

Abstract
Na+-NQR is a unique respiratory enzyme that couples the free energy of electron transfer reactions to electrogenic pumping of sodium across the cell membrane. This enzyme is found in many marine and pathogenic bacteria where it plays an analogous role to the H+-pumping complex I. It has generally been assumed that the sodium pump of Na+-NQR operates on the basis of thermodynamic coupling between reduction of a single redox cofactor and the binding of sodium at a nearby site. In this study, we have defined the coupling to sodium translocation of individual steps in the redox reaction of Na+-NQR. Sodium uptake takes place in the reaction step in which an electron moves from the 2Fe-2S center to FMNC, while the translocation of sodium across the membrane dielectric (and probably its release into the external medium) occurs when an electron moves from FMNB to riboflavin. This argues against a single-site coupling model because the redox steps that drive these two parts of the sodium pumping process do not have any redox cofactor in common. The significance of these results for the mechanism of coupling is discussed, and we proposed that Na+-NQR operates through a novel mechanism based on kinetic coupling, mediated by conformational changes.
Keywords: electron transfer, ion coupling, sodium transport
The sodium pumping NADH:quinone oxidoreductase (Na+-NQR) is the main entry site for electrons to the respiratory chain of Vibrio cholerae and other marine and pathogenic bacteria. As a redox-driven sodium pump, Na+-NQR plays an important role in both the bioenergetics and homeostasis of these organisms (1–3). This enzyme catalyzes the oxidation of NADH and the reduction of CoQ (4), analogous to the reaction catalyzed by complex I of mitochondria and of other prokaryotes. In contrast with complex I, which pumps H+, Na+-NQR specifically translocates Na+, with a stoichiometry of one Na+ per electron (5).
Na+-NQR is composed of six subunits and contains a noncovalently bound FAD, a 2Fe-2S center, two FMN’s covalently bound to the NqrB and NqrC subunits (FMNB and FMNC, respectively), as well as noncovalently bound riboflavin. Riboflavin as cofactor has not been reported in any other enzyme (6–13). The covalently bound FMN molecules can each give rise to anionic radicals (FMNB, anionic radical I and FMNC, anionic radical II), while riboflavin is found as a stable neutral radical (RibH•) in the oxidized form of the enzyme (as prepared) (1, 8). Studies carried out by our group with mutants that do not incorporate individual cofactors have shown that electrons move through the enzyme following the pathway: FAD → 2Fe-2S → FMNC → FMNB → Riboflavin (Fig. 1) (14).
Fig. 1.

Internal electron transfer pathway and Na+ uptake and release steps in Na+-NQR. 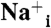 and
and  indicate the internal and external Na+.
indicate the internal and external Na+.
Although the coupling mechanism of most of the respiratory chain complexes and other redox pumps have been studied in detail (15–17), the fundamental mechanism of cation pumping in Na+-NQR might follow different rules because this is the only known example of a redox pump that translocates sodium instead of protons. The elucidation of this mechanism may open new windows for the understanding of cation pumping in other enzymes and could help to define general principles of biological transport as a whole.
A classical approach to study this question is to look for redox cofactors whose midpoint potentials vary as a function of the concentration of the pumped ion (18, 19). Interestingly, none of the equilibrium redox potentials in the Na+-NQR have been found to be dependent on bulk Na+ concentration (20, 21). This contrasts with the prediction of essentially all mechanisms that have been proposed for Na+-NQR, which would require changes in Na+ affinity to be directly coupled to the redox state of one of the cofactors. In these circumstances one promising approach is to study the kinetics of functional turnover using mutants that alter the translocation of sodium.
Here we show that Na+ uptake by Na+-NQR is a nonelectrogenic process that occurs upon electron transfer from the 2Fe-2S center to FMNC, while the electrogenic transport of Na+ across the membrane, and likely release into solution, occurs upon electron transfer from FMNB to riboflavin. These results, together with other recent findings in the field (21, 22), lead us to propose that Na+-NQR belongs to a distinct family of redox-driven ion pumps that operate through a conformationally mediated, kinetically coupled mechanism.
Results
ΔΨ Formation by Reconstituted Na+-NQR.
