


Abstract
Background
The hepatic stellate cell is the primary cell type responsible for the excessive formation and deposition of connective tissue elements during the development of hepatic fibrosis in chronically injured liver. Culturing quiescent hepatic stellate cells on plastic causes spontaneous activation leading to a myofibroblastic phenotype similar to that seen in vivo. This provides a simple model system for studying activation and transdifferentiation of these cells. The introduction of exogenous DNA into these cells is discussed controversially mainly due to the lack of systematic analysis. Therefore, we examined comparatively five nonviral, lipid-mediated gene transfer methods and adenoviral based infection, as potential tools for efficient delivery of DNA to rat hepatic stellate cells and their transdifferentiated counterpart, i.e. myofibroblasts. Transfection conditions were determined using enhanced green fluorescent protein as a reporter expressed under the transcriptional control of the human cytomegalovirus immediate early gene 1 promoter/enhancer.
Results
With the use of chemically enhanced transfection methods, the highest relative efficiency was obtained with FuGENE™6 gene mediated DNA transfer. Quantitative evaluation of representative transfection experiments by flow cytometry revealed that approximately 6% of the rat hepatic stellate cells were transfected. None of the transfection methods tested was able to mediate gene delivery to rat myofibroblasts. To analyze if rat hepatic stellate cells and myofibroblasts are susceptible to adenoviral infection, we have inserted the transgenic expression cassette into a recombinant adenoviral type 5 genome as replacement for the E1 region. Viral particles of this replication-deficient Ad5-based reporter are able to infect 100% of rat hepatic stellate cells and myofibroblasts, respectively.
Conclusions
Our results indicate that FuGENE™6-based methods may be optimized sufficiently to offer a feasible approach for gene transfer into rat hepatic stellate cells. The data further demonstrate that adenoviral mediated transfer is a promising approach for gene delivery to these hepatic cells.
Background
Transfection is the insertion of foreign molecules such as cDNAs or promoter constructs into eukaryotic cells. This method has become a powerful experimental tool for studying gene functions and to analyze the control of gene expression. Genes of interest can either be transfected transiently or stable into cultured mammalian cells. Detailed protocols for efficient gene transfer to various primary cells and continuous cell lines, irrespective whether these cells are grown as monolayers or in suspension have been established during the last decades. Many methods have been developed to overcome the low transfection efficiency in differentiated cells if classical approaches, such as calcium-phosphate-DNA coprecipitates [1], diethylaminoethyl (DEAE)-dextran [2] or polylysine mediated gene transfer [3] are applied. Transfection by electroporation, microinjection, biolistic particle delivery, activated dendrimers or cationic liposomes are useful for established cell lines [4,5,6,7,8]. However, in cells of primary culture most of these transfection systems proved to be inefficient. Direct introduction of genes into rat hepatic stellate cells (rHSCs) and their transdifferentiated phenotype, i.e. the rat myofibroblasts (rMFBs) is difficult to achieve, in part due to the quiescent and fragile phenotype of rHSCs or the extracellular matrix in which the rMFBs are embedded. In normal liver, quiescent HSCs (also called Ito cells, lipocytes, fat-storing cells) are the precursor cells for MFBs, which are responsible for the dramatic increase in the synthesis of extracellular matrix proteins in cirrhotic livers [9,10]. Upon fibrogenic stimuli, HSCs become activated, a process in which they loose vitamin A granules, proliferate, change morphologically into MFBs, and increase their synthesis of extracellular matrix proteins [11, 12]. Culture of quiescent HSCs on a plastic surface also results in spontaneous activation of these cells similar to that seen in liver fibrosis in vivo. Efficient gene delivery to cultured rHSCs and rMFBs would therefore be of great interest for studying the processes involved in hepatic fibrogenesis and for gene-therapeutic devices. To compare various transfection mediators for their potential to increase the efficiency of gene delivery to rHSC and rMFB we used the reporter plasmid pEGFP-C1 expressing the enhanced green fluorescent protein (EGFP) from the jellyfish Aequorea victoria as a reporter expressed under transcriptional control of the ubiquitously active human cytomegalovirus (CMV) immediate early gene 1 promoter. Transfections were performed with the commercially available cationic liposome reagents Effectene, LipofectAmine Plus, Superfect, a classical calcium phosphate based method with and without glycerol shock, and the lipid-based reagent FuGENE™6, respectively. Furthermore, we cloned an adenovirus type 5 reporter construct (Ad5-CMV-EGFP) harboring the CMV/EGFP transgene and showed that high levels of gene transfer can be achieved in rHSC/rMFB with recombinant replication-deficient viral particles generated thereof. Taken together, we conclude that (i) gene delivery to rHSC can be performed by transfection with FuGENE™6 as mediator and that (ii) Ad5-mediated gene delivery can serve as a useful tool for introduction foreign DNA into cultured rHSC/rMFB, particularly if high gene delivery rates are required.
