was treated identically, without protease.
RESULTS
The Tail Domain of Vinculin Inhibits Actin Filament Barbed End Elongation
We first showed in a previous report that a complex IpaA-vinculin behaves as a leaky barbed end capper by slowing down the addition of actin monomers at the barbed end by 40% (24). Because IpaA unmasks the F-actin-binding domain located in the tail, by disrupting the autoinhibitory interaction, we hypothesized that the capping activity of the IpaA-vinculin complex is located in the tail domain of vinculin. First, we tested the ability of a Vt construct (residues 879–1066) to inhibit actin filament barbed end elongation. Barbed end growth was measured by monitoring the increase in pyrenyl-labeled actin fluorescence upon polymerization in the presence of spectrin-actin seeds. Vt induced a total inhibition of barbed end elongation, in contrast with the partial inhibition recorded with the IpaA-vinculin complex (Fig. 1A).
FIGURE 1.

The tail domain of vinculin inhibits actin filament barbed end elongation. A, barbed end growth was measured in the presence of 100 pm spectrin-actin seeds (SP), 1.5 μm MgATP-G-actin (10% pyrenyl-labeled), and the indicated concentrations of Vt. B, actin was polymerized in the presence of 100 pm spectrin-actin seeds, 1.5 μm MgATP-G-actin, and the indicated concentrations of Vt for 30 min. The polymerized (P) and unpolymerized (S) fractions were separated by ultracentrifugation, resolved by 10% SDS-PAGE, detected by Coomassie Blue staining (inset), and quantified. C, barbed end growth was measured in the presence of 100 pm spectrin-actin seeds, 1.5 μm MgATP-G-actin (10% pyrenyl-labeled), and increasing concentrations of Vt in the absence or presence of 12 μm profilin. Pointed end growth was measured in the presence of 4 nm gelsolin-actin complex, 2 μm MgATP-G-actin (10% pyrenyl-labeled), and increasing concentrations of Vt. The initial rate of pointed end growth and the rate of barbed end growth at 25% polymerization were plotted versus the concentration of Vt. D, Vt increases the concentration of F-actin at steady state in the presence of gelsolin but has no effect in the absence of gelsolin. 1.4 μm MgATP-F-actin (10% pyrenyl-labeled) was incubated overnight with increasing amounts of Vt in the absence or presence of gelsolin. All of the experiments were performed three times with the same results. a.u., arbitrary units.
To distinguish between an effect of Vt on actin polymerization or pyrenyl-labeled actin fluorescence, we measured the amount of polymerized and unpolymerized actin as a function of Vt concentration by a high speed sedimentation assay (Fig. 1B). The unambiguous dose-dependent decrease in actin in the polymerized fraction demonstrated that Vt inhibited actin assembly. Fig. 1D (red points) also clearly showed that Vt had no effect on the fluorescence of prepolymerized pyrenyl-labeled F-actin. Altogether, our data demonstrate that the effect of Vt observed in Fig. 1A corresponds to an inhibition of actin assembly and rule out the quenching of pyrenyl-actin fluorescence by Vt recently proposed by others (34).
To distinguish between an inhibition of barbed end elongation and a sequestration of actin monomers, we performed two series of experiments. First we plotted the elongation rates of spectrin-actin seeds as a function of actin concentration in the absence or presence of Vt. The two plots were linear, indicating that the fraction of inhibition of barbed end elongation was not dependent on the concentration of actin monomers (supplemental Fig. S1A). Second, we measured the effect of Vt on the depolymerization of actin filament after dilution. In this condition, filaments depolymerize mainly from the barbed end. We can specifically measure pointed end depolymerization when filaments are capped at their barbed end. We compared the effect of Vt on the depolymerization of non-capped and gelsolin-capped filaments. We found that Vt inhibited more efficiently the depolymerization of non-capped than capped filaments (supplemental Fig. S1B). Altogether, these results clearly demonstrate that Vt acts on the filament barbed end rather than on the monomer.
Assuming that Vt binds to actin filament barbed ends, the plot of elongation rate versus Vt concentration gives a good estimate of the affinity of vinculin for the barbed end. Because the kinetics of barbed end growth in the presence of Vt showed a slightly delayed inhibition (Fig. 1A), suggesting slow kinetics of Vt interaction with barbed ends, we found it more accurate to use the elongation rate at 25% polymerization, instead of the initial rate of elongation. The best fit of the data gave a Kd of 0.2 μm (Fig. 1C). This inhibition was specific because, in the same range of concentrations, Vt did not inhibit (and even slightly enhanced) the pointed end growth from gelsolin-actin complexes (Fig. 1C).
Vinculin Tail Does Not Affect the Critical Concentration of Actin Assembly
These first results favored a barbed end capping function. Because such an activity is known to induce an increase in the critical concentration of actin assembly, we expected Vt to decrease the concentration of F-actin at steady state. However, we did not see any significant effect of Vt on the concentration of F-actin at steady state when filaments had free barbed ends (Fig. 1D). In contrast, when barbed ends were capped by gelsolin, the concentration of F-actin increased markedly as a function of Vt concentration (Fig. 1D). We also used the properties of profilin to test whether Vt blocks actin filament barbed ends. Because profilin-actin complexes do not assemble at the pointed end, profilin induces the depolymerization of actin filaments capped at their barbed ends at steady state. However, in contrast with control gelsolin-capped filaments, profilin failed to significantly depolymerize actin filaments in the presence of 3 μm Vt (data not shown). To rule out a competition between profilin and Vt for the barbed end, we established that the affinity of Vt for actin filament barbed ends was not significantly affected by a saturating amount of profilin (12 μm) (Fig. 1C). Altogether, our data indicate that Vt does not affect the critical concentration of actin assembly.