Wild-type Na+-NQR reconstituted in proteoliposomes is able to transport sodium (Fig. 2, trace i), and this process is insensitive to proton ionophores (CCCP) and abolished by sodium ionophores (ETH-157) (23). Surprisingly, the enzyme is also able to form an electrochemical gradient of sodium in the absence of CoQ (Fig. 2, trace ii), which indicates that the internal electron transfer process of the enzyme is itself electrogenic. This type of reaction is referred to as partial-turnover ΔΨ formation, although the membrane potential produced under these conditions could be equivalent to a single catalytic cycle. The gradient formation under partial turnover was abolished by the Na+ ionophore ETH-157 (5 μM, trace iv) but not by CCCP (trace iii). To confirm these results, the formation of ΔΨ was also measured using the fast-responding fluorescent probe RH421 with similar results (Fig. S1). The inhibitory effect of CoQH2 (see below) indicates that ΔΨ formation in the absence of CoQ is not the result of electrons leaking to oxygen at the Na+ pumping site of the enzyme. Also, the strict dependence of ΔΨ formation on Na+ and the direction of the gradient (positive inside the vesicles) rule out the possibility that the observed changes in membrane potential are due to internal electron movement within the enzyme.
Fig. 2.

Formation of ΔΨ by reconstituted Na+-NQR. ΔΨ was measured spectrophotometrically using oxonol VI, as described before (23). Unless otherwise stated the reaction buffer contained 100 μM NADH, 100 μM CoQ-1, 100 mM NaCl, 50 mM HEPES, 150 mM KCl, 1 mM EDTA, pH 7.5. Multiple (steady-state) turnover membrane potential is shown in trace i. For partial-turnover experiments CoQ-1 was added ∼10–12 s after NADH (trace ii). Traces iii and iv show the effect of CCCP (5 μM) and ETH-157 (5 μM), respectively, on ΔΨ formation. The effect of CoQH2 on the formation of ΔΨ was measured using 100 μM and 300 μM NADH (trace v and vi, respectively). The spectrophotometric signals were calibrated against the ΔΨ produced with valinomycin (1 μg/mL) and different concentrations of potassium, according to the Nernst equation.
To identify the specific redox step that is involved in this electrogenic process, ΔΨ formation was studied in two mutant enzymes that do not incorporate the FMN cofactors, which have been characterized previously (9, 14). In contrast with wild-type enzyme, NqrB-T236Y (which lacks FMNB) and NqrC-T225Y (which lacks FMNC) were unable to produce an electrochemical potential under either partial or multiple turnover conditions (Fig. S1), indicating that the steps in which FAD, the 2Fe-2S center, and FMNC become reduced are not electrogenic. The remaining candidates for the Na+ pumping step are the reduction of FMNB and the reduction of RibH•. These two possibilities could be distinguished by studying the partial-turnover ΔΨ formation in an enzyme that does not contain riboflavin. However, this cofactor cannot be eliminated by site-directed mutagenesis because its binding site is not currently known. Instead, riboflavin can be inactivated as a redox acceptor by incubating the enzyme with CoQH2, which specifically reduces RibH• to RibH2, thus halting the forward electron flow after FMNB. Stopped flow experiments confirmed that the preincubation of the enzyme with CoQH2 blocks the electron flow at FMNB, the electron donor of RibH• (Table S1, Fig. S2 A and B).
Traces v and vi (Fig. 2) show that partial-turnover ΔΨ formation by wild-type enzyme is almost completely blocked by incubation of the sample with 1 mM CoQH2, indicating that the redox step involved in Na+ ejection is the reduction of riboflavin. However, steady-state kinetic studies have shown that CoQH2 behaves as a competitive inhibitor against NADH, which could potentially account for the decrease in ΔΨ formation observed. (Fig. S3 A and B). A hallmark of competitive inhibition is that it can be overcome by increasing concentrations of the substrate. This effect was observed under multiple turnover conditions by increasing the concentration of NADH but not under partial turnover (Fig. 2, trace vi), which indicates that the specific effect of CoQH2 is to limit the electron flow to riboflavin. These results are consistent with the assignment of the reduction of RibH• as the redox step involved in the translocation of Na+ across the membrane. Further evidence for this proposal was obtained by analyzing the reduction and oxidation kinetics of the mutants that block the binding and the ejection of Na+ (see below).
Fast Kinetics of Oxidation and Reduction of NqrB-D346A and NqrB-D397A Mutants NqrB-D397A.