Results and discussion
To analyze the benefits and disadvantages of various transfection vehicles the efficiency of gene delivery to rHSC and rMFB was determined using the reporter plasmid pEGFP-C1 expressing the enhanced green fluorescent protein (EGFP) under transcriptional control of the human cytomegalovirus (CMV) promoter. We compared the cationic liposome reagents Effectene, LipofectAmine Plus, Superfect, a classical calciumphosphate based method with and without glycerol shock, and the lipid-based reagent FuGENE™6 applying essentially the protocols given by the manufacturers. As control for evaluation of the suitability of each transfection protocol we transfected NIH/3T3 cells in parallel under the same conditions. This continuous cell line of murine embryonic fibroblastic origin has been reported previously to be highly transfectable with all reagents used in this study. Forty-eight hours after transfection we analyzed the morphology and viability of cells. Phase contrast microscopy revealed that rHSCs/rMFBs remained viable and continued to grow normally. To estimate the rate of transfection we counted EGFP positive and negative cells in the fluorescence microscopy. Although transfection efficiencies for the established cell line NIH/3T3 was in the expected range (Effectene ~9.5%, LipofectAmine Plus ~7.5%, Superfect ~2.5%, calcium-phosphate/ -glycerol shock ~8%, calcium-phosphate/+glycerol shock ~12%, FuGENE™6 ~30%) there was obviously an overall low transfection rate of rHSCs and rMFBs.
Interestingly, only FuGENE™6 was able to mediate a significant gene transfer (~ 6%) to rHSCs (Fig. 1). None of the mediators tested was able to provide sufficient gene delivery in rMFBs. FACS analysis was used to estimate the percentage of transfected cells. EGFP has a single red-shifted excitation peak at 488 nm which is suitable for flow cytometric analysis by argon ion laser excitation and a standard FITC filter set [13]. The viability of transfected cells was proven by double staining with propidium iodide (PI), which selectively stains dead cells. Fluorescence signals were acquired using a 488 nm excitation and a 530 ± 30 nm emission fluorescence filter for EGFP and a 630 ± 11 nm emission fluorescence filter for PI, respectively. This "double staining" protocol allows to discriminate between viable but not transfected cells, non viable and not transfected cells, viable transfected cells and dead transfected cells. Representative results obtained after transfection with FuGENE™6 as vehicle are summarized in Fig. 2. In agreement with our microscopic results (see above) 6% of rHSC were found to be positive for EGFP. The transfected cells are viable as evidenced by the lack of PI staining. Furthermore, the flow cytometric analysis revealed that the non viable rHSC were negative for EGFP. The fraction of about 11% dead rHSC may be due to destructive effects of trypsinization and washing procedures on these fragile cells. rMFB were transfectable neither with FuGENE™6 nor with the other non viral transfection systems tested. The fraction of 30% transfected NIH/3T3 cells documents the correct handling of the transfection protocol.
Figure 1.
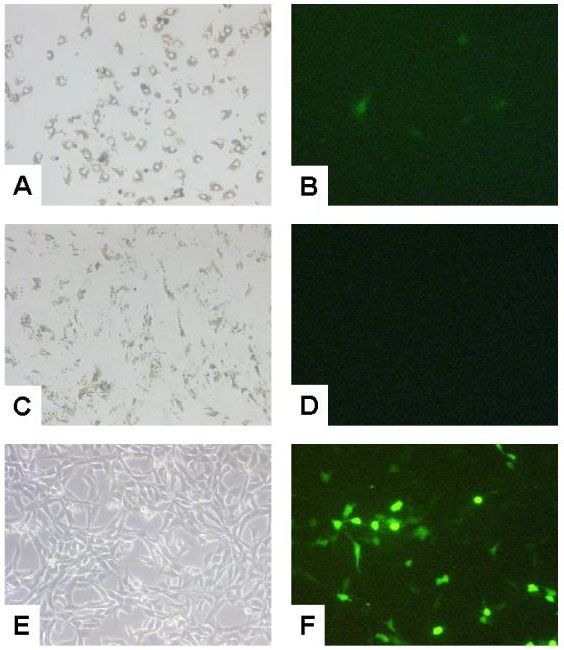
Representative analysis of transfection efficiencies of rat hepatic stellate cells and myofibroblasts using FuGENE™6 transfection procedure. Rat hepatic stellate cells (rHSCs) were isolated from adult male Sprague-Dawley rats by the pronase-collagenase method and a single-step density gradient centrifugation. Rat myofibroblasts (rMFBs) were prepared by subculturing primary rHSCs by trypsinization at day 7 after seeding following spontaneous activation on a plastic surface. The established murine cell line NIH/3T3 served as a reporter transfection cell line. Cells were transfected with 2 μg of reporter construct pEGFP-C1 and FuGENE™6 according to the suppliers instructions. After incubation for proposed time media were changed and transfection efficiencies were monitored 48 hours after transfection. Representative phase contrast microscopy (A, C, E) and fluorescence microscopy (B, D, F) of rHSCs (A, B), rMFBs (C, D) and NIH/3T3 cells (E, F) are shown. The intensity of fluorescence varies among transfected cells indicating various levels of reporter expression.
