

Abstract
Background
Atherosclerosis and coronary heart disease (CHD) are significant contributors to morbidity and mortality in developed countries. A noted exception is the low mortality of CHD in France, particularly the southwest region. This phenomenon, commonly referred to as the French paradox, may be associated with high consumption of red wine. We investigate whether the cardioprotective activity of red wine may involve the grape skin-derived polyphenol, resveratrol. We further test the possibility that resveratrol acts by modulating structural and functional changes in endothelial cells lining the blood vessel wall.
Results
Bovine pulmonary artery endothelial cells (BPAEC) were incubated with resveratrol, with and without concurrent exposure to simulated arterial shear stress. Resveratrol significantly affected proliferation and shape of BPAEC; growth was suppressed and cells became elongated, based on morphologic analysis of rhodamine-conjugated phalloidin stained F-actin by confocal microscopy. Using selective signaling inhibitors, we showed that the resveratrol-induced cellular phenotype was dependent on intracellular calcium and tyrosine kinase activities, and assembly of actin microfilaments and microtubules, but was unrelated to PKC activity. Exposure to simulated arterial flow revealed that, whereas controls cells easily detached from the culture support in a time-dependent manner, resulting in total cell loss after a 5 min challenge with simulated arterial flow conditions, a significant percentage of the treated cells remained attached to the cultured plastic coverslips under identical experimental conditions, suggesting that they adhered more strongly to the surface. Western blot analysis shows that whereas cells treated with 25 μM and 100 μM resveratrol had no change in total ERK1/2, treatment did result in an increase in phosphorylated ERK1/2, which probably involved stabilization of the active enzyme. An increase in nitric oxide synthase expression was detected as early as 6 h and persisted for up to 4 days of treatment.
Conclusions
Results of our studies show that resveratrol interacts with endothelial cells in vitro to elicit morphological and structural changes; the observed changes support the interpretation that resveratrol acts as a cardioprotective agent.
Background
Atherosclerosis and coronary heart disease (CHD) have long been considered major contributors to morbidity and mortality in developed countries [1,2,3]. One noted exception is the low mortality of CHD in France, particularly the southwest region [4,5,6]. This phenomenon, commonly referred to as the French paradox, may be associated with high consumption of red wine [7,8]. A negative correlation between CHD and alcohol consumption was first noted 25 years ago, and numerous studies since have confirmed a statistically significant inverse relationship between these two factors [9,10,11,12,13,14]. In vivo studies have shown that wine consumption, particularly red wine, is more effective in the prevention of CHD not seen with other forms of alcoholic beverages [15,16,17]. Accordingly, it has been postulated that naturally occurring components in wine might afford or contribute to its enhanced protection against CHD by targeting sites that participate in the etiology of CHD, including soluble blood components (LDL), cellular blood elements (platelets), or the vasculature itself (endothelium). Siemann and Creasy proposed that trans-resveratrol, a tri-hydroxy stilbene, in red wine exhibits cardioprotective properties [18] and can inhibit LDL oxidation [19], suppress smooth muscle proliferation [20], induce nitric oxide synthase expression [21] and block collagen-induced aggregation responses in washed platelets [22,23,24]. However, relatively few studies have been performed on the effects of resveratrol on vascular components such as the endothelial cells, which are known to play a critical role in maintaining the integrity and functioning of the vascular endothelium. Homeostasis of the vascular endothelium, both with respect to metabolic and physiologic activities, can be affected by overall nutritional status and by specific ingredients in the diet [25,26,27].
The aims of the present study were to determine changes resulting from resveratrol:endothelial cell interaction. We observed that resveratrol induced significant cellular and biochemical changes in endothelial cells, which were accompanied by altered functional responsiveness to conditions simulating arterial flow. Since previous studies have shown several signaling molecule changes during reorganization of the endothelial cell cytoskeleton accompanying arterial shear stress [28,29,30,31], we evaluated the same set of biochemical parameters in resveratrol-treated cells. Use of selective signaling pathway inhibitors allowed the demonstration that the cytoskeletal changes elicited by resveratrol depended on intracellular calcium and tyrosine kinase activity changes, and also appeared to be linked to integrity of actin microfilaments and microtubule network. Resveratrol treatment also led to activation of ERK1/2 MAP kinase, similar to changes induced by shear [30,31]. Thus resveratrol may act by a mechanism(s) closely resembling that triggered by shear stress.