NqrB-D397A was chosen to study the redox step involved in Na+ binding because the apparent affinity for Na+ is decreased several orders of magnitude in this mutant (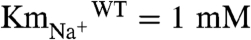 vs.
vs. 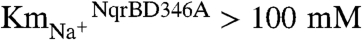 ) (23). Stopped flow experiments showed that the reduction of NqrB- D397A by NADH is very similar to the reduction of wild-type enzyme but does not show sensitivity to Na+, and the rate of RibH• → RibH2 is decreased by a factor of three (Table 1, Fig. 3). We have demonstrated that the RibH• → RibH2 transition is controlled by the rate of electron flow from the 2Fe-2S center to FMNC, the rate limiting step of the reaction (14). These results indicate that the rate of reduction of FMNC is the main feature modified in the NqrB-D397A mutant. This interpretation was corroborated by studying the kinetics of oxidation of the dithionite-reduced enzyme by CoQ. Wild-type Na+-NQR is completely oxidized in a single step, in which all the redox cofactors participate (Table 2 and Fig. S4), suggesting that the binding of CoQ and CoQH2 are under fast equilibrium and electron transfer from riboflavin to CoQ is the rate limiting step, so the oxidation of the enzyme is not limited by accessibility of the oxidant. In the cofactor deletion mutants, NqrB-T236Y and NqrC-T225Y, where the normal flow of electrons is interrupted at different points, the oxidation process takes place in two phases: a fast oxidation of the cofactors downstream of the missing FMN cofactor, followed by a slower oxidation of the upstream cofactors (Table 2 and Fig. S4). Similarly, in NqrB-D397A, the reaction takes place in two steps: rapid oxidation of riboflavin and the two FMNs, followed by oxidation of FAD, at a 16-fold lower rate (Table 2, Fig. 3). This agrees with our conclusion that the electron transfer between the 2Fe-2S center and FMNC is highly inhibited in the NqrB-D397A mutant.
) (23). Stopped flow experiments showed that the reduction of NqrB- D397A by NADH is very similar to the reduction of wild-type enzyme but does not show sensitivity to Na+, and the rate of RibH• → RibH2 is decreased by a factor of three (Table 1, Fig. 3). We have demonstrated that the RibH• → RibH2 transition is controlled by the rate of electron flow from the 2Fe-2S center to FMNC, the rate limiting step of the reaction (14). These results indicate that the rate of reduction of FMNC is the main feature modified in the NqrB-D397A mutant. This interpretation was corroborated by studying the kinetics of oxidation of the dithionite-reduced enzyme by CoQ. Wild-type Na+-NQR is completely oxidized in a single step, in which all the redox cofactors participate (Table 2 and Fig. S4), suggesting that the binding of CoQ and CoQH2 are under fast equilibrium and electron transfer from riboflavin to CoQ is the rate limiting step, so the oxidation of the enzyme is not limited by accessibility of the oxidant. In the cofactor deletion mutants, NqrB-T236Y and NqrC-T225Y, where the normal flow of electrons is interrupted at different points, the oxidation process takes place in two phases: a fast oxidation of the cofactors downstream of the missing FMN cofactor, followed by a slower oxidation of the upstream cofactors (Table 2 and Fig. S4). Similarly, in NqrB-D397A, the reaction takes place in two steps: rapid oxidation of riboflavin and the two FMNs, followed by oxidation of FAD, at a 16-fold lower rate (Table 2, Fig. 3). This agrees with our conclusion that the electron transfer between the 2Fe-2S center and FMNC is highly inhibited in the NqrB-D397A mutant.
Table 1.
Redox transitions and rate constants of the fast kinetics of reduction by NADH of wild-type Na+-NQR, NqrB-D346A and NqrB-D397A mutants
| Rate constants (s-1) |
|||||
| Enzyme | NaCl | K1 | K2 | K3 | K4 |
| WT | |||||
| 0 | 249.3 | 15.4 | 4.2 | 0.31 | |
| FAD → FADH2 | RibH• → RibH2 | 2(FMN → FMN•-) | 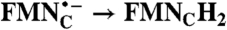 |
||
| 100 | 143.2 | 35.1 | 0.7 | ||
| FAD → FADH2; RibH• → RibH2 | 2(FMN → FMN•-) | 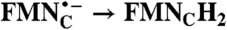 |
|||
| NqrB-D346A | |||||
| 0 | 235 | 20.1 | 3.6 | 0.3 | |
| FAD → FADH2 | 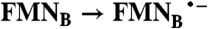 |
 ; Rib → RibH• ; Rib → RibH•
|
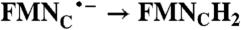 |
||
| 50 | 121.6 | 16.4 | 1.5 | 0.2 | |
FAD → FADH2; 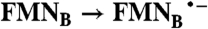
|
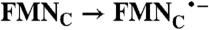 ; Rib → RibH• ; Rib → RibH•
|
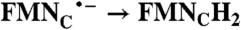 |
RibH• → RibH2 | ||
| NqrB-D397A | |||||
| 0 | 268 | 4.2 | 0.3 | ||
| FAD → FADH2 | RibH• → RibH2; 2(FMN → FMN•-) | 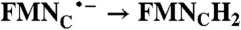 |
|||
| 50 | 270 | 4.6 | 0.35 | ||
| FAD → FADH2 | RibH• → RibH2; 2(FMN → FMN•-) | 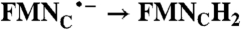 |
|||
| 200 | 258 | 5.1 | 0.4 | ||
| FAD → FADH2 | RibH• → RibH2; 2(FMN → FMN•-) | 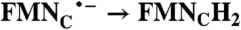 |
|||
Desalted samples were mixed with 250 μM NADH using different concentrations of sodium.