Figure 2.

Representative results of flow cytometric analysis of transfected rat hepatic stellate cells and rat myofibroblasts in comparison to NIH/3T3 cells using FuGENE™6 vehicle. For this experiment rHSCs, rMFBs and NIH/3T3 cells were transfected 2 days after seeding with 2 μg reporter plasmid complexed with 5 μl FuGENE™6. FACS analysis was performed 48 hours after transfection. Cultured cells were trypsinized under standard conditions and flow cytometric measurements were performed immediately after collection of cells. Fluorescence signals were recorded with a flow cytometer FACS-Calibur (Becton Dickinson, Sparks, MD) using a 488 nm excitation and a 530 ± 30 nm emission fluorescence filter for enhanced green fluorescence protein (EGFP) and a 630 ± 11 nm emission fluorescence filter for propidium iodide (PI), respectively. Data were acquired and analyzed with the CellQuest™ software version 3.1 (Becton Dickinson). Histograms of fluorescence intensities in EGFP (A, C, E) and PI (B, D, F) channels are shown for rHSC (A, B), rMFB (C, D) and reporter cell line NIH/3T3 (E, F), respectively. To establish background for fluorescence and to set gates for data acquisition, mock-transfected cells (not shown) were used. Mean fluorescence intensity was used to calculate levels of EGFP expression. Cells that took up PI were deemed nonviable. Nontransfected cells did not show fluorescence in EGFP channel.
It was previously shown that the lipid formulation FuGENE™6 is able to promote DNA uptake by primary thyrocytes and articular chondrocytes known to be refractory to most transfection systems [14, 15]. In our hands none of the five transfection systems tested was able to (>0.5%) provide significant gene delivery to rMFBs. Human MFBs isolated from outgrowths of cirrhotic liver pieces and cells from the established line CFSC [16] derived from cirrhotic rHSCs were highly transfectable with all mediators (not shown). A potential reason for the observed higher transfection susceptibility of human MFBs and immortalized rMFBs might be the difference in the rate of cell proliferation. A connection between effective gene transfer and mitotic activity was recently demonstrated in various cells and is also well documented in smooth muscle cells [17, 18].
To test if adenoviral gene delivery is a suitable alternative to overcome the observed insufficient gene delivery rates we analyzed the susceptibility of rHSCs and rMFBs to adenoviruses. To facilitate the insertion of the CMV/EGFP reporter cassette within an appropriate adenoviral shuttle vector we cut pEGFP-C1 with AsnI and SspI, filled in the resulting 1.66-kbp fragment with Klenow DNA polymerase and inserted this fragment into the filled in EcoRI site of vector pΔE1sp1A (Fig. 3A) [19]. The orientation of the reporter transgene and the cloning boundaries in the resulting vector pΔE1sp1A-CMV-EGFP were confirmed by digestion with multiple restriction endonucleases and sequencing. We then cotransfected vector pΔE1sp1A-CMV-EGFP and backbone vector pJM17 [20] into the packaging cell line 293 [21], which supplies the missing adenoviral E1 gene products necessary to generate recombinant viral particles (Fig. 3B and 3C). Based on reports describing that the integrated colinear Ad5-segment spanning bp 1 to bp 4344 of Ad5 genome [22] is transcriptionally silenced or lost frequently in 293 cultures at high passage numbers we first tested whether corresponding Ad5-genes (E1A and E1B) were actively transcribed in the 293 cells used in this study. Therefore, we performed a Northern blot analysis (Fig. 4) with total RNA of 293 cells and the 2.8-kbp EcoRI/HindIII fragment of vector pAd5SalIB containing bp 1 to bp 2804 of Ad5 genome (cf. GenBank Accession number X02996.1) [23]. The sizes of observed hybridization signals (1.3 kb and 2.6 kb) expressed at high levels are in agreement with the complexity of the primary transcripts for E1A (1134 nt) and E1B (2369 nt) genes if in addition 200 nt for poly A+-tail are estimated. In the murine control cell line NIH/3T3 transcripts of E1A or E1B are virtually absent due to the absence of Ad5 sequences in these cells. The Northern analysis therefore revealed that the 293 cells used in this study are able to produce relevant mRNAs for both trans-activator genes. Twenty four hours after cotransfection of pΔE1sp1A-CMV-EGFP and pJM17 approximately 40% of 293 cells were positive for EGFP (not shown). Six days later the typical viral focies which are able to produce recombinant viral particles were observed and 7-12 days after transfection all cells were infected by developing recombinant Ad5-CMV-EGFP particles. Viral particles were isolated, amplified once on 293 cells and purified by standard procedures.
Figure 3.