The abilities of Vt to enhance the elongation of gelsolin-capped filaments (Fig. 1C) and to stabilize gelsolin-capped filaments at steady state (Fig. 1D) were probably the results of the binding of this construct to the side of the filaments that stabilized the contacts between subunits at the pointed end. In support of this hypothesis, Vt inhibited the pointed end depolymerization of gelsolin-capped filaments (supplemental Fig. S1B). The inhibition of pointed end depolymerization also explains why Vt failed to increase the critical concentration of actin assembly. Indeed, this effect requires that actin filament barbed ends are almost saturated by Vt and that pointed ends are free to depolymerize. Therefore, we cannot observe the effect of Vt on the critical concentration because side binding and barbed end capping have opposite effects.
Vinculin Nucleates Actin Filaments Capped at Their Barbed Ends in Low Salt Conditions
Another common feature of capping proteins is to nucleate actin filaments capped at their barbed ends. In the physiological conditions used in the previous experiments (100 mm KCl, 1 mm MgCl2), Vt (4 μm) stimulated actin assembly very weakly (Fig. 2A). Interestingly, a recent work suggested that vinculin enhances actin nucleation in low salt conditions (34). We also found that the nucleation activity of 4 μm Vt was greatly enhanced when the concentration of KCl was decreased to non-physiological values (Fig. 2A). To test whether the increase of pyrenyl-actin fluorescence indeed corresponded to the formation and elongation of actin filaments, we observed the final reaction in fluorescence microscopy. We polymerized actin in 25 mm KCl and 1 mm MgCl2 in the absence and presence of 1 μm Vt and observed single actin filaments after staining and dilution with the F-actin-specific fluorescent probe Alexa488-labeled phalloidin. In agreement with nucleation behavior, the number of filaments dramatically increased in the presence of Vt (Fig. 2B). A severing effect was ruled out by the direct observation that growing filaments were not cleaved by Vt (see an example in Fig. 3 and supplemental Movie 2). The dose dependence indicates that Vt is a weak nucleator that only displays a significant nucleation activity at 1 μm (Fig. 2C). Although Vt is a very poor and probably non-physiological nucleator, we took advantage of these nucleating conditions to test the dynamics of Vt-nucleated actin filament barbed ends in a pyrenyl-actin polymerization assay. We showed that the addition of a 21 nm concentration of the recombinant mouse CP did not affect actin nucleation by Vt, suggesting that these filament barbed ends were already capped by Vt and elongated mainly from their pointed ends (Fig. 2D). As a control, we showed that in the same ionic strength conditions, the same concentration of CP totally inhibited the barbed end elongation of spectrin-actin seeds (Fig. 2D, inset).
FIGURE 2.

Vinculin tail nucleates actin filaments that are capped at their barbed ends. A, spontaneous nucleation of 1.5 μm MgATP-G-actin (10% pyrenyl-labeled) was measured in the presence of 4 μm Vt, in a buffer containing 5 mm Tris, pH 7.8, 0.2 mm ATP, 1 mm MgCl2, 0.1 mm CaCl2, 1 mm DTT, and 10 mm KCl supplemented with the indicated concentrations of KCl. B, observation of single actin filaments nucleated by Vt in fluorescence microscopy. The polymerization of 5 μm MgATP-G-actin was measured in a buffer containing 5 mm Tris, pH 7.8, 0.2 mm ATP, 1 mm MgCl2, 0.1 mm CaCl2, 1 mm DTT, and 25 mm KCl in the absence (left) or presence of 1 μm Vt (right). As soon as 90% polymerization was reached, actin filaments were stabilized and stained with a molar equivalent of Alexa488-labeled phalloidin, diluted, and observed by fluorescence microscopy. Scale bar, 10 μm. C, the polymerization of 1.5 μm MgATP-G-actin (10% pyrenyl-labeled) was measured in the presence of increasing concentrations of Vt in a buffer containing 5 mm Tris, pH 7.8, 0.2 mm ATP, 1 mm MgCl2, 0.1 mm CaCl2, 1 mm DTT, and 25 mm KCl. D, Vinculin remains bound to the barbed end of nucleated filaments at low ionic strength. 1.5 μm MgATP-G-actin (10% pyrenyl-labeled) was polymerized alone, in the presence of 2 μm Vt or 21.5 nm CP or Vt and CP, in a buffer containing 5 mm Tris, pH 7.8, 0.2 mm ATP, 1 mm MgCl2, 0.1 mm CaCl2, 1 mm DTT, and 25 mm KCl. Inset, as a control, the effect of 21.5 nm CP or 4 μm Vt or Vt and CP on the elongation of 337 pm spectrin-actin seeds (SP) is shown in the same buffer conditions. All of the experiments were performed three times with the same results. a.u., arbitrary units.
FIGURE 3.

Direct real-time observation of actin filament barbed end capping by vinculin tail. Conditions were as follows. 0.6 μm MgATP-G-actin (10% Alexa488-labeled) was polymerized in a flow chamber coated with NEM-myosin in 5 mm Tris, pH 7.8, 100 mm KCl, 0.2 mm ATP, 0.1 mm CaCl2, 1 mm MgCl2, 10 mm DTT, 0.2 mm 1,4-diazabicyclo(2,2,2)-octane, 0.2% methyl-cellulose. A, time lapse of the elongation of single actin filaments in the absence (top) or presence of 0.5 μm Vt (bottom). See also the corresponding supplemental Movies 1 and 2. Scale bar, 5 μm. B, kymographs of the single actin filaments shown in A. Actin filament fluorescence intensity was measured along its length (vertical axis) for each frame of the time lapse (horizontal axis). The filaments are oriented with the non-growing pointed end to the bottom and the growing barbed end to the top. In contrast with the continuous elongation observed for control filaments (top), barbed end growth was occasionally interrupted by pauses in the presence of Vt (bottom). C–F, quantification of the association and dissociation rate constants that characterized the interaction between Vt and actin filament barbed ends using TIRF microscopy. C and E, periods of growth and arrest were measured on kymographs corresponding to the elongation of actin filaments in the presence of 0.6 μm MgATP-G-actin (10% Alexa488-labeled) and 0.5 μm Vt. The exponential fit of the time distribution of the periods of growth (C) and arrest (E) gave the association and dissociation rates, respectively. The association rates (D) and dissociation rates (F) were plotted versus increasing concentrations of Vt. Error bars, S.E. G, measurement of the dissociation rate of vinculin from actin filament barbed ends after dilution by using a fluorescence bulk assay. A reaction in which spectrin-actin seeds have been pre-elongated in the presence of 2 μm actin and 2 μm Vt was diluted 20 times in a solution containing 2 μm MgATP-G-actin, 10% pyrenyl-labeled (red line). As a control, we performed the dilution of the same reaction, but we let Vt dissociate for 15 min before adding G-actin (blue line). H, the subtraction of the dissociation kinetics from the linear fit of the control kinetics shown in G gave the kinetics of dissociation of Vt from the barbed end. The exponential fit of the curve gave a dissociation rate constant k− = 0.014 s−1. All of the experiments were performed three times with the same results. a.u., arbitrary units.