Fig. 3.

(A) Reduction of the NqrB-D397A mutant by 250 μM NADH in the absence and presence of 100 mM NaCl. (B) Kinetic phases of the oxidation of the NqrB-D397A mutant. In A and B blue lines show the difference spectra of the reduction steps associated with each transition, obtained by data fitting. Green lines show the difference spectra of the assigned redox steps using in wild-type Na+-NQR as reported before (14). Bottom panels: Time course at the absorbance maximum (450 nm) of Na+-NQR (C) Scheme showing the redox transitions for the reduction and oxidation kinetics in the NqrB-D397A mutant. Open squares represent the oxidized state of the cofactor, and black squares the reduced form. The phases highlighted in gray show the transitions slowed down in the mutant enzyme.
Table 2.
Redox transitions and rate constants for the oxidation of Na+-NQR by CoQ of the dithionite-reduced wild type, NqrB-D346A, and NqrB-D397A mutants
| Rate constants (s-1) |
||
| Enzyme | K1 | K2 |
| WT | 163.2 ± 5.4 | |
FADH2 → FAD; 2(FMN•- → FMN); 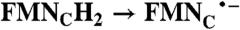 ; RibH2 → RibH• ; RibH2 → RibH•
|
||
| NqrB-D346A | 80.4 ± 12.3 | 6.4 ± 3.6 |
| RibH2 → RibH• | FADH2 → FAD; 2(FMN•- → FMN); 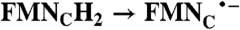
|
|
| NqrB-D397A | 133.8 ± 12.4 | 8.3 ± 1.56 |
2(FMN•- → FMN); 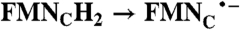 ; RibH2 → RibH• ; RibH2 → RibH•
|
FADH2 → FAD | |
Enzyme samples were reduced with dithionite and mixed with 500 μM CoQ-1 and 100 mM NaCl.
It remained possible that this two-step oxidation process was caused by inhibition of electron transfer into the 2Fe-2S center. Reduction of the 2Fe-2S can be easily masked by the flavins of the enzyme (14). However, there is evidence from steady-state kinetics that the properties of the 2Fe-2S center are not altered in this mutant. Hayashi et al. (6) have shown that Na+-NQR is capable of transferring electrons from NADH to the artificial electron acceptor menadione, probably through the 2Fe-2S center. Indeed, the NqrF-C76A mutant, which lacks this cofactor, shows a 90% decrease in menadione reductase activity (Fig. S5). On the other hand, the rate of reduction of the 2Fe-2S center in NqrB-D397A is not affected because the menadione reductase activity was similar to that of the wild-type enzyme (Fig. S5). Taken together these results indicate that the redox step involved in Na+ binding is the reduction of FMNC, consistent with our earlier results with the NqrB-T236Y mutant (14).
NqrB-D346A.