Construction of a replication-defective recombinant adenovirus expressing enhanced green fluorescent protein under transcriptional control of the human cytomegalovirus immediate-early gene 1 promoter/enhancer.(A) In the mammalian reporter vector pEGFP-C1 the enhanced green fluorescent protein (EGFP) is expressed under control of the human cytomegalovirus immediate-early gene 1 promoter (PCMV). Abbreviations are: Kan/Neo, kanamycin/neomycin resistance genes; Amp, ampicillin resistance gene; pUC ori, E. coli origin of replication; SV40 ori, simian virus 40 origin of replication; SV40 polyA, simian virus 40 polyadenylation signal; HSVTK polyA, herpes simplex thymidine kinase polyadenylation signal. The adenoviral shuttle vector pΔE1sp1A contains Ad5 sequences from bp 22 (0 mu) to bp 5790 (16.1 mu) with a deletion of E1 sequences (ΔE1) from bp 342 to bp 3523 (1.0 - 9.8 mu) and a selectable ampicillin resistance gene (Amp). A multiple cloning site (MCS) containing unique restriction sites for ClaI, BamHI, XhoI, XbaI, EcoRV, EcoRI, HindIII, SalI, and BglII is embedded in the Ad5-sequences. For construction shuttle vector pΔE1sp1A-CMV-EGFP the 1656-bp AsnI/SspI fragment from plasmid pEGFP-C1 was filled in by Klenow DNA polymerase and cloned into the EcoRI digested and filled in pΔE1sp1A vector.(B) For integration of the reporter cassette from pE1Δsp1A-CMV-EGFP into the Ad5 backbone plasmid vector pJM17 both plasmids were cotransfected into human embryo kidney cell line 293 leading to homologous recombination between common Ad5 regions. Generation of recombinant viral particles were visualized by an increase of EGFP-positive cells and by viral focus formation in fluorescence microscopy. (C) Released replication-defective viral particles are infectious and are capable to deliver the CMV-EGFP cassette to target cells. The nucleotide sequence of the cloned vector pΔE1sp1A-CMV-EGFP is deposited in GenBank (Accession number AF288620).
Figure 4.

Expression of E1A and E1B transactivators in human embryo kidney cell line 293. A Northern blot analysis using equal amounts (30 μg) of total cellular RNA from human embryo kidney 293 cells (HEK293) and NIH/3T3 cells is shown. The blotted RNAs were hybridized with 3.5 × 107 cpm of a [α-32P]dCTP-labelled 2.8-kbp EcoRI/HindIII fragment of clone pAd5SalIB23 containing complete E1A-gene and partial E1B-gene of Ad5. The autoradiograph was exposed for 3 hours using an intensifying screen. The RNAs were rehybridized with 1.5 × 107 cpm of a [α-32P]dCTP-labelled GAPDH-specific cDNA probe, and filter was re-exposed for 3 hours.
To test the susceptibility of cultured rHSC/rMFB we infected them two days after seeding using a multiplicity of infection (MOI) of 10. Analysis of the cells for forty-eight hours after infection revealed that 100% of the cells (rHSCs and rMFBs) were positive for EGFP providing evidence that adenoviral based vectors are very effective in gene delivery to these cells in culture (Fig. 5). However, it was surprising that the use of adenovirus at a MOI of 10 was sufficient for complete infection. It is well established that the internalization of adenoviral particles is promoted by integrins [24]. Also, it was previously demonstrated that HSC and MFB express high levels of various types of integrins [25, 26]. Therefore, it is most likely that the observed high adenoviral susceptibility of HSC and MFB may be caused by high-level expression of integrins. Interestingly, also the relative intensities of fluorescence signals in these cells were extremely high compared to those observed after chemically enhanced gene delivery (compare Fig. 5B and Fig. 1B). Infected cells continued to express the product of the transgene (EGFP) for at least 2 weeks. It is most likely that the termination of transgene expression after 2 weeks is related to the loss of episomal transgene, which is not incorporated into the host genome and, therefore, is lost with cell division. Partial cellular disintegration occurred (not shown) at later stages most likely due to the intrinsic cytotoxicity of high EGFP content or the overexpression of some viral proteins [27]. Independent experiments revealed further that infection of these sinusoidal hepatic cells is highly reproducible, irrespective whether infection is performed in the presence of 2%, 5% or 10% fetal calf serum (not shown).
Figure 5.

Adenoviral infection of rHSCs and rMFBs with Ad5-CMV-EGFP. For infection of rHSCs/rMFBs with Ad5-CMV-EGFP 105 cells were seeded in 2 ml medium and infected 2 days later with 500 μl viral stock containing approximately 106 plaque forming units. Representative phase-contrast microscopy (A, C) and fluorescence microscopy (B, D) of rHSC (A, B) and rMFB (C, D) 48 hours after adenoviral infection are shown. Mock infected rHSCs or rMFBs are negative for EGFP-expression (not shown).