Results
Resveratrol induces morphologic change in endothelial cells
Sterilized plastic 18 mm × 21 mm coverslips were seeded with BPAEC and treated 24 h later with various concentrations of resveratrol. Following an additional 2 day of culture, the coverslips containing the attached BPAECs were fixed, stained with rhodamine-phalloidin, and mounted onto slides for analysis by confocal microscopy. The polyphenol (100 μM) had a distinct morphologic effect, changing the cells from a boxy, cobblestone-like appearance (panel A, Fig. 1) to an elongated, ellipsoidal shape with long, tortuous processes (panel B). As little as 25 μM resveratrol sufficed to induce these cellular changes, in passage 4-7 cells (compare panel C with panel A, Fig. 2). To quantify the percentages of cells with the elongated morphology, a minimum of 300 cells in representative microscopic field were counted and the results presented in Fig. 3. The percentage of cells showing the elongated phenotype is proportional to the concentration of resveratrol added to the culture. Treatment with resveratrol also suppressed growth of BPAEC (data not shown), as we previously reported [21].
Figure 1.

Resveratrol treatment produces elongation of BPAEC. Panel A. Example of stellar, cobblestone-like morphology characteristic of normal BPAECs grown in culture. Panel B. Example of elongated, spindle-shaped morphology characteristic of resveratrol-treated cells. Here cells were treated with 100 μM resveratrol and were viewed with 20X objective. Six experiments, each involving a different preparation of BPAEC, were performed with similar results.
Figure 2.

Morphology of passage 6 BPAEC treated with the following concentrations of resveratrol: A) 0 μM; B) 10 μM; C) 25 μM; D) 50 μM. Cells were stained with rhodamine-phalloidin and viewed under 10X confocal microscopy. The same experiment was repeated three times, using passages 6-7 cells from two different preparation of BPAEC.
Figure 3.

Resveratrol treatment causes a dose-dependent increase in BPAEC elongation. Cells from representative confocal microscopy fields from passages 5 and 6 were visually evaluated for overall change in cell morphology. A minimum of 300 cells were scored for each treatment condition.
Resveratrol treatment leads to increased adherence of BPAEC under simulated arterial flow conditions
To test whether functional changes may accompany the modified morphology, control and 2 day and 100 μM resveratrol-treated cells were exposed to 0 min (control), 2 min, or 5 min of simulated arterial shear stress using a parallel plate perfusion chamber (see Materials and Methods). Following the shear challenge, cells remaining attached were scored by staining with rhodamine-phalloidin, as described. Since the cells were evenly distributed throughout the area of the coverslip exposed to flow, representative pictures were taken of the confocal microscopy fields located directly in the center of the coverslip, and the number of cells were counted. As can be seen in Fig. 4, compared to the 0 min sample, a significant percentage of resveratrol-treated BPAEC remained attached to the plastic coverslips after 2 min and 5 min flow challenge. In comparison, under identical experimental conditions, cells adhering to the coverslips decreased dramatically in controls, with virtually no cells remaining after a 5 min simulated flow. These results suggest that a functional alteration, measured by greater adherence of cells under simulated arterial flow conditions, accompanied the change in cellular phenotype, following treatment with resveratrol.
Figure 4.
