

Abstract
MicroRNAs, by modulating gene expression, have been implicated as regulators of various cellular and physiological processes, including differentiation, proliferation, and cancer. Here, we study the role of microRNAs in Schwann cell (SC) differentiation by conditional removal of the microRNA processing enzyme Dicer1. We reveal that both male and female mice lacking Dicer1 in SC (Dicer1 conditional knock-outs) display a severe neurological phenotype resembling congenital hypomyelination. Ultrastructural analyses show that many SC lacking Dicer1 are stalled in differentiation at the promyelinating state and fail to myelinate axons. Gene expression analyses reveal a failure to extinguish genes characteristic of the undifferentiated state such as Sox2, Jun, and Ccnd1. Sox2 and Jun are well characterized negative regulators of SC differentiation. Consistent with Sox2/Jun maintenance, Egr2, a master regulator of the myelinating program, is drastically downregulated and likely accounts for the myelination defect. We posit a model wherein microRNAs are critical for downregulation of antecedent programs of gene expression. In SC differentiation, this is particularly relevant in the key developmental transition from a promyelinating to myelinating SC.
Introduction
Cell differentiation is a multistage process, the progression from one stage to the next being driven by underlying alterations in genetic programs. In the Schwann cell (SC) lineage, multiple molecularly and morphologically distinct transitional stages have been described (Jessen and Mirsky, 2005). Embryonically, neural crest cells give rise to SC precursors, which then transition to a cell that is committed to the SC lineage, the immature SC. A large number of immature SCs, originally contacting multiple axons, establish a one-to-one relationship with a single axon, to reach the promyelinating stage. Finally, proliferation ceases and myelination commences (Jessen and Mirsky, 2005).
Critical for developmental transitions from one stage to the next is the repression of genes involved in the antecedent stage, as well as the activation of new genes that will define the succeeding stage (Jessen and Mirsky, 2005; D'Antonio et al., 2006). These genetic cascades have been best described between the promyelinating and myelinating states. Developmental expression analysis reveals cohorts of genes being coordinately increased (e.g., Egr2, Mpz, and Mbp) and others coordinately extinguished (e.g., Sox2, Jun, and Ccnd1) during the promyelinating–myelinating transition.
The Egr2 transcription factor is key for the developmental progression to a myelinating state. SC with reduced or no Egr2 fail to myelinate axons (Topilko et al., 1994; Zorick et al., 1999; Le et al., 2005a). Subsequent molecular analyses reveal that Egr2 is a direct transcriptional activator of genes encoding key myelin proteins such as Mpz (LeBlanc et al., 2006). Interestingly, Egr2 also serves as a transcriptional repressor for some promoters (Mager et al., 2008). In conjunction with Nab1/2 corepressors, Egr2 represses a subset of genes that are characteristic of immature SCs (Le et al., 2005b; Desmazieres et al., 2008). Furthermore, in the Egr2 mutant, several genes of the immature/promyelinating state, including Sox2 and Pou3f1, fail to be extinguished (Zorick et al., 1999; Le et al., 2005a). Repression of genes of the antecedent stage is critical for the transition to a myelinating SC. Thus, artificially sustained expression of genes characteristic of immature/promyelinating cells, such as Sox2, Jun, or POU3f1, prevents normal SC myelination (Le et al., 2005a; Ryu et al., 2007; Parkinson et al., 2008; Woodhoo et al., 2009).
MicroRNAs (miRNAs, miRs) are ∼23 bp, noncoding RNAs that bind to target mRNAs harboring a short stretch of complementary sequence, resulting in posttranscriptional silencing of target gene expression (Bartel and Chen, 2004; Bartel, 2009). We reasoned that miRNAs may play a role in facilitating the timely repression of antecedent genes, toward the normal progression of SC differentiation. To evaluate the role of miRNAs in SC differentiation, we ablated Dicer1, a key miRNA processing enzyme, from developing SCs. In Dicer1 conditional knock-outs (cKOs), most SCs fail to myelinate, are arrested in an undifferentiated state, and continue to proliferate. Molecular analyses reveal a massive reduction of myelination-related genes, including Egr2, but the maintained expression of genes characterizing the undifferentiated SC state, such as Sox2, Jun, and Ccnd1. Finally, developmental profiling, bioinformatics, and in vitro analysis identify miR138 as one potential repressor of these genes.
Materials and Methods
Generation and genotyping of mice.
P0::Cre, Dicer1Del/Wt mice were bred to Dicer1F/F mice harboring an R26R floxed lacZ allele (Soriano, 1999), on a mixed FVB/B6 background. Mice were killed at various time points, and their sciatic nerves were dissected and processed for the appropriate assay. When necessary, 50 mg/kg bromodeoxyuridine (BrdU) was administered 2 h before dissection, as described previously (Joksimovic et al., 2009). For BrdU quantification, ∼500 4′,6′-diamidino-2-phenylindole-positive (DAPI+) nuclei were counted from representative sections of control and Dicer1 cKO nerves.
Electron microscopy.
Mouse sciatic nerves were dissected from control and Dicer1 cKO mice and immediately fixed in 2.0% glutaraldehyde in 0.12 m phosphate buffer. Semithin section analysis and electron microscopy were performed as described previously (Feltri et al., 2002).
RNA extraction and real-time PCR.
Mouse sciatic nerves were dissected, immediately snap frozen in liquid nitrogen, and stored until RNA extraction. Total RNA, including small RNAs, was extracted using the mirVana (Ambion) kit. For mRNA PCR, a cDNA library was synthesized using the SuperScript II Reverse Transcriptase (Invitrogen), followed by SYBR green, real-time PCR (iQ SYBR Green Supermix; Bio-Rad) performed with MyiQ (Bio-Rad). Primers are listed in supplemental Figure S1 (available at www.jneurosci.org as supplemental material).