We have previously found that the redox reaction in this mutant is significantly inhibited and the Na+ sensitivity of CoQ reduction is completely abolished, while the apparent affinity for Na+ (KmNa+), measured during steady-state turnover, is essentially unchanged. On this basis, we proposed that the mutation blocks Na+ ejection from a binding site within the enzyme (23). To better understand the connection between the redox reaction and Na+ transport in the enzyme, we have studied the kinetics of the reduction of this mutant by NADH, by using stopped flow methods (Table 1, Fig. 4). The first redox step is the sodium-insensitive reduction of FAD, as in the wild-type enzyme (14, 24). The effects of the mutation appear at the second step, where one FMN (probably FMNB) becomes reduced, whereas reduction of riboflavin is observed in the corresponding phase in the wild-type enzyme (Table 2, Fig. 4). These changes are remarkably similar to those observed when the wild-type enzyme is preincubated with CoQH2 (Table S1), blocking reduction of RibH•. This suggests that in NqrB-D346A electron transfer between FMNB and riboflavin is inhibited. The remaining phases in the reaction of this mutant with NADH are very slow but, surprisingly, include formation of the neutral riboflavin semiquinone (Rib → RibH•) a species that is usually present in the oxidized enzyme (as prepared). X-band EPR spectra confirm that the oxidized form of the mutant contains only a small amount of RibH• while the reduced enzyme exhibits the typical contributions of the 2Fe-2S center and the anionic flavosemiquinone radical (Fig. S6). We have previously shown that the flavin content of this mutant is normal (23). This, together with the fact that formation of RibH• is observed during the reduction of this enzyme, indicates that the small amount of neutral flavosemiquinone initially present is not due to an insufficient quantity of the riboflavin cofactor. Instead, the riboflavin is present but in its fully oxidized state. The reduction kinetics of NqrB-D346A suggest that the electron transfer from FMNB to the riboflavin is inhibited and only takes place when the enzyme is almost completely reduced. This may explain the low occupancy of the neutral radical in the oxidized enzyme (as prepared).
Fig. 4.

(A) Reduction of the NqrB-D346A mutant by 250 μM NADH in the absence and presence of 100 mM NaCl. (B) Kinetic phases of the oxidation of the NqrB-D346A mutant. In A and B blue lines show the difference spectra of the reduction steps associated with each transition, obtained by data fitting. Green lines show the difference spectra of the assigned redox steps using wild-type Na+-NQR as reported before (14). Bottom panels: Time course at the absorbance maximum (450 nm) of Na+-NQR (C) Scheme showing the redox transitions for the reduction and oxidation kinetics in the NqrB-D346A mutant. Open squares represent the oxidized state of the cofactor, and black squares the reduced form. The phases highlighted in gray show the transitions slowed down in the mutant enzyme.
To further investigate electron transfer in this mutant, the oxidation of the dithionite-reduced enzyme by CoQ was studied (Table 2, Fig. 4). The oxidation process is divided in two phases. The first phase consists of the fast oxidation of riboflavin; during the second phase all the other cofactors are oxidized, at a thirteen-times slower rate. This clearly shows that electron flow from FMNB to riboflavin is impaired. It should be pointed out that the complete oxidation the riboflavin (RibH• → Rib) was not observed over the course of several hours (Fig. S7), suggesting that the stability of RibH• is comparable to that in the wild-type enzyme.
The kinetics of oxidation and reduction of this mutant are consistent with the idea that NqrB-D346A is not able to eject Na+ and therefore the electrons are stuck in the cofactor immediately upstream of the step responsible for the electrogenic activity.
Discussion
Several models of the sodium pumping mechanism of Na+-NQR have been proposed (2, 3, 25, 26). Most of these models are based on the localized coupling concept, in which Na+ affinity is modulated by the redox state of the cofactors. The original thermodynamic hypothesis of Rich of a center whose reduction is directly involved in Na+ capture and release was especially attractive, coupling the electroneutral uptake of sodium and electrons into the hydrophobic core of the membrane (25). Later, Dimroth (26) and Bogachev (3) suggested that a semiquinone radical, originating from ubiquinone or flavin molecules, could be the essential redox cofactor responsible for sodium translocation. The thermodynamic hypothesis can be tested by analyzing the midpoint potentials of the redox cofactors of the enzyme. Redox titrations have identified three relatively exergonic redox steps that could be involved in driving Na+ pumping (21, 22). However, none of these steps stands out as having a particularly large ΔEm, which could identify it as an important driving reaction. Moreover, none of the cofactor redox potentials showed significant dependence on the bulk Na+ concentration, in contrast to what would be expected for redox–Bohr coupling (18, 19). Verkhovsky and Bogachev (27) have cited their recent 23NaNMR measurements, showing an apparent change in Na+ affinity upon reduction of the enzyme, as evidence for a thermodynamically coupled mechanism. However, it is also possible that these measurements could reflect changes in the kinetics of accessibility of a sodium-binding site rather than changes in affinity per se.