Based on these data we conclude that a MOI of 10 is sufficient to achieve an infection of 100% in cultured rHSC and rMFB, respectively, under most experimental conditions.
Conclusions
In agreement with other reports on gene delivery to cultured rHSCs by adenoviral based techniques our data show that this method is straightforward particularly when high efficiency of gene transfer is required [28,29,30,31]. Furthermore, our report indicates that introduction of foreign DNA even into rMFBs is possible by use of adenoviral based vector systems. Because of the considerable interest on the rHSC/rMFB transition as a cell culture model for liver fibrogenesis the improvement of efficiency of gene delivery to these cells should facilitate applications such as reproducible reporter vector assays, or bulk expression of signalling proteins for biochemical or cell biological assays.
Additional studies will be required to determine the optimal in vivo conditions for adenoviral gene transfer to rHSCs/rMFBs.
Material and Methods
Isolation and culture of rat hepatic stellate cells
All animal protocols were in full compliance with the guidelines for animal care and were approved by the Animal Care Committee from the government. Adult male Sprague-Dawley rats (Harlan Winkelmann GmbH, Borchen, Germany), which had free access to altromin chow (Altromin GmbH, Lage, Germany) and water were anaesthetized with an intramuscular injection of 100 mg/kg body weight ketaminhydrochloride (Ketavet®; SANOFI-CEVA GmbH, Düsseldorf, Germany) and 7 mg/kg body weight xylazinhydrochloride (Rompun®; BayerVital, Leverkusen, Germany). rHSCs were isolated by the pronase-collagenase method, a two-step method for preparation of rat liver cells comprising a first step in which the liver is pre-perfused with a Ca2+-free medium in order to remove intercellular Ca2+, and a second step in which the liver is perfused with a Ca2+ requiring collagenase mixture [32, 33, 34]. Briefly, livers were perfused at 10 ml/minute with 200 ml Hanks balanced salt solution (HBSS) without Ca2+and Mg2+ (PAA Laboratories GmbH, Linz, Austria), 100 ml solution E [0.35% (w/v) pronase E in HBSS with Ca2+ and Mg2+] followed by recirculating perfusion for approximately 30 minutes with 60 ml solution F [0.015% (w/v) collagenase in HBSS with Ca2+ and Mg2+]. Thereafter, livers were dissected under permanent pH control (pH 7.3) in 100 ml HBSS with Ca2+ and Mg2+ containing 10 mg DNase, type II (Roche, Mannheim, Germany). The cell suspension was subsequently filtered through nylon mesh (250 μm and 100 μm) and centrifuged for 7 minutes at 450 × g at 4°C. The resulting pellet was washed in ice cold solution G containing 0.25% (w/v) bovine serum albumin in HBSS/+Ca2+/+Mg2+. rHSCs were purified by single-step density gradient centrifugation with 8.25% (w/v) Nycodenz® (Nycomed Pharma AS, Oslo, Norway) as described in detail elsewhere [33]. rHSCs were carefully collected by aspiration of the white top layer of the gradient. The purity of cell preparations was assessed by light microscopic appearance, vitamin A autofluorescence of the cells and positive immunofluorescence stainings [35]. rMFBs were prepared by subculturing primary rHSCs by trypsinization at day 7 after seeding following spontaneous activation on the plastic surface. rHSCs and rMFBs were cultured in Dulbecco's modified Eagle medium (DMEM; BioWhittaker Europe, Verviers, Belgium) containing 4 mM L-glutamine and 10% fetal calf serum (Seromed, Biochrom KG, Berlin, Germany). Additionally, all culture media were supplemented with penicillin (100 IU/ml) and streptomycin (100 μg/ml), respectively. All cultures were maintained at 37°C and 5% CO2 in a humidified atmosphere.
The murine fibroblast cell line NIH/3T3 [36] was purchased from the American Type Culture Collection (Rockville, MD) and maintained in culture at 37°C in DMEM supplemented with 10% fetal calf serum, 4 mM L-glutamine, penicillin (100 IU/ml) and streptomycin (100 μg/ml). All transfections of NIH/3T3 cells with various transfection agents were performed at approximately 60% confluency.
Transfection of hepatic stellate cells
The reporter construct pEGFP-C1 (GenBank Accession number U55763) was purchased from CLONTECH (Palo Alto, CA). rHSCs were transfected two days after seeding, rMFBs cells two days after subculturing (see above) with 2 μg plasmid DNA. The DNA was isolated by a standard alkaline extraction method or a cesium chloride gradient purification protocol. Transfections were performed with the commercially available cationic liposome reagents Effectene (Qiagen, Hilden, Germany), LipofectAmine Plus (Gibco Life Technologies, Karlsruhe, Germany), Superfect (Qiagen), the lipid-based reagent FUGENE™6 (Roche), and a classical calciumphosphate based method (Promega, Madison, WI) with and without glycerol shock, following essentially the instructions of the manufacturers. For glycerol shock the medium was removed, cells were rinsed in HBSS/-Ca2+/-Mg2+ three times and overlayed with 15% glyerol in HBSS/-Ca2+/-Mg2+for 2 minutes at 37°C. Thereafter, glycerol solution was removed and cells were washed three times with HBSS/-Ca2+/-Mg2+prior addition of regular growth medium. Transfection efficiencies were monitored 48 hours later by fluorescence microscopy and flow cytometry analysis.