For miRNA analyses, RNA was extracted as described, and Taqman PCR (Applied Biosystems) was performed for miR29a, miR134, miR138, miR138*, miR150, and miR338 from control and Dicer1 cKO mice. Small nucleolar RNA (Sno-202) was used to normalize the data. For developmental analysis, Swiss Webster wild-type nerves were analyzed for miR138 expression, relative to Sno-202.
Immunofluorescence.
Tissue-Tek OCT compound-embedded fresh frozen sciatic nerves were cut longitudinally into 8 μm sections. The sections were postfixed for 5 min in 2.0% paraformaldehyde and blocked for 1 h in the appropriate incubation buffer. Nerves were incubated with rabbit polyclonal Sox2 (1:10,000; Millipore Bioscience Research Reagents), rat monoclonal Mbp (1:500; Millipore Bioscience Research Reagents), or rat monoclonal BrdU (1:500; Serotec) antibodies in 20% FBS plus 0.1% Triton X-100 in PBS. Rabbit polyclonal Pou3f1 (1:1000; a kind gift from John Bermingham, McLaughlin Research Institute, Great Falls, MT) was incubated in 1% nonfat dried milk, 1% BSA, and 0.1% Triton X-100 in PBS. Rabbit polyclonal Ki67 (Novocastra) and rabbit polyclonal Caspase (Apo Active 3; Cell Signaling Technology) were incubated in 5% normal donkey serum and 0.1% Triton X-100 in PBS. Before BrdU incubation, slides were treated with antigen unmasking solution (Vector Laboratories). All primary antibodies were incubated for 14–18 h at 4°C, followed by a 2 h room temperature incubation with the respective secondary antibody [donkey anti-rabbit Alexa 555 or donkey anti-rat Alexa 488 (1:250; Invitrogen)]. For teased nerve fiber staining, sciatic nerves were dissected and immediately teased in PBS. They were then stained for Mbp as described above.
Luciferase assays.
The stem loop of mmu (mouse)–miR138-1 (GenBank accession number MI0000722) plus ∼100 bp 5′ and 3′ flanking sequence was cloned into the pCAG–RFP–int vector (Addgene plasmid 19822). The region of each target gene predicted to contain an mmu–miR138-1 seed match [identified with TargetScan (Lewis et al., 2005), microCosm (Griffiths-Jones et al., 2008), or, in the case of Sox2, the rna22 algorithm (Miranda et al., 2006), identified two sites in the coding sequence as described previously (Tay et al., 2008)] was PCR purified from genomic DNA using primers, including XbaI restriction sites (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Each fragment was cloned into the pmirGLO vector (E1330; Promega). The plasmids were cotransfected into 24-well plates of HEK293 cells using Lipofectamine 2000 (catalog #11668; Invitrogen). After 48 h, the cells were harvested for a luciferase assay (E1910; Promega) and measured with the Clarity Luminescence Microplate Reader (Bio-Tek). Each Firefly reading was normalized to Renilla, and all samples were replicated 10 times.
Gene expression microarray analysis.
RNA samples were run on a MouseWG-6 version 2 Expression BeadChip (Illumina). We used the Bioconductor Illumina package to preprocess the Illumina data with default settings. Probes with all samples “Absent” (lower or around background levels) were removed from additional analysis. To identify differentially expressed genes, we applied routines implemented in Illumina package to fit linear models to the normalized expression values. The variance used in the t score calculation was corrected by an empirical Bayesian method for better estimation under small sample size. Genes identified in the comparison of experimental samples with baseline had fold change >2.0 and p value (false discovery rate adjusted) <0.05 for the differences to be considered significant. The definition of fold change is that, when we compare controls with Dicer1 cKOs, the value is always the ratio of maximum to minimum of expression values of Dicer1 cKOs and controls. The data discussed in this publication have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus and are accessible through Gene Expression Omnibus Series accession number GSE16741 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE16741).
MicroRNA microarrays.
Sciatic nerve mRNA was harvested from three postnatal day 1 (P1) and three P14 rats and subject to microarray analysis (LC Biosciences).
Results
We used a previously characterized allele of Dicer1 that included loxP sites flanking the critical RNaseIII domain (Harfe et al., 2005). To obtain SC deletions, we used a P0::Cre strain that is active in the SC lineage from midgestational time points (13.5 d postcoitum) (Feltri et al., 1999, 2002) (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) and thus would allow the study of most stages of SC differentiation. Accordingly, in P7 Dicer1 cKO sciatic nerve, mRNA representing the floxed allele is almost undetectable, thus verifying efficient recombination of the Dicer1 locus (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Dicer1 cKO mice appeared externally normal during the first few postnatal days. Between P7 and P14, movements were visibly impaired, and, by P21, these mice displayed severe clenching and sporadic hindlimb paralysis (supplemental video, available at www.jneurosci.org as supplemental material). By P28, limb movements were severely impaired, and loss of muscle mass was observed in the distal regions of the hindlimb. Thus, the behavioral phenotype is similar to mouse models of congenital hypomyelination.
To investigate the pathological changes underlying these gross abnormalities, we performed semithin (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) and ultrastructural analyses on sciatic nerves of control and Dicer1 cKO mice at P4, P14, P21, and P28 (Fig. 1). As early as P4, severe morphological differences were visible between Dicer1 cKO mice and control littermates. At this stage, in the control nerve, radial sorting of axons was almost complete and few unsorted bundles remained. Some promyelinating SCs were observed that had established one-to-one relationships, and the majority of SCs were associated with thin myelin sheaths. In the Dicer1 cKO nerves, promyelinating SCs and myelin sheaths were few, and large numbers of unsorted axon bundles remained.