Bundling and Barbed End Capping
Vt is known to cross-link actin filaments into bundles (11–13). We confirmed the bundling activity of Vt by using both a low speed sedimentation assay and a direct observation in TIRF microscopy (supplemental Fig. S2). To determine whether the bundling activity contributes to the capping activity, we compared the concentration range of Vt in which these two activities occur. We already determined that 0.2 μm Vt blocked half of the barbed ends (Fig. 1C). In addition, the quantification of the low speed sedimentation assay indicated that 0.8–1.6 μm Vt induced the bundling of half of F-actin (supplemental Fig. S2, A and B). The direct observation of actin filament bundles in TIRF microscopy confirmed that 0.6 μm Vt did not cross-link actin filaments significantly. However, characteristic bundles were clearly formed in the presence of 5 and 10 μm Vt (supplemental Fig. S2C). The significant difference between the ability of Vt to cap actin filament barbed ends and to bundle actin filaments indicates that these activities are independent.
Direct Real Time Observation of Actin Filament Barbed End Capping by Vinculin Tail
To test whether Vt acts as a conventional barbed end-capping protein, we compared the kinetics of elongation of single actin filaments in the absence or presence of Vt by using TIRF microscopy. In the absence of Vt, actin filaments showed a constant rate of elongation (Fig. 3, A and B (top panels), and supplemental Movie 1). In the presence of Vt, actin filaments displayed a binary mode of growth with phases of continuous growth followed by phases during which the barbed end growth is totally arrested (Fig. 3, A and B (bottom panels), and supplemental Movie 2). The kymographs corresponding to the elongation of single filaments clearly showed the periods of growth and arrest (Fig. 3B, bottom panels).
This behavior is in agreement with a barbed end capping activity and rules out a monomer sequestering activity. Indeed, the sequestration of monomers would result in a slower but still linear elongation rate, reflecting the gradual saturation of the monomers.
We also wanted to determine whether Vt is in equilibrium with the barbed end or whether Vt follows barbed end growth in a processive manner. Using TIRF microscopy, we observed the growth of actin filaments at the surface of a glass coverslip coated with Vt. Because Vt is immobile on the surface, a processive movement with the elongating barbed end, combined with a capping activity, would result in the backward movement of the filament, with the appearance of stops. We never observed this behavior. Instead, side binding allowed the filaments to grow on the surface until the barbed ends bound to an immobilized Vt and stopped (supplemental Fig. S3, A and B). Although the filaments were flexible enough between the vinculin-bound barbed end and a second anchor point (supplemental Fig. S3C), we never saw the elongation of a loop resulting from an insertional polymerization between Vt and the barbed end (supplemental Movie 3), as described previously for formins (35). These observations rule out a processive movement of Vt with the barbed end.
Assuming that a growth arrest event represents the binding of Vt to the barbed end of an actin filament, we defined the times at which the pauses ended as the dissociation times of Vt from actin filament barbed ends and the times at which the pauses started as the association times. The exponential fit of the time distribution of the periods of growth and arrest gave values for the association and dissociation rates, respectively (Fig. 3, C and E). As expected, the association rate increases linearly with the concentration of Vt (Fig. 3D), whereas the dissociation rate remains constant in the same range of concentration (Fig. 3F). The y axis intercept and the slope of the association rate plot gave the dissociation rate constant (k− = 0.0017 s−1) and the association rate constant (k+ = 0.0023 μm−1. s−1), respectively (Fig. 3D). The ratio k−/k+ gave a Kd = 0.73 μm that is in reasonable agreement with the value obtained from solution experiments (Kd = 0.2 μm; Fig. 1C). The direct measurement of the dissociation rate constant gave an average value (k− = 0.0038 s−1; Fig. 3F), that is also in reasonable agreement with the k− extrapolated from the association rate plot (k− = 0.0017 s−1; Fig. 3D).
To confirm the values of the dissociation rate constant derived from TIRF microscopy, we measured the dissociation rate of Vt from actin filament barbed ends after dilution by using an independent bulk assay. Because the concentration of barbed ends is extremely low, this cannot be observed directly. Therefore, a reaction, in which spectrin-actin seeds have been pre-elongated in the presence of G-actin (2 μm) and capped by the addition of Vt (2 μm), was diluted 20 times in a solution containing 2 μm G-actin. The kinetics of polymerization showed a slow increase in the elongation rate that finally reached a constant and maximum value (Fig. 3G, red curve). As a control, we performed the dilution of the same reaction, let Vt dissociate during 15 min, added G-actin, and observed kinetics that immediately displayed a constant and maximum rate of elongation (Fig. 3G, blue curve). Assuming that the slow increase in the elongation rate observed in the first experiment represented the formation of free barbed ends made available for growth by dissociation of Vt, the subtraction of the two fluorescence time courses gave the kinetics of dissociation of Vt from the barbed end (Fig. 3H). A dissociation rate constant k− = 0.014 s−1 was derived from the exponential fit of the curve. Although 3.5–8-fold higher than the values derived from TIRF microscopy, this number is in reasonable agreement with measurements on individual filaments. The fact that we used Alexa488-labeled actin in TIRF microscopy and pyrenyl-labeled actin in the bulk assay could explain this difference.