Microscopic data documentation
Microscopic images shown are from representative experiments. For visual observation of transfected and infected cells expressing EGFP, we typically used a 40 × objective lens and a 10 × eyepiece lens. Documentation of representative eye fields had been performed with a HV-C20 digital camera (HITACHI Denshi Ltd., Tokyo, Japan) and the DISKUS software package version 4.14 (Hilgers, Königswinter, Germany).
Flow Cytometry
Flow cytometric measurements were performed immediately after collection of cultured cells. Cells were trypsinized under standard conditions and washed once with HBSS/-Ca2+/-Mg2+. To get informations about viability, collected cells were additionally stained with propidium iodide (PI) (Sigma, Deisenhofen, Germany). Fluorescence signals were recorded with a flow cytometer FACS-Calibur (Becton Dickinson, Sparks, MD) using a 488 nm excitation and a 530 ± 30 nm emission fluorescence filter for EGFP and a 630 ± 11 nm emission fluorescence filter for PI, respectively. Data were acquired and analyzed with the CellQuest™ software version 3.1 (Becton Dickinson).
Construction of replication-defective recombinant adenoviruses
Plasmids pΔE1sp1A [19] and pJM17 [20] for construction of adenoviral vector were obtained from Microbix Biosystems Inc. (Toronto, Ontario, Canada). For construction the adenoviral reporter shuttle vector pΔE1sp1A-CMV-EGFP the 1656-bp AsnI/SspI fragment from plasmid pEGFP-C1 was filled in by Klenow DNA polymerase and cloned into the EcoRI digested and Klenow filled in pΔE1sp1A. The orientation of the CMV-EGFP expression cassette in pΔE1sp1A-CMV-EGFP and the integrity of cloning boundaries were verified by restriction analysis and sequencing with flanking primers 5'-d(GCGTAACCGAGTAAGAATTTG)-3' and 5'-d(GGCGACCATCAATGCTGGAG)-3', respectively. Primers were obtained from MWG- Biotech AG (Ebersberg, Germany) and sequence reactions were done with the ABI PRISM BigDye® termination reaction kit (PE Applied Biosystems, Weiterstadt, Germany). Integration of vector sequences from pΔE1sp1A-CMV-EGFP into adenoviral backbone vector pJM17 for production of replication-deficient virus particles were performed by in vitro homologous recombination in the human embryo kidney cell line 293, which constitutively express the E1 gene products required for propagation of recombinant adenoviruses [21]. Briefly, approximately 5 × 105 293 cells were seeded in six-well dishes and cotransfected with 2 μg of plasmid pΔE1sp1A-CMV-EGFP, 2 μg pJM17 and 10 μl of the FuGENE™6 reagent. After 16 hours at 37°C, the media containing the transfection mix was removed, and 2 ml of growth medium was added and cells were cultured for 10-12 days with subsequent addition of fresh medium. Generation of recombinant viral particles were visualized by increase of EGFP-positive cells and by viral foci formation in fluorescence microscopy. After total infection viral particles were released from cells by three rounds of a freeze-thaw cycle. Viral particles were separated from cell debris by centrifugation at 3000 rpm for 10 minutes. To generate higher titer viral stocks, 293 cells were re-infected at a multiplicity of infection (MOI) of 1 and grown for 3-4 days, at which time viruses were harvested as described above.
Adenoviral infection
Viral stock solutions were titered on 293 cells in growth medium containing 10% fetal calf serum following standard procedures [37] and kept frozen at -20°C until use. For infection of rHSCs/rMFBs with adenoviral particles 105 cells were seeded in six-well dishes with 2 ml medium and cells were infected 2 days later with 500 μl viral stock containing approximately 10 6 plaque forming units of Ad5-CMV-EGFP.
RNA isolation and Northern analysis
Cells from human cell line 293 and murine cell line NIH/3T3 were rinsed with ice cold HBSS/-Ca2+/-Mg2+, and then lysed in 1 ml per 106 cells of lysis buffer containing 25 mM sodium acetate pH 6.0, 4 M guanidine thiocyanate and 0.835% (v/v) β-mercaptoethanol. The lysate was adjusted to 8 ml with lysis buffer, pressed three times through a 20-g needle, layered onto a 4-ml cushion of a solution containing 25 mM sodium acetate pH 6.0 and 5.7 M cesium chloride, and then centrifuged for approximately 24 h at 21°C and 25000 rpm in a Beckman SW41-type rotor. RNA pellets were resuspended in 300 mM sodium acetate (pH 6.0), ethanol precipitated and resuspended in water. The concentration of RNA was determined by absorbance at 260 nm.