Figure 1.
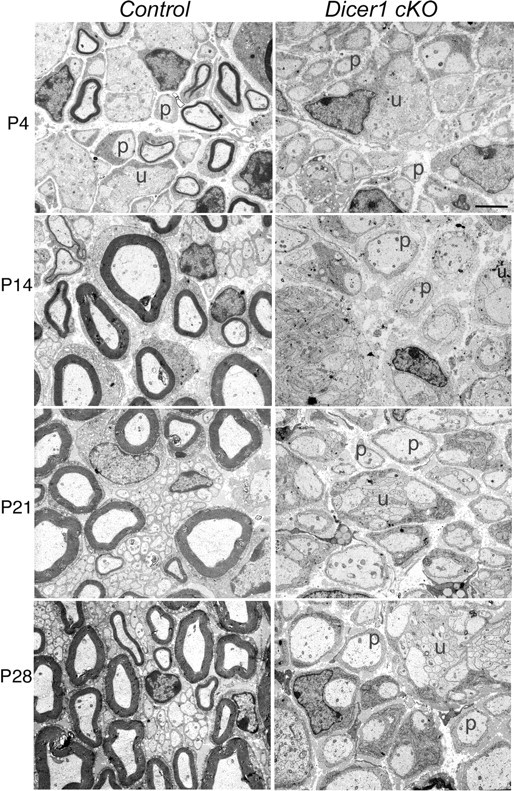
Ultrastructural analyses of control and Dicer1 cKO nerves at different postnatal stages reveals an arrest in SC differentiation. In controls, unsorted bundles (u) and promyelinating cells (p) are mainly observed at P4. In contrast, Dicer1 cKO SCs, throughout development, show persistence of promyelinating cells (p) in a one-to-one relation with axons, several unsorted bundles (u), and very few myelin sheaths (not shown in these fields) (n = 3 except for P28, in which n = 2).
By P14, the control nerve had very few unsorted bundles and promyelinating cells (Fig. 1). At this stage, myelin sheaths were thicker than at P4. In Dicer1 cKO nerves, many groups of amyelinated large axons (two to eight) were present and ensheathed by SCs in a rather disorganized way (as at P4). Even if many cells appeared to have established one-to-one relationships, few myelin sheaths were observed, and these sheaths appeared normally compacted. These myelin sheaths likely occur because a few SCs did not efficiently recombine the Dicer1 locus. At P21 and P28, many SCs appeared to remain arrested at the promyelinating stage; some unsorted bundles and few myelin sheaths were also observed. Onion bulbs or tomacula were not observed at any age examined (i.e., up to 4 weeks), consistent with a congenital hypomyelinating phenotype. Thus, in SCs lacking Dicer1, the most prominent phenotype is that SC differentiation is stalled at the immature/promyelinating stages.
To evaluate whether the aberrant developmental transitions observed in our ultrastructural analyses were accompanied by corresponding gene expression changes, we performed microarray analysis on P14 control and Dicer1 cKO sciatic nerves (Fig. 2 A) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Such analyses revealed a marked downregulation of most myelin genes, including Mpz, Pmp22, Mbp, Mag, Prx, Plp1, Gjb1, and Egr2 in Dicer1 cKO nerves. In contrast, genes characteristic of undifferentiated SCs, including Egr1, Egr3, Sox2, Ngfr, Jun, and Ncam1, were increased. In addition, genes associated with proliferation, including Ccnd1, were also drastically increased in Dicer1 cKO nerves. In contrast, Sox10, a key Egr2 regulator, appeared unchanged (supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
Figure 2.

Gene expression analyses reveal a reduction of myelin-related genes and an upregulation of immature SC-specific genes. A, List of select genes that were altered according to microarray analysis (n = 3). B, Quantitative RT-PCR analysis for select genes in control and Dicer1 cKO nerves at P7 (top; n = 4) and P14 (bottom; n = 5). In B, myelin-related genes are presented separately from immature/promyelinating genes. Black bars, Control; gray bars, Dicer1 cKOs. MT-WT, Mutant versus wild type fold change.
We confirmed these microarray results by performing quantitative reverse transcription (RT)-PCR on sciatic nerve samples from individual P7 and P14 control and Dicer1 cKO mice (Fig. 2 B). At P7 and P14, myelin genes, including Mpz and Pmp22, were dramatically reduced in Dicer1 cKOs; Plp1 was reduced to a lesser degree. Egr2 was decreased by ∼90%. This fold decrease in Egr2 is comparable with that described in the Egr2 Lo/Lo mouse (Le et al., 2005a) and therefore is sufficient to explain the reduction in myelin gene expression and lack of myelination in SCs lacking Dicer1. mRNAs characteristic of immature SCs, including Egr1, Egr3, Sox2, and Ngfr, were increased in Dicer1 cKOs at P7, although Jun was unchanged. By P14, the fold increase in Egr1, Egr3, Sox2, and Ngfr was further exacerbated, and, at this point, Jun and Pou3f1 also showed a modest increase. The fold increase in Sox2 is similar to that reported in Egr2 deficient SCs (i.e., approximately sixfold to sevenfold) (Le et al., 2005a). These mRNA profile data are consistent with a blockade of SC differentiation observed in our morphological analyses.