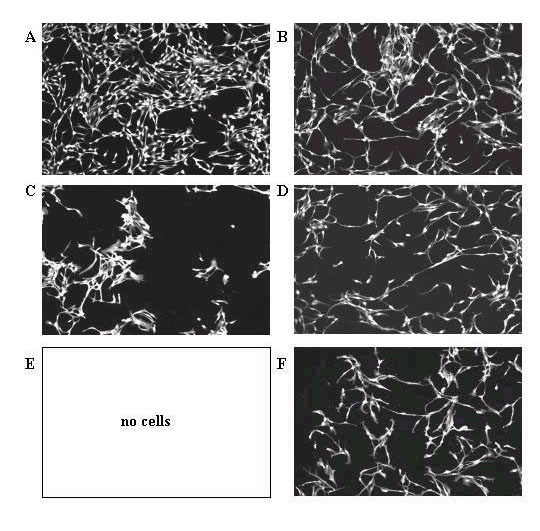
Passage 7 BPAEC were treated with the following concentration of resveratrol as described in Materials and Methods and then subjected to simulated arterial flow durations, respectively. A) 0 μM, 0 min (control); B) 100 μM; C) 0 μM, 2 min; D) 100 μM, 2 min; E) 0 μM, 5 min; F) 100 μM, 5 min. Similar results were obtained using passage 6 BPAEC (data not shown). Four experiments, using passages 6-7 cells from two different preparation of BPAEC, were performed.
Effect of cell signaling inhibitors on cell elongation induced by resveratrol
Further insights into the nature of the resveratrol-induced morphologcial changes came from studies using selective inhibitors for different signaling pathways. These include: quin2-AM (intracellular [Ca2+]), herbimycin A (tyrosine kinases), chelerythrine (PKC), cytochalasin D (actin microfilaments), and nocodazole (disassembly of microtubules). Cells were treated with inhibitors, at concentrations indicated in Materials and Methods, and then exposed to resveratrol. Representative confocal microscopy fields for both control and resveratrol-treated cells were counted for elongated and non-elongated cells. The inhibitors used noticeably suppressed cell growth (particularly treatment with cytochalasin D and nocodazole), particularly controls not treated with resveratrol. The degree of growth inhibition varied, depending on preparations of primary BPAEC. Interestingly, cells treated with resveratrol, which had to be reduced to 40 h because of adverse responses of control cells to inhibitors (see Materials and Methods), afforded better growth in samples containing the inhibitors. Table 1 summarized results averaged from 4 separate experiments, each performed with a different preparation of primary BPAEC. Since growth of control cells as well as their response to resveratrol and addition of inhibitors differed between BPAEC preparations, cells showing the elongated shape in each experiment were calculated as a percent of total cells counted (ranging from 60-700). The numbers obtained in this manner were used to calculate the mean (%)± SD, as shown. Addition of chelerythrine, an inhibitor of protein kinase C, caused a significant increase in percentage of elongated cells alone and did not affect morphologic response of cells to resveratrol. Inhibitors of intracellular [Ca2+] and tyrosine kinase activity effectively reduced the percentage of elongated cells elicited by resveratrol. Results in Table 1 further showed that cytoskeletal changes elicited by resveratrol also appeared to be linked to integrity of actin microfilaments and microtubule network as the morphologic changes were also substantially lowered in cells treated with cytochalasin D and nocodazole, which inhibit formation of actin microfilaments and microtubule network, respectively.
Table 1.
Use of inhibitors to probe the basis for elongation of BPAEC induced by resveratrol. These experiments used passages 4-5 BPAEC cells. The cells on cover slips were incubated with inhibitors for 24 h, before 100 μM resveratrol was added and incubation was continued for an additional 40 h. An exception was quin2-AM, which was applied 1 h prior to resveratrol treatment.
| Cellular | Controlª | Cells treated with 100 μM resveratrola |
| Component | ||
| affected | % elongated cells | % elongated cells |
| Control | 12.90 ± 5.56 | 33.80 ± 7.50 |
| Intracellular | 15.80 ± 1.42 | 11.73 ± 1.89 |
| [Ca2+] | ||
| PKC | 22.47 ± 3.58 | 37.63 ± 6.17 |
| Tyrosine kinases | 9.65 ± 1.05 | 10.55 ± 1.05 |
| Actin | ||
| Microfilaments | 10.40 ± 2.60 | 12.25 ± 3.75 |
| Actin | ||
| Microtubules | 4.17 ± 4.94 | 8.60 ± 6.53 |
a Results in this table were averaged from 4 separate experiments, each performed with a different preparation of primary BPAEC's. Since growth of control cells as well as their response to resveratrol and addition of inhibitors differed between BPAEC preparations, cells showing the elongated shape in each experiment were calculated as a percent of total cells counted (ranging from 60-700). The numbers obtained in this manner were used to calculate the mean (%)± SD, as shown.