For Northern analysis, 30 μg of total cellular RNAs were separated by electrophoresis in a 1.2% (w/v) denaturing agarose gel, transferred to a Hybond-N membrane (Amersham Pharmacia Biotech, Freiburg, Germany) and fixed by baking for 2 h at 80°C. Blots were pre-hybridized for at least 3 h, and then hybridized for 16 h at 37°C to [α-32P]dCTP-labelled probes (Multiprime DNA labelling system; Amersham) in a buffer containing 50% (v/v) formamide, 6 × SSC, 5 × Denhardt's solution [0.1% (w/v) Ficoll 400, 0.1% (w/v) BSA, 0.1% polyvinylpyrrolidone], 5 mM EDTA, 0.5% (w/v) SDS, and 100 μg/ml sheared denatured herring sperm DNA (Roche). Filters were washed once at 55°C for 20 minutes in a solution containing 2 × SSC, 1 mM EDTA, and 0.1% (w/v) SDS, then twice at 50°C for 20 minutes in a solution containing 0.4 × SSC, 1 mM EDTA, and 0.1% (w/v) SDS. Autoradiographs were exposed for indicated times to Kodak X-OMAT AR films at -80°C using intensifying screens. As an internal standard (loading control) the blots were re-hybridized with a GAPDH specific cDNA.
Acknowledgments
Acknowledgements
The authors are grateful for helpful comments from Prof. Walter Doerfler (Institute of Genetics, University of Cologne, Germany) during the initial planning of the project and Dr. F. Fallaux (Department of Molecular Cell Biology, Leiden University, The Netherlands) for sending clone pAd5SalIB. Experiments dealing with adenoviral constructs are covered by permission of the Landesumweltamt Nordrhein-Westfalen (Az. 521-K-1.59/99), solely indicating this fact. This work was supported by grants from the Deutsche Forschungsgemeinschaft to RW (We2554-1) and to AMG (SFB-542).
Sequence data from this article have been deposited with the GenBank Data Library under accession number AF288620.
Contributor Information
Ralf Weiskirchen, Email: rweiskirchen@post.klinikum.rwth-aachen.de.
Jens Kneifel, Email: rweiskirchen@post.klinikum.rwth-aachen.de.
Sabine Weiskirchen, Email: weiskirchen@t-online.de.
Eddy van de Leur, Email: eddy.vandeleur@t-online.de.
Dagmar Kunz, Email: dagmar.kunz@post.klinikum.rwth-aachen.de.
Axel M Gressner, Email: gressner@rwth-aachen.de.
References
- Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Vaheri A, Pagano JS. Infectious poliovirus RNA: a sensitive method of assay. Virology. 1965;27:434–436. doi: 10.1016/0042-6822(65)90126-1. [DOI] [PubMed] [Google Scholar]
- Zhou XH, Klibanov AL, Huang L. Lipophilic polylysines mediate efficient DNA transfection in mammalian cells. Biochim Biophys Acta. 1991;1065:8–14. doi: 10.1016/0005-2736(91)90003-Q. [DOI] [PubMed] [Google Scholar]
- Neumann E, Schaefer-Ridder M, Wang Y, Hofschneider PH. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982;1:841–845. doi: 10.1002/j.1460-2075.1982.tb01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capecchi MR. The new mouse genetics: altering the genome by gene targeting. Trends Genet. 1989;5:70–76. doi: 10.1016/0168-9525(89)90029-2. [DOI] [PubMed] [Google Scholar]
- Burkholder JK, Decker J, Yang NS. Rapid transgene expression in lymphocyte and macrophage primary cultures after particle bombardment-mediated gene transfer. J Immunol Methods. 1993;165:149–156. doi: 10.1016/0022-1759(93)90340-D. [DOI] [PubMed] [Google Scholar]
- Tang MX, Redemann CT, Szoka FC., Jr In vitro gene delivery by degraded polyamidoamine dendrimers. Bioconjug Chem. 1996;7:703–714. doi: 10.1021/bc9600630. [DOI] [PubMed] [Google Scholar]
- Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci USA. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston.The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. New Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- Gressner AM. Perisinusoidal lipocytes and fibrogenesis. Gut. 1994;35:1331–1333. doi: 10.1136/gut.35.10.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressner AM, Bachem MG. Molecular mechanisms of liver fibrogenesis - A homage to the role of activated fat-storing cells. Digestion. 1995;56:335–346. doi: 10.1159/000201257. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Uyttersprot N, Costagliola S, Miot F. A new tool for efficient transfection of dog and human thyrocytes in primary culture. Mol Cell Endocrin. 1998;142:35–39. doi: 10.1016/S0303-7207(98)00122-1. [DOI] [PubMed] [Google Scholar]