Egr2 and Sox2 are thought to negatively regulate each other. Thus, in Egr2-depleted SCs, Sox2 is increased. Conversely, Sox2 is sufficient to reduce Egr2 levels and suppress myelination (Le et al., 2005a; Kao et al., 2009). In Dicer1 cKO SCs, Sox2 mRNA levels are significantly elevated, possibly leading to this drastic differentiation phenotype. We therefore examined Sox2 expression by immunofluorescence labeling, in the developing control and Dicer1 cKO sciatic nerve (Fig. 3). In P1 control and Dicer1 cKO nerves, most SC nuclei were Sox2+ (Fig. 3 C,D). By P7 in the control nerve, a few bright Sox2+ nuclei and several faintly stained nuclei were observed. In contrast, in the Dicer1 cKO nerve, large numbers of bright Sox2+ nuclei were observed (Fig. 3 E,F). By P14, few Sox2+ nuclei were observed in the control, whereas large numbers of nuclei continued to express Sox2 in the Dicer1 cKO (Fig. 3 G,H). Similar to Sox2, analyses of Pou3f1 showed several more Pou3f1+ nuclei in the Dicer1 cKO compared with the control nerve; the difference was more pronounced at P22 (than at P14; data not shown), because at this age, fewer control SC nuclei still expressed Pou3f1 (Fig. 3 I,J). Mbp+ cells were fewer at P1, P16, and P21 (Fig. 3 A,B) (supplemental Fig. S5, available at www.jneurosci.org as supplemental material) (data not shown). The data illustrate that the timely developmental downregulation of Sox2 and Pou3f1 fails to occur in SCs lacking Dicer1.
Figure 3.
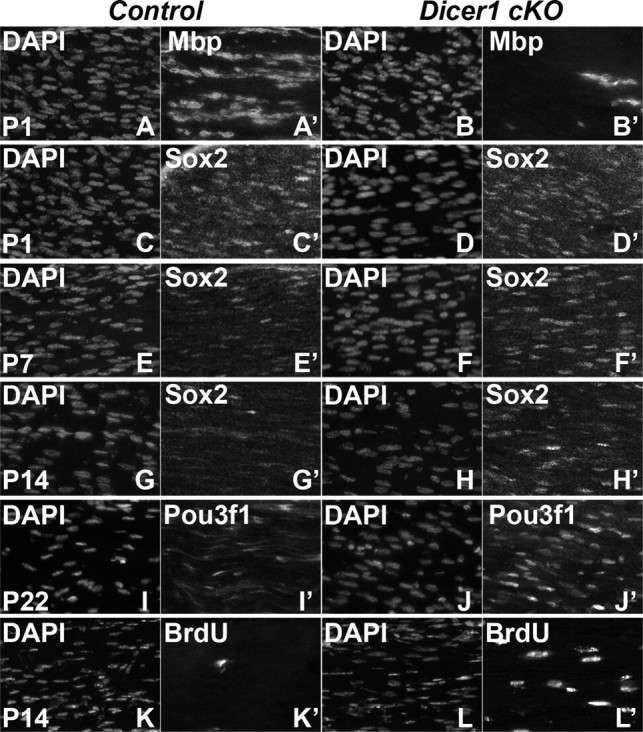
Failure of Sox2 downregulation, impaired cell cycle exit, and lack of myelination in Dicer1 cKO nerves. Longitudinal cryosections of sciatic nerve were immunolabeled for Mbp, Sox2, Pou3f1, and BrdU. A, B, Less Mbp+ cells are observed in the P1 (n = 3) Dicer1 cKO (B) compared with control (A). C–H, In contrast, whereas Sox2 expression is similar in Dicer1 cKO and control nerve at P1 (n = 3) (C, D) and P7 (n = 3), and, at P14 (n = 5), its expression is maintained in the Dicer1 cKO (F, H) but downregulated in control mice (E, G). I, J, Similarly, Pou3f1 staining at P22 (n = 4) reveals maintained expression in the Dicer1 cKO (J) compared with the control (I). K, L, Finally, a 2 h pulse of BrdU incorporation at P14 (n = 4) shows an increase in proliferation in the Dicer1 cKO (L) compared with the control (K).
Gene expression analysis revealed an increase in cell-cycle-related genes in Dicer1 cKO nerves. Accordingly, short-pulse BrdU analysis revealed that proliferation was increased at P7 in Dicer1 cKO nerves compared with controls (data not shown). At P14, this increase was exacerbated because controls showed very few BrdU+ cells at this stage (controls, <0.1% BrdU+ nuclei; Dicer1 cKOs, 7.7 ± 0.85% BrdU+ nuclei) (Fig. 3 K,L). Ki67, a marker of proliferating cells, labeled large numbers of SCs (even more than BrdU labeling) in Dicer1 cKOs compared with controls (supplemental Fig. S6C,D, available at www.jneurosci.org as supplemental material). Together, these data suggest that many SCs in this mutant fail to exit the cell cycle. Because microarray analyses showed elevated cell death genes, we assayed for apoptotic cell death using an activated Caspase 3 antibody. Indeed, some apoptotic cells were observed in P21 Dicer1 cKO nerves (supplemental Fig. S6A,B, available at www.jneurosci.org as supplemental material).