Effect of resveratrol on ERK1/2 activation and eIF4E, eNOS expression
Biochemical changes in resveratrol-treated BPAEC were assessed by western blot analyses. Fig. 5 shows that in control cells, level of active ERK1/2-P remained relatively constant up to 48 h but showed a precipitous decline from 48 to 96 h (panel A). Under the same conditions, levels of total ERK1/2 (panel B), eIF4E (panel C), a downstream effector of ERK1/ERK2, and actin (panel E) showed no substantial change at all time points assayed, in control cells. Treatment with 25 μM and 100 μM resveratrol prevented the decrease in active ERK1/2-P seen at 96 h (panel A, last 3 lanes). In addition, these concentrations of resveratrol markedly increased eNOS expression as early as 6-h, which reached maximum induction at 48-h, and remained substantially elevated at 96-h (panel D). Quantification of relative changes of total ERK1/2, active ERK1/2-P, and eNOS as a function of time of treatment with two concentrations of resveratrol is depicted in Figure 6.
Figure 5.

Biochemical changes in resveratrol-treated BPAEC. BPAEC were treated with the indicated concentrations of resveratrol and lysed after 6, 48, and 96 h, respectively. Lysates were run under 10% SDS-PAGE, and probed with antibodies for the various target proteins. Panel A. Changes in activated ERK1/2-P (representative of 3 experiments). Panel B. Changes in ERK1/2 (representative of 2 experiments). Panel C. Changes in eIF-4E. Panel D. Changes in eNOS. Panel E. Changes in actin. Lanes 1,4,7 correspond to control at 6, 48, and 96 h; lanes 2,5,8 correspond to BPAEC treated with 25 μM resveratrol for 6, 48, and 96 h; lanes 3,6,9 correspond to BPAEC treated with 100 μM resveratrol for 6, 48, and 96 h. The results represent the average of two experiments, each analyzed in duplicate or triplicate.
Figure 6.
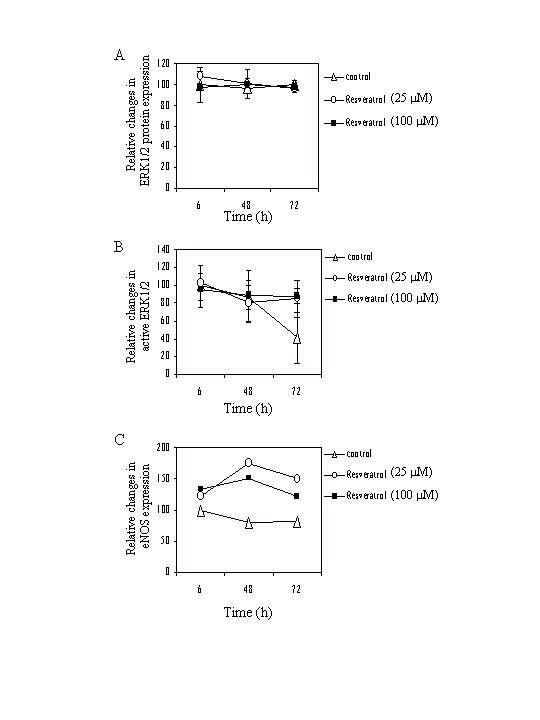
Time-dependent relative changes in expression of total ERK1/2, active ERK1/2, and eNOS. Intensity of signals corresponding to total ERK1/2 (panel A), active ERK1/2 (panel B), and eNOS (panel C) obtained by western blot analysis, similar to those shown in Figure 5, was quantified by actin-adjusted image analysis and plotted as a function of time of treatment with 25 μM and 100 μM resveratrol. The control value at 6 h was set at 100. The results represent the average of two experiments, each analyzed in duplicate or triplicate.