The C-terminal Arm of Vinculin Tail Is Required for Actin Filament Barbed End Capping but Not for Side Binding
Interestingly, the combination of cryoelectron microscopy reconstitution, probability of binding, and mutagenesis showed that the F-actin-binding domain located in Vt is divided into two spatially distinct clusters that contact two consecutive actin subunits (11, 13). The lower patch, proximal to the barbed end, includes residues of helices 3 and 4 and the C-terminal arm following helix 5 of the tail domain. The upper patch contains residues of helices 2 and 3 (13). We designed C-terminal deletions of the tail domain to remove the residues of the lower patch. From a vinculin tail construct (residues 812–1066), we deleted the last 22 amino acids, corresponding to the C-terminal arm (vinculin 812–1044) and the last 88 amino acids, including helices 4 and 5 and the C-terminal arm (vinculin 812–978) (Fig. 4, A and B). The study of these constructs showed that both deletions resulted in a 20-fold reduction of the barbed end capping activity (Fig. 4C). To demonstrate that the deletion of the C-terminal arm only inhibited the capping function and did not alter the structure of the five-helix bundle of Vt, we compared the affinity of the tail constructs for the side of actin filaments by using a F-actin co-sedimentation assay. Vinculin 812–1066, 812–1044, and 812–978 bound actin filaments with Kd = 0.75, 0.99, and 0.81 μm, respectively, showing that vinculin 812–1044 and 812–978 did not significantly lose affinity for the side of actin filaments (Fig. 4D).
Actin Filament Barbed End Capping and Side Binding Activities Are Both Autoinhibited but Regulated Differently
The discovery of the capping activity of vinculin located in Vt raises the question of whether this activity is regulated by the D1 head subdomain like the F-actin side-binding domain or whether it is constitutive. Fig. 4, C and D, and our previous study clearly showed that full-length vinculin alone did not exhibit significant capping activity or F-actin side binding (24). To test whether the absence of capping activity is simply due to the known D1-Vt autoinhibitory interaction, we generated a construct of vinculin deleted of the D1 subdomain (vinculin 253–1066 or ΔD1-vinculin). We compared the capping activity and the actin filament side binding of full-length vinculin and ΔD1-vinculin. A previous report already showed that a different deletion mutant of vinculin (residues 153–1066) binds actin filaments constitutively (13). We confirmed this observation by showing that, in contrast with full-length vinculin, ΔD1-vinculin bound actin filaments. We extended this observation by showing that ΔD1-vinculin binds F-actin with a Kd of 1.1 μm, which is similar to the value (Kd = 0.75 μm) found for the isolated vinculin tail domains (Fig. 4D). However, like full-length vinculin, ΔD1-vinculin did not show a significant actin filament barbed end capping activity (Fig. 4C). This observation demonstrates that the capping activity of Vt is inhibited by intramolecular contacts other than the D1-Vt contacts. These contacts could involve the head subdomains D3 or D4 that also interact with Vt. To confirm this hypothesis, we decided to measure the contribution of the head subdomains in the autoinhibition of the capping activity. We first showed that, in a kinetic assay, the capping activity of Vt is trans-inhibited by a construct corresponding to the full-length head domain (Vh 1–851) that comprises the four subdomains D1–D4 (Figs. 4A and 5A). Using this assay, we compared the ability of the isolated D1 (Vh 1–258) and the full-length head domain (Vh 1–851) to inhibit the capping activity of the isolated Vt domain. We first assumed that the plot of the fraction of inhibition of Vt versus Vh concentration gives a good estimate of the affinity of Vh for the capping domain located in Vt. We found that D1 (Vh 1–258) has a 6-fold lower affinity for Vt than Vh 1–851, indicating a contribution of at least one of the other subdomains (D3 or D4) in the autoinhibition of the capping activity (Fig. 5B).
FIGURE 5.

Actin filament side binding and barbed end capping are both autoinhibited but regulated differently. A, vinculin head domain 1–851 (D1–D4) inhibits the capping activity of Vt. Barbed end growth was measured in the presence of 275 pm spectrin-actin seeds (SP), 2 μm MgATP-G-actin (10% pyrenyl-labeled), 0.3 μm Vt, and 2.2 μm vinculin head (residues 1–851). B, vinculin head domain 1–851 inhibits the capping activity of Vt more efficiently than Vh 1–258. Barbed end growth was measured in the presence of 100 pm spectrin-actin seeds, 2 μm MgATP-G-actin (10% pyrenyl-labeled), 0.75 μm Vt, and increasing amounts of vinculin head 1–258 and 1–851. The fraction of inhibition of Vt by Vh was plotted versus the concentration of each Vh construct. The best fit of the data gave the affinity of the head domain for the capping domain located in Vt. C, the VBS1 domain of talin binds to full-length vinculin with low affinity. The increase in fluorescence anisotropy of 3 μm IAEDANS-labeled VBS1 observed in the presence of increasing concentrations of full-length vinculin was taken as a measure of the VBS1-vinculin complex formation. The fraction of VBS1 bound to vinculin was plotted versus the total concentration of vinculin, and the best fit of the data gave a Kd of 24 μm. Inset, the affinity of full-length vinculin for VBS1 is enhanced by F-actin. The fraction of vinculin bound to VBS1 was obtained by measuring the fraction of vinculin (2 μm) that co-sedimented with F-actin (12 μm) in the presence of increasing concentrations of VBS1. The fraction of vinculin bound to VBS1 was plotted versus the concentration of VBS1, and the best fit of the data gave a Kd of 5 μm. D, the capping domain of vinculin, cleaved by V8 protease, is unmasked by the binding of the VBS1 domain of talin. Barbed end growth was measured in the presence of 100 pm spectrin-actin seeds, 2 μm MgATP-G-actin (10% pyrenyl-labeled), 5 μm intact full-length vinculin (blue points), or V8-cleaved vinculin (red points) and increasing concentrations of VBS1. All of the experiments were performed three times with the same results. a.u., absorbance units.