We next evaluated miRNAs that were present in sciatic nerve that could contribute to this drastic phenotype. We reasoned that miRNAs that are involved in this phenotype are likely to match three key criteria. First, they are likely to be significantly increased during development at a time when previous programs are being repressed. Second, they should be drastically reduced in the Dicer1 cKO. Third, they should be bioinformatically predicted to bind mRNAs that are derepressed in the Dicer1 cKO, such as Ccnd1, Jun, and Sox2. Toward indentifying such candidate miRNAs, we performed miRNA arrays, sampling 1024 miRNAs, on P1 and P14 rat sciatic nerve. Sixty-two miRNAs were changed more than twofold. Five miRNAs were changed more than 10-fold, but, of these, miR138 was the only one that increased during development (Fig. 4 A). miR138*, the reverse strand of miR138-1, was also increased, albeit to a lesser extent. Additionally, miR29a, a previously identified SC miRNA, appeared to increase during development, in contrast to a previous study (Verrier et al., 2009). Of miRNAs known to be key regulators of the oligodendrocyte (OL) lineage, miR219 (Lau et al., 2008) was not identified in SC arrays and miR338 (Lau et al., 2008) was only modestly induced. Last, of miRNAs published or bioinformatically predicted to target Sox2, miR134 (five binding sites in the coding region) (Tay et al., 2008), miR145 (Xu et al., 2009), and miR150, were either only modestly increased or, in fact, decreased. Because miR138 showed a prominent developmental increase, we confirmed the array results with quantitative RT-PCR on developing mouse sciatic nerves. Indeed, miR138 showed a developmental increase between P2 and P21, with a peak at P14 (Fig. 4 B).
Figure 4.
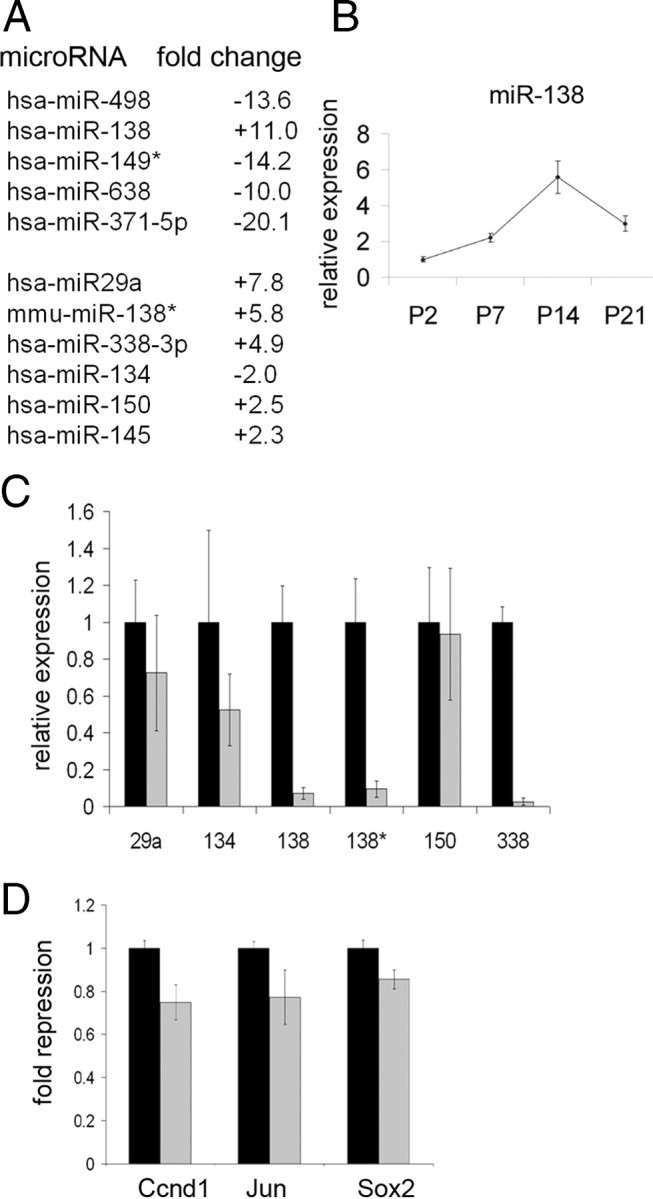
mmu–miR138-1 is sufficient to repress several immature SC genes. A, Microarray analyses on P1 and P14 rat sciatic nerve. Top, miRNAs that are greater than 10-fold changed between P1 and P14. Bottom, Select miRNAs that have been identified previously in SCs or OLs (miR29a and miR338) or as repressors of Sox2 (miR134, miR145, and miR150). B, Quantitative RT-PCR of miR138 showing a developmental increase in expression from P2 to P14 in control nerves (n = 4–5). C, Quantitative RT-PCR showing expression of six miRNAs in control (black bars) and Dicer1 cKO (gray bars) nerves at P7 (n = 4–6). Note that not all miRNAs show a drastic decrease in the Dicer1 cKOs (p < 0.05 for miR138, miR138*, and miR338). D, Cotransfection assays in HEK293 cells show that mmu–miR138-1 (gray bars) is sufficient to modestly repress luciferase constructs containing Ccnd1, Jun, or Sox2 sequences relative to vector alone (black bars) (p = 0.008 for Ccnd1, p = 0.083 for Jun, and p = 0.025 for Sox2). hsa, Human.
To determine which miRNAs were reduced in Dicer1 cKOs, we used Taqman array miRNAs cards (Applied Biosystems) on P7 control and Dicer1 cKO nerves (data not shown). Twenty-five miRNAs of 518 unique mouse miRNAs showed a greater than 10-fold reduction. These included miR138, miR138*, and miR338. Interestingly, of the other relevant miRNAs, miR219 was not detected and miR29a, miR134, miR145, and miR150 were not drastically reduced during the period of myelination in the Dicer1 cKO. Residual miRNAs in Dicer1 cKO nerve are likely, at least in part, attributable to the extraordinary stability of some miRNAs (Fineberg et al., 2009) and have been observed in some other Dicer1 mutants (O'Rourke et al., 2007; Damiani et al., 2008). These results were confirmed by individual quantitative RT-PCR assays on P7 control and Dicer1 cKO sciatic nerve (Fig. 4 C).