Hirst (28) has pointed out that locally coupled mechanisms are not consistent with the understood requirements of Na+-binding to proteins and cation selectivity of the pump. In all cases where structures are known, Na+-binding sites consist of six negatively charged amino acids arranged in an octahedral geometry (29). Localized coupling does not appear to be consistent with the highly hydrophilic environments in which sodium typically binds. We have shown that in Na+-NQR at least three acid residues play important roles in Na+-binding (NqrB-D397, NqrD-D133, NqrE-E95), while four additional glutamate or aspartate residues participate in later steps of transport (NqrB-E28, NqrB-E144, NqrB-D346, NqrD-D88) (23). Also, the lack of pH dependence of the cofactor midpoint potentials strongly indicates that the cofactors are not accessible to the aqueous environment and, consequently, to sodium (21).
In this work we have used an approach based on the kinetic characterization of wild type and of specific mutants to dissect the redox steps involved in Na+ translocation by Na+-NQR. Two experimental strategies were followed to study this question: (i) characterization of partial-turnover ΔΨ formation to identify the redox step involved in translocation of Na+ across the membrane and (ii) analysis of the reduction and oxidation kinetics of the NqrB-D346A and NqrB-D397A mutants. We have previously shown that NqrB-D397 forms part of a sodium binding site in the protein and that NqrB-D346 is likely to be involved in a sodium exit channel (23). The study of these mutants provided us important clues about the redox steps involved in sodium binding and release. Our results indicate that sodium uptake takes place in the reaction step in which an electron moves from the 2Fe-2S center to FMNC, while the translocation of sodium across the membrane dielectric (and probably its release into the external medium) occurs in the reaction step in which an electron moves from FMNB to riboflavin. In contrast with previous models, none of the redox cofactors participate in both of these steps, which indicates that a direct or localized coupling mechanism is unlikely and suggests that Na+-NQR operates through a novel type of mechanism for a redox-driven ion pump. We propose that redox events in the enzyme drive conformational changes, which in turn lead to switching the accessibility of the binding site between the two sides of the membrane.
Compared to Na+-NQR, the analogous H+-pumping NADH:quinone oxidoreductase, complex I, is a much a more complicated enzyme with up to 45 subunits and 10 cofactors (30). The relative simplicity of Na+-NQR, which has no apparent homology to complex I, has greatly benefited the current study. This has made it possible to assign the redox coupling sites for Na+-NQR or other type of NADH dehydrogenases and to shed light on the mode of coupling between the redox reaction and ion transport. These results will also contribute to our understanding of the general principles governing redox-driven ion translocation.
Materials and Methods
Methods for the following procedures have been published previously: mutant strains (9, 12, 23), cell culture, and protein purification (1). Na+-NQR concentration was determined by measuring the net flavin content of enzyme preparations under denaturing conditions at 450 nm using a molar absorptivity of 48.4 mM-1 cm-1 for wild type, NqrF-C76A, NqrB-D346A and NqrB-D397A, and 36.3 4 mM-1 cm-1 for NqrB-T236Y and NqrC-T225Y.
Na+-NQR Reconstitution.
Wild-type Na+-NQR was mixed with Escherichia coli phospholipids using a phospholipid:protein ratio of 5∶1 in the presence of octylglucoside (detergent/ratio=1.3) (23). Detergent was slowly removed by adding SM Biobeads, and proteoliposomes were concentrated and washed twice as reported before (23).
Menadione Reductase Activity.
Menadione reductase activity was measured spectrophotometrically as reported by Hayashi et al. (31).
Membrane Potential (ΔΨ).
Membrane potential (ΔΨ) was measured spectrophotometrically at 625 minus 587 nm in proteoliposomes containing Na+-NQR, using 3 μM oxonol VI (19). The reaction buffer contained 100 μM NADH, 100 μM CoQ-1, 100 mM NaCl, 50 mM HEPES, 150 mM KCl, 1 mM EDTA, pH 7.5 (23). Alternatively, ΔΨ formation was measured with the fluorescent indicator RH421 (100 nM) at λex = 500 nm and λem = 650 nm (32). In both cases signals were calibrated against the ΔΨ produced with valinomycin and potassium (23).
Fast Kinetics Experiments.
The reduction kinetics of the mutants were studied in buffer containing 250 μM K2-NADH in the absence and in the presence of 100 mM or 200 mM NaCl (final concentrations). In the case of the oxidation reaction, the enzyme (10–20 μM) was reduced with dithionite in the presence of 100 mM NaCl, and the reaction was measured by mixing the sample with buffer containing 500 μM of CoQ-1. The raw data were averaged and analyzed as reported previously (14). The kinetic phases of the redox reactions were assigned by comparing the difference spectra obtained from data fitting with those of redox transitions described recently in V. cholerae Na+-NQR (14).
Supplementary Material
Acknowledgments.