- Madry H, Trippel SB. Efficient lipid-mediated gene transfer to articular chondrocytes. Gene Therapy. 2000;7:286–291. doi: 10.1038/sj.gt.3301086. [DOI] [PubMed] [Google Scholar]
- Greenwel P, Schwartz M, Rosas M, Peyrol S, Grimaud JA, Rojkind M. Characterization of fat-storing cell lines derived from normal and CCl4-cirrhotic livers. Differences in the production of interleukin-6. Lab Invest. 1991;65:644–653. [PubMed] [Google Scholar]
- Mortimer I, Tan P, MacLachlan I, Graham RW, Saravolac EG, Joshi PB. Cationic lipid-mediated transfection of cells in culture requires mitotic activity. Gene Therapy. 1999;6:403–411. doi: 10.1038/sj.gt.3300837. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Gal D, Leclerc G, Pickering JG, Riessen R, Weir L, Isner JM. Increased gene expression after liposome-mediated arterial gene transfer associated with intimal smooth muscle cell proliferation. In vitro and in vivo findings in a rabbit model of vascular injury. J Clin Invest. 1994;93:652–661. doi: 10.1172/JCI117017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett AJ, Haddara W, Prevec L, Graham FL. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrory WJ, Bautista DS, Graham FL. A simple technique for the rescue of early region 1 mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Louis N, Evelegh C, Graham FL. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology. 1997;233:423–429. doi: 10.1006/viro.1997.8597. [DOI] [PubMed] [Google Scholar]
- Bernards R, Houweling A, Schrier PI, Bos JL, Van der Eb AJ. Characterization of cells transformed by Ad5/Ad12 hybrid early region 1 plasmids. Virology. 1982;120:422–432. doi: 10.1016/0042-6822(82)90042-3. [DOI] [PubMed] [Google Scholar]
- Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- Carloni V, Romanelli RG, Pinzani M, Laffi G, Gentilini P. Expression and function of integrin receptors for collagen and laminin in cultured human hepatic stellate cells. Gastroenterology. 1996;110:1127–1136. doi: 10.1053/gast.1996.v110.pm8613002. [DOI] [PubMed] [Google Scholar]
- Iwamoto H, Sakai H, Nawata H. Inhibition of integrin signaling with Arg-Gly-Asp motifs in rat hepatic stellate cells. J Hepatol. 1998;29:752–759. doi: 10.1016/S0168-8278(98)80256-0. [DOI] [PubMed] [Google Scholar]
- Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to the living cells? Biochem Biophys Res Commun. 1999;260:712–717. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- Hellerbrand C, Jobin C, Iimuro Y, Licato L, Sartor RB, Brenner DA. Inhibition of NFκB in activated rat hepatic stellate cells by proteasome inhibitors and an IκB super-repressor. Hepatology. 1998;27:1285–1295. doi: 10.1002/hep.510270514. [DOI] [PubMed] [Google Scholar]
- Lang A, Schoonhoven R, Tuvia S, Brenner DA, Rippe RA. Nuclear factor κB in proliferation, activation, and apoptosis in rat hepatic stellate cells. J Hepatol. 2000;33:49–58. doi: 10.1016/S0168-8278(00)80159-2. [DOI] [PubMed] [Google Scholar]
- Stefanovic B, Hellerbrand C, Brenner DA. Regulatory role of the conserved stem-loop structure at the 5' end of collagen α1(I) mRNA. Mol Cell Biol. 1999;19:4334–4342. doi: 10.1128/mcb.19.6.4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Shao R, Qian HS, George SE, Rockey DC. Gene transfer of the neuronal NO synthase isoform to cirrhotic rat liver ameliorates portal hypertension. J Clin Invest. 2000;105:741–748. doi: 10.1172/JCI7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw AM, McCarthy SP, Geerts A, Knook DL. Purified rat liver fat-storing cells in culture divide and contain collagen. Hepatology. 1984;4:392–403. doi: 10.1002/hep.1840040307. [DOI] [PubMed] [Google Scholar]
- Schafer S, Zerbe O, Gressner AM. The synthesis of proteoglycans in fat storing-cells of rat liver. Hepatology. 1987;7:680–687. doi: 10.1002/hep.1840070411. [DOI] [PubMed] [Google Scholar]
- Seglen PO. Preparation of rat liver cells, II. Effects of ions and chelators on tissue dispersion. Exp Cell Res. 1973;76:25–30. doi: 10.1016/0014-4827(73)90414-x. [DOI] [PubMed] [Google Scholar]
- Zerbe O, Gressner AM. Proliferation of fat storing cells is stimulated by secretions of Kupffer cells from normal and injured liver. Exp Mol Pathol. 1988;49:87–101. doi: 10.1016/0014-4800(88)90023-8. [DOI] [PubMed] [Google Scholar]
- Todaro GJ, Green MD. Quantitative studies of the growth of mouse embryo cells in culture and their development into established line. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]