Not only did miR138 match our first two criteria, the major and minor products from the mmu–miR138-1 stem loop (miR138 and miR138*) are bioinformatically predicted to bind Ccnd1 (miR138), Sox2 (miR138), and Jun mRNAs (miR138*) (supplemental Fig. S7, available at www.jneurosci.org as supplemental material). To determine whether mmu–miR138-1 could repress Ccnd1, Jun, and Sox2, we cotransfected into HEK293 cells an mmu–miR138-1 expression construct with luciferase constructs containing the predicted binding regions of the above genes. Indeed, miR138, but not the empty vector, repressed luciferase constructs harboring Ccnd1, Sox2, and Jun sequences (Fig. 4 D). In all cases, repression was modest, in accordance with most miRNA experiments in heterologous cells (Bartel and Chen, 2004; Baek et al., 2008; Lau et al., 2008; Zhao et al., 2010). Together, these studies suggest that miR138 could be, at least in part, responsible for dampening immature SC gene expression, although it is likely that other miRNAs may abet this effort.
Discussion
The role of miRNAs has been evaluated by removal of the key miRNA processing enzyme Dicer in various model systems. The phenotypes range from mild to catastrophic. Zebrafish lacking zygotic and maternal dicer undergo fairly normal early embryonic development, including axis formation and the differentiation of multiple cell types, suggesting that miRNAs are dispensable for these early stages of development (Giraldez et al., 2005). Likewise, when Dicer1 was genetically deleted from the zone of polarizing activity of embryonic mouse limbs, no remarkable phenotype was observed. However, when Dicer1 was deleted from the entire limb mesoderm, the limb failed to develop to its normal size, although patterning was still normal (Harfe et al., 2005). When Dicer1 was deleted in lung primordium, catastrophic defects in lung morphogenesis were observed (Harris et al., 2006). In dopamine neuron-specific Dicer1 cKOs, progressive but delayed neurodegeneration was observed, characterized by neuronal death (Kim et al., 2007). In developing and adult oligodendrocyte-specific Dicer1 cKOs, myelination defects were observed (Shin et al., 2009; Dugas et al., 2010; Zhao et al., 2010). More recently, Shkumatava et al. (2009) have suggested that derepression of genes in a Dicer1 null background is an accurate method of identifying bona fide miRNA targets. To determine the role of miRNAs in SC differentiation and to define relevant putative miRNA targets, we ablated Dicer1 specifically in developing SC. We reveal that many of these SCs are stalled at a key juncture of SC differentiation and, at least in part, phenocopy human as well as rodent models of congenital hypomyelination.
Humans and rodents deficient for the master regulatory transcription factor Egr2 display severe hypomyelination (Topilko et al., 1994; Warner et al., 1998; Zorick et al., 1999; Le et al., 2005a; Decker et al., 2006; Desmazieres et al., 2008). The SCs in these models are dramatically stalled in a promyelinating state and predominantly fail to myelinate axons (Zorick et al., 1999; Le et al., 2005a). Thus, the transition from a promyelinating cell to a myelinating cell appears to be a critical juncture in SC differentiation that requires Egr2 activity. Molecular analyses have revealed that Egr2 may be required for activating certain genes and extinguishing other genes (Zorick et al., 1999; Parkinson et al., 2004; Le et al., 2005a; Mager et al., 2008). Thus, in Egr2-deficient models, several immature/promyelinating genes are derepressed, including Sox2 and Pou3f1 (Zorick et al., 1999; Le et al., 2005a). Repression of antecedent genes is critical: indeed, artificially sustained expression of Sox2 results in transcriptional downregulation of Egr2 via its myelin-specific enhancer (Ghislain et al., 2002; Kao et al., 2009) and myelination defects (Le et al., 2005a). One plausible model of these results is that the stoichiometry of these mutually antagonistic transcription factors at any given time determines the phenotype of the SC. Thus, in immature SCs, Egr2 and Sox2 are coexpressed, but the Egr2 protein is present at reduced levels, whereas Sox2 is abundant. At this time, Egr2 is driven off a proximal promoter sequence active mainly in immature SCs (Ghislain et al., 2002); this promoter is insensitive to Sox2-mediated repression, at least in the absence of calcineurin/Nfat signaling (Kao et al., 2009). Egr2 directly or indirectly represses Sox2 [and other immature SC transcription factors, such as Jun (Parkinson et al., 2004)]. This reduced Sox2/Sox10 ratio would likely favor transcriptional upregulation of Egr2 via its myelin-specific enhancer (Kao et al., 2009), modeling a feedforward loop. When Egr2 accumulates to sufficient levels, expression of Sox2 (and other immature SC transcription factors, such as Jun) is extinguished, proliferation ceases, and myelination proceeds. Our results reveal that miRNAs may be an integral component of this loop, possibly acting in concert with these transcriptional circuits. We posit a model in which miRNAs are required for downregulation of immature/promyelinating SC transcription factors, such as Sox2 and Jun. Loss of miRNAs results in elevated immature/promyelinating SC transcription factor levels that result in drastic suppression of Egr2 levels. The feedforward loop that normally results in Egr2 accumulation and myelination is thus disrupted. Consequently, SCs lacking Dicer1, at least in part, phenocopy the congenital hypomyelinating Egr2-deficient mouse mutants.