To test the hypothesis that at least two Vh-Vt contacts must be disrupted to relieve the autoinhibition of the barbed end capping activity of vinculin, we tried to unmask the capping domain of vinculin by destabilizing both D1-Vt and D3D4-Vt interactions. The D1-Vt interaction is disrupted in the presence of a saturating amount of one of the numerous VBSs of talin (36, 37). However, a protein that disrupts specifically the contacts between Vt and the other subdomains, D3 and D4, has never been reported to our knowledge. To overcome this problem, we used the Staphylococcus aureus V8 protease that cleaves vinculin polyproline linker after amino acids 851 and 857, between D4 and Vt (38). This cleavage must weaken the D3-Vt and D4-Vt contacts, changing the intramolecular interactions into bimolecular interactions.
To relieve the D1-Vt autoinhibition, we used a bacterially expressed VBS1 domain of talin (residues 482–636) that is known to interact with the isolated Vh domain with high affinity (39) and to induce the helical bundle conversion of D1 that disrupts the D1-Vt interaction (39, 40). We first measured the affinity of an IAEDANS-labeled VBS1 for full-length vinculin by using fluorescence anisotropy and found a Kd of 24 μm (Fig. 5C). Because the binding of VBS1 to vinculin induces the binding of vinculin to actin filaments, we also used a F-actin co-sedimentation assay to measure the affinity of VBS1 within the ternary F-actin-vinculin-VBS1 complex and found a Kd of 5 μm (Fig. 5C, inset). Based on these affinity measurements, we expected to activate half of the vinculin and to inhibit half of the barbed end elongation with 5–24 μm VBS1 in an actin polymerization assay. However, vinculin did not inhibit barbed end elongation in the presence of concentrations of VBS1 as high as 50 μm (Fig. 5D). In addition to the lack of capping activity of ΔD1-vinculin (Fig. 4C), these observations confirm the view that the disruption of the D1-Vt interaction is not sufficient to unmask the barbed end-capping domain.
The cleavage of vinculin by the V8 protease did not induce the barbed end capping activity of vinculin, indicating that the Vh-Vt interaction remains strong (Fig. 5D, first point of the red curve, without VBS1). However, in contrast to the activity of intact vinculin, the capping activity of V8-cleaved vinculin was activated by VBS1 in a dose-dependent manner (Fig. 5D). In these conditions, the rate of barbed end growth was inhibited by 60%, which reflects the partial saturation of vinculin by VBS1 or the existence of residual Vh-Vt contacts. Altogether, our data demonstrate that the autoinhibition of the capping activity of vinculin involves contacts between Vh and Vt in addition to the talin-regulated D1-Vt interface.
Vinculin Tail and the Formin mDia1 Compete for Actin Filament Barbed Ends
Several studies demonstrated a role for the formin mDia1 in the regulation of actin dynamics in focal adhesion (41–43). Because the FH1FH2 domain of mDia1 catalyzes rapid filament growth from profilin-actin, by remaining processively bound to the elongating barbed end (44), we wanted to determine whether mDia1-FH1FH2 and Vt compete for the barbed end. Using TIRF microscopy, we determined the ability of Vt to block the elongation of actin filament barbed ends in the presence of the FH1FH2 domain of mDia1. We performed the experiments in the presence of profilin for several reasons. First, we already determined that Vt blocked the elongation of actin filament barbed ends in the presence of profilin (Fig. 1C). Second, mDia1-FH1FH2-bound barbed ends are easy to detect in the presence of profilin because barbed end elongation is dramatically enhanced (Fig. 6, A and B) (44). We confirmed that Vt blocked actin filament elongation efficiently in the presence of profilin (Fig. 6, A and C). In contrast, Vt did not provoke any growth arrest in the presence of mDia1-FH1FH2 and profilin (Fig. 6, A–C, and corresponding supplemental Movies 4–7). Altogether, our data demonstrate that vinculin and mDia1 compete for the barbed end.
FIGURE 6.

Vinculin tail and mDia1 compete for actin filament barbed ends. Conditions were as follows. 0.6 μm MgATP-G-actin (10% Alexa488-labeled) was polymerized in a flow chamber coated with NEM-myosin in 5 mm Tris pH 7.8, 100 mm KCl, 0.2 mm ATP, 0.1 mm CaCl2, 1 mm MgCl2, 10 mm DTT, 0.2 mm 1,4-diazabicyclo(2,2,2)-octane, 0.2% methyl-cellulose. A, representative kymographs of growing single actin filaments in the presence of 6 μm profilin (top left; see corresponding supplemental Movie 4); 6 μm profilin and 1 μm Vt (top right; see corresponding supplemental Movie 5); 20 nm mDia1-FH1FH2 and 6 μm profilin (bottom left; see corresponding supplemental Movie 6); or 20 nm mDia1-FH1FH2, 6 μm profilin, and 1 μm Vt (bottom right; see corresponding supplemental Movie 7). B, average elongation rates between the pauses in the indicated conditions. C, percentage of time in growth and pause in the indicated conditions. Error bars, S.E.
DISCUSSION
In this study, we identified a barbed end capping activity in the tail domain of vinculin. We also showed that actin filaments nucleated by Vt in low salt conditions are capped at their barbed ends. The interaction between vinculin and the barbed end of actin filaments is characterized by a weak affinity and slow association and dissociation rate constants. The barbed end capping activity requires the C-terminal arm of vinculin that is dispensable for actin filament side binding. The actin filament side binding and barbed end capping activities are autoinhibited. However, in contrast to the actin filament side-binding domain, the barbed end-capping domain is not unmasked by the binding of the VBS1 domain of talin to the D1 subdomain of Vh and requires the dissociation of an additional autoinhibitory interaction. Finally, we showed that mDia1-bound actin filament barbed ends are protected against vinculin capping.
Regulation of the Actin Filament Side Binding and Barbed End Capping Activities of Vinculin
We confirmed that the binding of the VBS1 domain of talin is sufficient to disrupt the D1-Vt interaction, allowing the binding of Vt to the side of actin filaments (Fig. 7, states 1–3) (20, 29). We did not confirm that the VBS1 domain of talin interacts with high affinity with full-length vinculin, as previously measured by surface plasmon resonance (Kd = 77 nm) (29). We found that VBS1 has a weak affinity for vinculin in solution (Kd = 24 μm). This low affinity was increased by F-actin binding to vinculin (Kd = 5 μm). Both results are in reasonable agreement with previous reports (20, 45).