We thank Dr. Robert Gennis and Michel Shea for their helpful comments. This work was supported by the National Institutes of Health (Grant GM069936 to B.B.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1002866107/-/DCSupplemental.
References
- 1.Barquera B, et al. Purification and characterization of the recombinant Na(+)-translocating NADH:quinone oxidoreductase from Vibrio cholerae. Biochemistry. 2002;41:3781–3789. doi: 10.1021/bi011873o. [DOI] [PubMed] [Google Scholar]
- 2.Unemoto T, Hayashi M. Na+-translocating NADH-quinone reductase of marine and halophilic bacteria. J Bioenerg Biomembr. 1993;25:385–391. doi: 10.1007/BF00762464. [DOI] [PubMed] [Google Scholar]
- 3.Bogachev AV, Verkhovsky MI. Na+- translocating NADH:quinone oxidoreductase: progress achieved and prospecst of investigations. Biochemistry-Moscow. 2005;70:143–149. doi: 10.1007/s10541-005-0093-4. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi M, Unemoto T. Characterization of the sodium-dependent respiratory chain NADH: quinone oxidoreductase of the marine bacterium, Vibrio alginolyticus, in relation to the primary sodium pump. Biochim Biophys Acta. 1984;767:470–477. [Google Scholar]
- 5.Bogachev AV, Murtazina RA, Skulachev VP. The Na+/e- stoichiometry of the Na+-motive NADH:quinone oxidoreductase in Vibrio alginolyticus. FEBS Lett. 1997;409:475–477. doi: 10.1016/s0014-5793(97)00536-x. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi M, Nakayama Y, Unemoto T. Recent progress in the Na(+)-translocating NADH-quinone reductase from the marine Vibrio alginolyticus. Biochim Biophys Acta. 2001;1505:37–44. doi: 10.1016/s0005-2728(00)00275-9. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi M, et al. FMN is covalently attached to a threonine residue in the NqrB and NqrC subunits of Na(+)-translocating NADH-quinone reductase from Vibrio alginolyticus. FEBS Lett. 2001;488:5–8. doi: 10.1016/s0014-5793(00)02404-2. [DOI] [PubMed] [Google Scholar]
- 8.Barquera B, Zhou W, Morgan JE, Gennis RB. Riboflavin is a component of the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. Proc Natl Acad Sci. 2002;99:10322–10324. doi: 10.1073/pnas.162361299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barquera B, Ramirez-Silva L, Morgan JE, Nilges MJ. A new flavin radical signal in the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae: An EPR/ENDOR investigation of the role of the covalently bound flavins in subunits B and C. J Biol Chem. 2006;281:36482–36491. doi: 10.1074/jbc.M605765200. [DOI] [PubMed] [Google Scholar]
- 10.Barquera B, Hase CC, Gennis RB. Expression and mutagenesis of the NqrC subunit of the NQR respiratory Na(+) pump from Vibrio cholerae with covalently attached FMN. FEBS Lett. 2001;492:45–49. doi: 10.1016/s0014-5793(01)02224-4. [DOI] [PubMed] [Google Scholar]
- 11.Barquera B, et al. X- and W-band EPR and Q-band ENDOR studies of the flavin radical in the Na+-translocating NADH:quinone oxidoreductase from Vibrio cholerae. J Am Chem Soc. 2003;125:265–275. doi: 10.1021/ja0207201. [DOI] [PubMed] [Google Scholar]
- 12.Barquera B, et al. Mutagenesis study of the 2Fe-2S center and the FAD binding site of the Na(+)-translocating NADH:ubiquinone oxidoreductase from Vibrio cholerae. Biochemistry. 2004;43:12322–12330. doi: 10.1021/bi048689y. [DOI] [PubMed] [Google Scholar]
- 13.Juarez O, Nilges MJ, Gillespie P, Cotton J, Barquera B. Riboflavin is an active redox cofactor in the Na+-pumping NADH:quinone oxidoreductase (Na+-NQR) from Vibrio cholerae. J Biol Chem. 2008;283:33162–33167. doi: 10.1074/jbc.M806913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juárez O, Morgan JE, Barquera B. The electron transfer pathway of the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. J Biol Chem. 2009;284:8963–8972. doi: 10.1074/jbc.M809395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirst J. Towards the molecular mechanism of respiratory complex I. Biochem J. 2009;425:327–329. doi: 10.1042/BJ20091382. [DOI] [PubMed] [Google Scholar]