The remarkable feature of this mouse model compared with other catastrophic Dicer1 mutants is that large numbers of SCs seem to be stalled at a precise hinge point of development, with apparent similarity to Egr2 mutants. This suggests that, of the many possible roles of miRNAs in the SC lineage, one key role of miRNAs is to overcome the promyelinating–myelinating transition, by repressing pivotal genes such as Sox2. Because Sox2 is a potent transcription factor that defines the progenitor state and is key for even reprogramming fibroblasts to stem cells, it is a likely target for miRNA regulation. Indeed, miRNA manipulations targeted to depress Sox2 levels were sufficient to promote embryonic stem cell differentiation (Tay et al., 2008; Xu et al., 2009). In addition to Sox2, it is likely that derepression of other genes also contributes to the SC phenotype described here. For example, increased Ccnd1 levels could lead to augmented transcriptional activation of Notch1 (Bienvenu et al., 2010), which could then negatively regulate myelination (Woodhoo et al., 2009). In support of this possibility, Notch1 as well as Hes1 mRNAs are elevated in the Dicer1 cKO (supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
We have identified miR138 as one potential repressor of key immature SC genes, including Ccnd1, Jun, and Sox2. miR138-mediated repression, at least in a heterologous context, is modest but in accordance with that described for most miRNAs. It is possible that experiments in a SC milieu, containing other cooperatively acting miRNAs, would reveal more potent repression. We hypothesize that miR138-mediated repression of these genes, and possibly others, will facilitate myelination. Additional work will be required to test whether this miRNA targets these genes in vivo and whether miR138 is necessary and/or sufficient for myelination. Interestingly, a recent study on OLs also identified miR138 as an miRNA that facilitates early steps in OL differentiation (Dugas et al., 2010). The possibility that OLs and SCs use similar miRNAs to achieve differentiation suggests that there is likely some overlap in the pathways that need to be repressed for both cell types to differentiate. Consistent with this notion, in both OLs and SCs, Sox genes and Notch pathway genes are negative regulators of differentiation (Le et al., 2005a; Liu et al., 2006; Potzner et al., 2007; Woodhoo et al., 2009) and likely targets of miRNA regulation (Dugas et al., 2010; Zhao et al., 2010; this study).
The first miRNA discovered, lin4, was shown to play a role in suppressing a key target, lin28, and thereby controlling the timing of postembryonic cell divisions and fates in Caenorhabditis elegans (Pasquinelli and Ruvkun, 2002; Ruvkun, 2008). Based on these pioneering studies, it has been suggested that miRNA circuits may underlie various aspects of heterochrony or phylogenetic variation in the timing of developmental events. Here we reveal that miRNAs are critical to repress genes from antecedent stages in the SC lineage for normal myelination. Conceptually, it is possible that these molecular circuits underlie the marked differences in the relative timing of myelination observed in different mammalian species. Thus, miRNAs, such as miR138, by exerting control over the stoichiometry of key transcription factors, may control, at least in part, the observed heterochrony in SC myelination.
Footnotes
This work was supported by National Institutes of Health Grants R21NS063138 (R.A.), R01NS055256 (L.W.), and R01NS45630 (L.F.) and Telethon Italia Grants GGP071100 (L.W.) and GGP08021 (L.F.). We thank John Bermingham for the Pou3f1 antibody, John Kamholz for the Mbp antibody, and Brian Harfe and Michael McManus for the Dicer1 floxed mice. We thank Nadereh Jaffery, Simon Lin, and Gilbert Feng from the genomics and bioinformatics core facilities for assistance with microarrays. We thank Desirée Zambroni for superb technical assistance.
References
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, Geng Y, Zagozdzon A, Jecrois M, Young RA, Liu XS, Cepko CL, Gygi SP, Sicinski P. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010;463:374–378. doi: 10.1038/nature08684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani D, Alexander JJ, O'Rourke JR, McManus M, Jadhav AP, Cepko CL, Hauswirth WW, Harfe BD, Strettoi E. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci. 2008;28:4878–4887. doi: 10.1523/JNEUROSCI.0828-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antonio M, Michalovich D, Paterson M, Droggiti A, Woodhoo A, Mirsky R, Jessen KR. Gene profiling and bioinformatic analysis of Schwann cell embryonic development and myelination. Glia. 2006;53:501–515. doi: 10.1002/glia.20309. [DOI] [PubMed] [Google Scholar]
- Decker L, Desmarquet-Trin-Dinh C, Taillebourg E, Ghislain J, Vallat JM, Charnay P. Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression. J Neurosci. 2006;26:9771–9779. doi: 10.1523/JNEUROSCI.0716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmazières A, Decker L, Vallat JM, Charnay P, Gilardi-Hebenstreit P. Disruption of Krox20–Nab interaction in the mouse leads to peripheral neuropathy with biphasic evolution. J Neurosci. 2008;28:5891–5900. doi: 10.1523/JNEUROSCI.5187-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, Zamanian JL, Foo LC, McManus MT, Barres BA. Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron. 2010;65:597–611. doi: 10.1016/j.neuron.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999;883:116–123. [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, Mueller U, Wrabetz L. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199–209. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineberg SK, Kosik KS, Davidson BL. MicroRNAs potentiate neural development. Neuron. 2009;64:303–309. doi: 10.1016/j.neuron.2009.10.020. [DOI] [PubMed] [Google Scholar]