FIGURE 7.

Working model for the regulation of vinculin. In this scheme, each subdomain of vinculin is represented as a cylinder, according to Refs. 20 and 45. The proline-rich region that links Vh and Vt is represented as an orange dotted line. The C-terminal arm is represented as a black line (1). Vinculin exists in an autoinhibited conformation in which the F-actin binding site located in Vt is buried by contacts with the head subdomain D1. However, according to the crystal structure of vinculin and affinity measurements, Vt also interacts with the subdomains D3 and D4 (2). The low affinity binding of the VBS1 domain of talin is sufficient to disrupt the D1-Vt interaction (red arrow). However, other contacts between Vt and Vh remain (3). The exposed Vt domain binds to the side of actin filaments. In this conformation, vinculin allows barbed end elongation. Contacts between Vh and Vt still exist (4). In addition to the binding of VBS1 to D1, the disruption of unidentified contacts, probably between subdomains D3-D4 and Vt, leads to the full dissociation of Vt from Vh. This dissociation allows the C-terminal arm of Vt to cap the barbed end of the filament or change its structure to prevent the association of actin monomers.
Our results, showing that ΔD1-vinculin and the VBS1-vinculin complex bound to the side of actin filaments but did not cap their barbed ends, demonstrated that the D1 head subdomain only masks the F-actin side-binding domain of Vt, whereas additional contacts mask the newly discovered barbed end capping activity of Vt (Fig. 7, state 3). This view is also supported by the fact that the full-length Vh domain inhibits more efficiently the capping activity of Vt than the isolated D1 subdomain. Our data are in good agreement with the crystal structure and affinity measurements showing that additional contacts exist between Vt and the D3 and D4 subdomains (18, 27, 28). The mutation N773A/E775A, which disrupts the D4-Vt interaction (28), was not sufficient to unmask the capping domain of vinculin, even in the presence of VBS1, suggesting that additional contacts between Vt and D3 mask the capping domain (data not shown). Our results imply that vinculin, activated by VBS1, binds to the side of actin filaments but remains partially autoinhibited (Fig. 7, state 3). Structural studies confirm that the contacts made by D4 and D3 with Vt do not interfere with actin filament binding (13, 18, 27).
Mechanism of Inhibition of Barbed End Elongation
We demonstrated that Vt inhibits actin filament barbed end elongation. The deletion of the C-terminal arm of Vt abolished this capping function but did not affect actin filament side binding. Therefore, the C-terminal arm is required for the capping activity, whereas the side-binding domain is not sufficient. Although it is not sufficient, the actin filament side-binding domain located in Vt could also play a role in the capping activity. In support to this hypothesis, we showed that the isolated D1 subdomain, which inhibits side binding by Vt, also inhibits the capping activity of Vt. Therefore, it is possible that the capping mechanism involves the binding of Vt to the side of the last subunits at the barbed end. In addition to the side binding, the C-terminal arm of vinculin could directly cap the barbed end or induce a conformational change at the barbed end that inhibits monomer addition (Fig. 7, state 4).
Potential Cellular Activators of the Barbed End Capping Function of Vinculin
So far, we did not find a cellular mechanism that reveals the capping activity of vinculin. In contrast with a single VBS domain of talin, a short fragment containing two VBSs from the Shigella protein IpaA forms a complex with vinculin that inhibits barbed end growth (22–24). The study of the subversion of signaling pathways by pathogens has always been powerful at predicting the cellular mechanisms by which autoinhibited proteins are activated. However, structural studies that aimed to compare the activation of vinculin by IpaA and talin only documented the helical bundle conversion of D1 upon VBS binding and did not address a possible propagation of this structural change to the other neighboring subdomains of Vh that are probably responsible for the inhibition of the capping activity. Therefore, it is difficult to understand why the VBSs of IpaA activate the capping function of vinculin, whereas the VBS1 from talin does not. However, the discovery that IpaA contains two VBSs that bind two distinct sites of D1 could explain the activity of IpaA (46). The higher affinity VBS of IpaA induces a helical bundle conversion of the N-terminal part of D1, whereas the lower affinity VBS binds to the adjacent C-terminal part of D1, without affecting the Vt-D1 interaction (46). Interestingly, talin also contains VBSs that interact with both the N-terminal part of D1 (VBS1) and the C-terminal part of D1 (VBS33) (46). However, a combination of synthetic peptides made of VBS1 and VBS33 of talin failed to reveal the capping activity of vinculin in an actin assembly assay (data not shown).
Whether, among the 11 VBSs of talin, there exists a combination that requires a particular orientation and spacing of VBSs to activate the capping function of vinculin is not known. Such a mechanism cannot be tested easily because most of the VBSs of talin are buried and require the mechanical stretching of the protein to be exposed (47).
Because mechanical force is thought to play a major role in the activation of autoinhibited focal adhesion proteins, an attractive possibility for vinculin is that the force provided by the acto-myosin tension generated by stress fibers could, alone or in combination with a VBS, disrupt all of the contacts between Vh and Vt and expose the capping domain.
Finally, the C-terminal arm of vinculin is involved in the binding of Vt to the phosphatidylinositol 4,5-bisphosphate (48, 49). Although phosphatidylinositol 4,5-bisphosphate disrupts the intramolecular interaction Vh-Vt (50), it is unlikely that it unmasks the capping domain of Vt, because it also inhibits the binding of Vt to actin filaments (51). The interaction of phosphatidylinositol 4,5-bisphosphate with the C-terminal arm is also known to control the turnover of focal adhesions (52). Whether the regulation of the capping function by phosphatidylinositol 4,5-bisphosphate contributes to the regulation of focal adhesion turnover is an interesting open issue.