- 16.Kim YC, Wikström M, Hummer G. Kinetic gating of the proton pump in cytochrome c oxidase. Proc Natl Acad Sci USA. 2009;106:13707–13712. doi: 10.1073/pnas.0903938106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crofts AR, Lhee S, Crofts SB, Cheng J, Rose S. Proton pumping in the bc1 complex: A new gating mechanism that prevents short circuits. Biochim Biophys Acta. 2006;1757:1019–1034. doi: 10.1016/j.bbabio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Gelles J, Blair DF, Chan SI. The proton-pumping site of cytochrome c oxidase: A model of its structure and mechanism. Biochim Biophys Acta. 1986;853:205–236. doi: 10.1016/0304-4173(87)90002-4. [DOI] [PubMed] [Google Scholar]
- 19.Krab K, Wikstrom M. Principles of coupling between electron transfer and proton translocation with special reference to proton-translocation mechanisms in cytochrome oxidase. Biochim Biophys Acta. 1987;895:25–39. doi: 10.1016/s0304-4173(87)80015-0. [DOI] [PubMed] [Google Scholar]
- 20.Bogachev AV, Belevich NP, Bertsova YV, Verkhovsky MI. Primary steps of the Na+-translocating NADH:ubiquinone oxidoreductase catalytic cycle resolved by the ultrafast freeze-quench approach. J Biol Chem. 2009;284:5533–5538. doi: 10.1074/jbc.M808984200. [DOI] [PubMed] [Google Scholar]
- 21.Bogachev AV, Bloch DA, Bertsova YV, Verkhovsky MI. Redox properties of the prosthetic groups of Na(+)-translocating NADH:quinone oxidoreductase. 2. Study of the enzyme by optical spectroscopy. Biochemistry. 2009;48:6299–6304. doi: 10.1021/bi900525v. [DOI] [PubMed] [Google Scholar]
- 22.Bogachev AV, et al. Redox properties of the prosthetic groups of Na(+)-translocating nadh:quinone oxidoreductase. 1. Electron paramagnetic resonance study of the enzyme. Biochemistry. 2009;48:6291–6298. doi: 10.1021/bi900524m. [DOI] [PubMed] [Google Scholar]
- 23.Juarez O, Athearn K, Gillespie P, Barquera B. Acid residues in the transmembrane helices of the Na+-pumping NADH:quinone oxidoreductase (Na+-NQR) from Vibrio cholerae involved in sodium translocation. Biochemistry. 2009;48:9516–9525. doi: 10.1021/bi900845y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogachev AV, Bertsova YV, Barquera B, Verkhovsky MI. Sodium-dependent steps in the redox reactions of the Na+-motive NADH:quinone oxidoreductase from Vibrio harveyi. Biochemistry. 2001;40:7318–7323. doi: 10.1021/bi002545b. [DOI] [PubMed] [Google Scholar]
- 25.Rich PR, Meunier B, Ward FB. Predicted structure and possible ionmotive mechanism of the sodium-linked NADH-ubiquinone oxidoreductase of Vibrio alginolyticus. FEBS Lett. 1995;375:5–10. doi: 10.1016/0014-5793(95)01164-a. [DOI] [PubMed] [Google Scholar]
- 26.Dimroth P. Primary sodium ion translocating enzymes. Biochim Biophys Acta. 1997;1318:11–51. doi: 10.1016/s0005-2728(96)00127-2. [DOI] [PubMed] [Google Scholar]
- 27.Verkhovsky M, Bogachev AV. (Sodium-translocating NADH:quinone oxidoreductase as a redox-driven ion pump. Biochim Biophys Acta. doi: 10.1016/j.bbabio.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 28.Hirst J. Towards the molecular mechanism of respiratory complex I. Biochem J. 2009;425:327–339. doi: 10.1042/BJ20091382. [DOI] [PubMed] [Google Scholar]
- 29.Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J Mol Biol. 1996;256:228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- 30.Brandt U. Energy converting NADH:quinone oxidoreductase (complex I) Annu Rev Biochem. 2006;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi M, Hasegawa K, Oguni Y, Unemoto T. Characterization of FMN-dependent NADH-quinone reductase induced by menadione in Escherichia coli. Biochim Biophys Acta. 1990;1035:230–236. doi: 10.1016/0304-4165(90)90122-d. [DOI] [PubMed] [Google Scholar]
- 32.Bühler R, Stürmer W, Apell HJ, Läuger P. Charge translocation by the Na,K-pump: I. Kinetics of local field changes studied by time-resolved fluorescence measurements. J Membr Biol. 1991;121:141–161. doi: 10.1007/BF01870529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.