- Ghislain J, Desmarquet-Trin-Dinh C, Jaegle M, Meijer D, Charnay P, Frain M. Characterisation of cis-acting sequences reveals a biphasic, axon-dependent regulation of Krox20 during Schwann cell development. Development. 2002;129:155–166. doi: 10.1242/dev.129.1.155. [DOI] [PubMed] [Google Scholar]
- Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308:833–838. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A. 2005;102:10898–10903. doi: 10.1073/pnas.0504834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KS, Zhang Z, McManus MT, Harfe BD, Sun X. Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci U S A. 2006;103:2208–2213. doi: 10.1073/pnas.0510839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Joksimovic M, Yun BA, Kittappa R, Anderegg AM, Chang WW, Taketo MM, McKay RD, Awatramani RB. Wnt antagonism of Shh facilitates midbrain floor plate neurogenesis. Nat Neurosci. 2009;12:125–131. doi: 10.1038/nn.2243. [DOI] [PubMed] [Google Scholar]
- Kao SC, Wu H, Xie J, Chang CP, Ranish JA, Graef IA, Crabtree GR. Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science. 2009;323:651–654. doi: 10.1126/science.1166562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA, feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau P, Verrier JD, Nielsen JA, Johnson KR, Notterpek L, Hudson LD. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J Neurosci. 2008;28:11720–11730. doi: 10.1523/JNEUROSCI.1932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci U S A. 2005a;102:2596–2601. doi: 10.1073/pnas.0407836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Svaren J, LaPash C, Araki T, Schmidt RE, Milbrandt J. Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci. 2005b;8:932–940. doi: 10.1038/nn1490. [DOI] [PubMed] [Google Scholar]
- LeBlanc SE, Jang SW, Ward RM, Wrabetz L, Svaren J. Direct regulation of myelin protein zero expression by the Egr2 transactivator. J Biol Chem. 2006;281:5453–5460. doi: 10.1074/jbc.M512159200. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Liu A, Li J, Marin-Husstege M, Kageyama R, Fan Y, Gelinas C, Casaccia-Bonnefil P. A molecular insight of Hes5-dependent inhibition of myelin gene expression: old partners and new players. EMBO J. 2006;25:4833–4842. doi: 10.1038/sj.emboj.7601352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager GM, Ward RM, Srinivasan R, Jang SW, Wrabetz L, Svaren J. Active gene repression by the Egr2.NAB complex during peripheral nerve myelination. J Biol Chem. 2008;283:18187–18197. doi: 10.1074/jbc.M803330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- O'Rourke JR, Georges SA, Seay HR, Tapscott SJ, McManus MT, Goldhamer DJ, Swanson MS, Harfe BD. Essential role for Dicer during skeletal muscle development. Dev Biol. 2007;311:359–368. doi: 10.1016/j.ydbio.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Droggiti A, Dickinson S, D'Antonio M, Mirsky R, Jessen KR. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J Cell Biol. 2004;164:385–394. doi: 10.1083/jcb.200307132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR. c-Jun is a negative regulator of myelination. J Cell Biol. 2008;181:625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE, Ruvkun G. Control of developmental timing by micrornas and their targets. Annu Rev Cell Dev Biol. 2002;18:495–513. doi: 10.1146/annurev.cellbio.18.012502.105832. [DOI] [PubMed] [Google Scholar]
- Potzner MR, Griffel C, Lütjen-Drecoll E, Bösl MR, Wegner M, Sock E. Prolonged Sox4 expression in oligodendrocytes interferes with normal myelination in the central nervous system. Mol Cell Biol. 2007;27:5316–5326. doi: 10.1128/MCB.00339-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvkun G. The perfect storm of tiny RNAs. Nat Med. 2008;14:1041–1045. doi: 10.1038/nm1008-1041. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Wang JY, Le N, Baloh RH, Gustin JA, Schmidt RE, Milbrandt J. Misexpression of Pou3f1 results in peripheral nerve hypomyelination and axonal loss. J Neurosci. 2007;27:11552–11559. doi: 10.1523/JNEUROSCI.5497-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Shin JY, McManus MT, Ptácek LJ, Fu YH. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann Neurol. 2009;66:843–857. doi: 10.1002/ana.21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkumatava A, Stark A, Sive H, Bartel DP. Coherent but overlapping expression of microRNAs and their targets during vertebrate development. Genes Dev. 2009;23:466–481. doi: 10.1101/gad.1745709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–1128. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- Verrier JD, Lau P, Hudson L, Murashov AK, Renne R, Notterpek L. Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia. 2009;57:1265–1279. doi: 10.1002/glia.20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner LE, Mancias P, Butler IJ, McDonald CM, Keppen L, Koob KG, Lupski JR. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998;18:382–384. doi: 10.1038/ng0498-382. [DOI] [PubMed] [Google Scholar]
- Woodhoo A, Alonso MB, Droggiti A, Turmaine M, D'Antonio M, Parkinson DB, Wilton DK, Al-Shawi R, Simons P, Shen J, Guillemot F, Radtke F, Meijer D, Feltri ML, Wrabetz L, Mirsky R, Jessen KR. Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat Neurosci. 2009;12:839–847. doi: 10.1038/nn.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- Zhao X, He X, Han X, Yu Y, Ye F, Chen Y, Hoang T, Xu X, Mi QS, Xin M, Wang F, Appel B, Lu QR. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron. 2010;65:612–626. doi: 10.1016/j.neuron.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorick TS, Syroid DE, Brown A, Gridley T, Lemke G. Krox-20 controls SCIP expression, cell cycle exit and susceptibility to apoptosis in developing myelinating Schwann cells. Development. 1999;126:1397–1406. doi: 10.1242/dev.126.7.1397. [DOI] [PubMed] [Google Scholar]