Potential Role of the Barbed End Capping Function of Vinculin in Vivo
The role of a soluble capping protein like CP is to block the majority of actin filament barbed ends, which increases the concentration of monomeric actin at steady state to the value of the critical concentration of the pointed end, allowing the non-capped filaments to grow faster (33). It is very unlikely that vinculin enhances actin dynamics in a CP-like fashion because we showed that Vt does not increase the concentration of monomeric actin at steady state. The binding of vinculin to actin filament barbed ends is characterized by a slow association rate constant (k+ = 0.0023 μm−1 s−1), compared with CP for example (k+ = 6 μm−1 s−1) (53) and a slow dissociation rate constant (k− = 0.0017–0.014 s−1). These parameters suggest that vinculin allows transient periods of barbed end growth in focal adhesions, in agreement with the partial coupling between vinculin and actin observed in focal adhesions (7–9).
In addition to vinculin, several actin-binding proteins control actin dynamics in focal adhesions (25). Tensin caps actin filament barbed ends (54). The formin mDia1 and VASP nucleate and maintain the fast processive growth of actin filament barbed ends (43, 44, 55). We already showed that mDia1-bound actin filament barbed ends are protected against vinculin capping. Whether vinculin and mDia1 bind to actin filament barbed ends at different stages during the maturation of focal adhesions or whether they regulate the dynamics of different populations of actin filaments remains to be elucidated.
Understanding how all of these individual activities are coordinated to mechanically couple a dynamic actin network to the cell substrate will require in depth cell biological and biochemical studies as well as the development of new biomimetic assays.
Supplementary Material
Acknowledgments
We thank the Carlier group members, Guy Tran Van Nhieu, and Dorit Hanein for helpful discussions. We thank Tina Izard and Philippe Bois for the gift of the PET-vinculin plasmid and the HUGE protein database for the gift of the Talin1 cDNA. We thank Emmanuèle Helfer for her help with TIRF microscopy.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3 and Movies S1–S7.
- Vh
- vinculin N-terminal globular head
- Vt
- vinculin C-terminal tail
- IAEDANS
- N-iodoacetyl-N′-(5-sulfo-1-naphthyl)ethylenediamine
- CP
- capping protein
- NEM
- N-ethylmaleimide
- VBS
- vinculin binding site
- TIRF
- total internal reflection fluorescence
- DTT
- dithiothreitol.
REFERENCES
- 1.Ziegler W. H., Liddington R. C., Critchley D. R. (2006) Trends Cell Biol. 16, 453–460 [DOI] [PubMed] [Google Scholar]
- 2.Zamir E., Geiger B. (2001) J. Cell Sci. 114, 3583–3590 [DOI] [PubMed] [Google Scholar]
- 3.Coll J. L., Ben-Ze'ev A., Ezzell R. M., Rodríguez Fernández J. L., Baribault H., Oshima R. G., Adamson E. D. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 9161–9165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volberg T., Geiger B., Kam Z., Pankov R., Simcha I., Sabanay H., Coll J. L., Adamson E., Ben-Ze'ev A. (1995) J. Cell Sci. 108, 2253–2260 [DOI] [PubMed] [Google Scholar]
- 5.Xu W., Coll J. L., Adamson E. D. (1998) J. Cell Sci. 111, 1535–1544 [DOI] [PubMed] [Google Scholar]
- 6.Saunders R. M., Holt M. R., Jennings L., Sutton D. H., Barsukov I. L., Bobkov A., Liddington R. C., Adamson E. A., Dunn G. A., Critchley D. R. (2006) Eur J. Cell Biol. 85, 487–500 [DOI] [PubMed] [Google Scholar]
- 7.Hu K., Ji L., Applegate K. T., Danuser G., Waterman-Storer C. M. (2007) Science 315, 111–115 [DOI] [PubMed] [Google Scholar]
- 8.Brown C. M., Hebert B., Kolin D. L., Zareno J., Whitmore L., Horwitz A. R., Wiseman P. W. (2006) J. Cell Sci. 119, 5204–5214 [DOI] [PubMed] [Google Scholar]
- 9.Ji L., Lim J., Danuser G. (2008) Nat. Cell Biol. 10, 1393–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphries J. D., Wang P., Streuli C., Geiger B., Humphries M. J., Ballestrem C. (2007) J. Cell Biol. 179, 1043–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hüttelmaier S., Bubeck P., Rüdiger M., Jockusch B. M. (1997) Eur. J. Biochem. 247, 1136–1142 [DOI] [PubMed] [Google Scholar]
- 12.Menkel A. R., Kroemker M., Bubeck P., Ronsiek M., Nikolai G., Jockusch B. M. (1994) J. Cell Biol. 126, 1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janssen M. E., Kim E., Liu H., Fujimoto L. M., Bobkov A., Volkmann N., Hanein D. (2006) Mol. Cell 21, 271–281 [DOI] [PubMed] [Google Scholar]
- 14.Johnson R. P., Craig S. W. (1994) J. Biol. Chem. 269, 12611–12619 [PubMed] [Google Scholar]
- 15.Johnson R. P., Craig S. W. (1995) Nature 373, 261–264 [DOI] [PubMed] [Google Scholar]
- 16.Burridge K., Mangeat P. (1984) Nature 308, 744–746 [DOI] [PubMed] [Google Scholar]
- 17.Wachsstock D. H., Wilkins J. A., Lin S. (1987) Biochem. Biophys. Res. Commun. 146, 554–560 [DOI] [PubMed] [Google Scholar]
- 18.Bakolitsa C., Cohen D. M., Bankston L. A., Bobkov A. A., Cadwell G. W., Jennings L., Critchley D. R., Craig S. W., Liddington R. C. (2004) Nature 430, 583–586 [DOI] [PubMed] [Google Scholar]
- 19.Izard T., Evans G., Borgon R. A., Rush C. L., Bricogne G., Bois P. R. (2004) Nature 427, 171–175 [DOI] [PubMed] [Google Scholar]
- 20.Bass M. D., Patel B., Barsukov I. G., Fillingham I. J., Mason R., Smith B. J., Bagshaw C. R., Critchley D. R. (2002) Biochem. J. 362, 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izard T., Tran Van Nhieu G., Bois P. R. (2006) J. Cell Biol. 175, 465–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourdet-Sicard R., Rüdiger M., Jockusch B. M., Gounon P., Sansonetti P. J., Nhieu G. T. (1999) EMBO J. 18, 5853–5862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourdet-Sicard R., Egile C., Sansonetti P. J., Tran Van Nhieu G. (2000) Microbes Infect. 2, 813–819 [DOI] [PubMed] [Google Scholar]
- 24.Ramarao N., Le Clainche C., Izard T., Bourdet-Sicard R., Ageron E., Sansonetti P. J., Carlier M. F., Tran Van Nhieu G. (2007) FEBS Lett. 581, 853–857 [DOI] [PubMed] [Google Scholar]
- 25.Le Clainche C., Carlier M. F. (2008) Physiol. Rev. 88, 489–513 [DOI] [PubMed] [Google Scholar]
- 26.Cohen D. M., Kutscher B., Chen H., Murphy D. B., Craig S. W. (2006) J. Biol. Chem. 281, 16006–16015 [DOI] [PubMed] [Google Scholar]
- 27.Borgon R. A., Vonrhein C., Bricogne G., Bois P. R., Izard T. (2004) Structure 12, 1189–1197 [DOI] [PubMed] [Google Scholar]
- 28.Cohen D. M., Chen H., Johnson R. P., Choudhury B., Craig S. W. (2005) J. Biol. Chem. 280, 17109–17117 [DOI] [PubMed] [Google Scholar]
- 29.Bois P. R., O'Hara B. P., Nietlispach D., Kirkpatrick J., Izard T. (2006) J. Biol. Chem. 281, 7228–7236 [DOI] [PubMed] [Google Scholar]
- 30.Kuhn J. R., Pollard T. D. (2007) J. Biol. Chem. 282, 28014–28024 [DOI] [PubMed] [Google Scholar]
- 31.Pollard T. D. (1986) J. Cell Biol. 103, 2747–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schafer D. A., Jennings P. B., Cooper J. A. (1996) J. Cell Biol. 135, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pantaloni D., Le Clainche C., Carlier M. F. (2001) Science 292, 1502–1506 [DOI] [PubMed] [Google Scholar]
- 34.Wen K. K., Rubenstein P. A., DeMali K. A. (2009) J. Biol. Chem. 284, 30463–30473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kovar D. R., Pollard T. D. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 14725–14730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bass M. D., Smith B. J., Prigent S. A., Critchley D. R. (1999) Biochem. J. 341, 257–263 [PMC free article] [PubMed] [Google Scholar]
- 37.Gingras A. R., Ziegler W. H., Frank R., Barsukov I. L., Roberts G. C., Critchley D. R., Emsley J. (2005) J. Biol. Chem. 280, 37217–37224 [DOI] [PubMed] [Google Scholar]
- 38.Gimona M., Small J. V., Moeremans M., Van Damme J., Puype M., Vandekerckhove J. (1988) EMBO J. 7, 2329–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papagrigoriou E., Gingras A. R., Barsukov I. L., Bate N., Fillingham I. J., Patel B., Frank R., Ziegler W. H., Roberts G. C., Critchley D. R., Emsley J. (2004) EMBO J. 23, 2942–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Izard T., Vonrhein C. (2004) J. Biol. Chem. 279, 27667–27678 [DOI] [PubMed] [Google Scholar]
- 41.Watanabe N., Kato T., Fujita A., Ishizaki T., Narumiya S. (1999) Nat Cell Biol. 1, 136–143 [DOI] [PubMed] [Google Scholar]
- 42.Riveline D., Zamir E., Balaban N. Q., Schwarz U. S., Ishizaki T., Narumiya S., Kam Z., Geiger B., Bershadsky A. D. (2001) J. Cell Biol. 153, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hotulainen P., Lappalainen P. (2006) J. Cell Biol. 173, 383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romero S., Le Clainche C., Didry D., Egile C., Pantaloni D., Carlier M. F. (2004) Cell 119, 419–429 [DOI] [PubMed] [Google Scholar]
- 45.Chen H., Choudhury D. M., Craig S. W. (2006) J. Biol. Chem. 281, 40389–40398 [DOI] [PubMed] [Google Scholar]
- 46.Nhieu G. T., Izard T. (2007) EMBO J. 26, 4588–4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.del Rio A., Perez-Jimenez R., Liu R., Roca-Cusachs P., Fernandez J. M., Sheetz M. P. (2009) Science 323, 638–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fukami K., Endo T., Imamura M., Takenawa T. (1994) J. Biol. Chem. 269, 1518–1522 [PubMed] [Google Scholar]
- 49.Bakolitsa C., de Pereda J. M., Bagshaw C. R., Critchley D. R., Liddington R. C. (1999) Cell 99, 603–613 [DOI] [PubMed] [Google Scholar]
- 50.Gilmore A. P., Burridge K. (1996) Nature 381, 531–535 [DOI] [PubMed] [Google Scholar]
- 51.Steimle P. A., Hoffert J. D., Adey N. B., Craig S. W. (1999) J. Biol. Chem. 274, 18414–18420 [DOI] [PubMed] [Google Scholar]
- 52.Chandrasekar I., Stradal T. E., Holt M. R., Entschladen F., Jockusch B. M., Ziegler W. H. (2005) J. Cell Sci. 118, 1461–1472 [DOI] [PubMed] [Google Scholar]
- 53.Wear M. A., Yamashita A., Kim K., Maéda Y., Cooper J. A. (2003) Curr. Biol. 13, 1531–1537 [DOI] [PubMed] [Google Scholar]
- 54.Chuang J. Z., Lin D. C., Lin S. (1995) J. Cell Biol. 128, 1095–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Breitsprecher D., Kiesewetter A. K., Linkner J., Urbanke C., Resch G. P., Small J. V., Faix J. (2008) EMBO J. 27, 2943–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.