Discussion
Results of our studies suggest that resveratrol, an abundant red wine polyphenol, interacts with endothelial cells in vitro to result in morphologic and functional changes. The elongated shape, interspersed with long, tortuous projections displayed by resveratrol-treated BPAEC (Fig. 1,2,3) has apparent functional significance since these cells resisted detachment from the plastic coverslips under simulated arterial shear stress conditions (Fig. 4). The resveratrol-induced cellular changes could represent a mechanism of minimizing endothelial damage by shearing forces in vivo. Moreover, resistance to detachment could also make endothelial cells less likely to dislodge and become part of a growing thrombotic plug.
Modulation of endothelial cell shape by resveratrol
Size, shape, mutual orientation, and intercellular contacts in endothelium are not incidental and statistic events, but are precisely and dynamically regulated. Reversible change of endothelial cell shape, mutual orientation of cells in certain directions, changes in intercellular contacts, are controlled by a number of factors, such as, cooperative stimulation of the receptors or the systems of second messengers [32,33], dietary ingredients, e.g., retinoids [34], exercise [35,36], and fluid-imposed shear stress [37,38,39]. For example, endothelial cells are known to elongate and reorient their cytoskeletons in the direction of flow as a normal physiological response to prolonged shear stress [29]. Results of the present studies show that treatment with resveratrol induces a morphologic change from a stellar to an elongated shape (Figure 1). It is tempting to speculate that appearance of such cellular phenotype involves formation of stress fibers, which would rearrange the cytoskeleton in ways that facilitate better anchoring of the BPAEC to the culture substratum and contribute to the ability of BPAEC to resist simulated arterial flow challenge. Only studies in the future would validate such a possibility. It is also interesting to note that the observed changes in resveratrol-treated BPAEC are similar to that described for EC subjected to arterial shear. We propose that this could represent a physiologically-relevant mechanism by which resveratrol, as a polyphenolic constituent of red wine, contributes to cardioprotection by inducing resistance to potential damage by shearing forces.
The mechanism underlying this cytoskeletal rearrangement due to shear stress is ill-defined, but it has been found to be linked to tyrosine kinase activity, levels of intracellular calcium, intact actin microfilaments, and functional microtubule network, but is independent of protein kinase C, intermediate filaments, and stretch- and shear-activated mechanosensitive K+ channels [28]. We therefore sought to determine whether a similar mechanism could account for the resveratrol-mediated endothelial shape change. Our approach involved using a number of selective inhibitors for various signaling pathway. These studies showed that quin2-AM ([Ca2+] inhibitor), added together with resveratrol, clearly abolished the endothelial cell shape change induced by the polyphenol. In contrast, chelerythrine, a PKC inhibitor, which added alone triggered a significant shape change in BPAEC, had no effect on resveratrol-elicited morphological change (Table 1). Analyzed as a whole, these experiments suggest that the change in cellular phenotype, as a consequence of resveratrol:EC interaction, is dependent on Ca2+, tyrosine kinases, and intact actin microfilament and microtubules. These results also raise the possibility that there is cellular heterogeneity within endothelial cells used in the studies, based on the fact that a subset displayed exquisite sensitivity to chelerythrine. Overall, these results support the notion that resveratrol and shear stress induce elongation of the EC cytoskeleton via an overlapping outside-in signaling mechanism.
Prolonged shear stress leading to mechanotransduction signaling has been shown to activate the MAP kinase pathway in EC. Activation of ERK1 and ERK2, the major components of the MAPK pathway, presumably induces shear-specific transcription factors, such as c-fos/c-jun and NFκB, ultimately leading to changes in gene expression [30,31]. Western blot analysis of control and 25 μM and 100 μM resveratrol-treated BPAEC showed that there was no increase in active ERK 1/2 at the 6 h and 2 day time points. Rather, the decline in activated ERK 1/2 seen from days 2 to 4 of control cells, was effectively suppressed by resveratrol, suggesting that the polyphenol may act by affecting the stability of active ERK1/2 (Figure 6). This may involve modulation of MAPK phosphatase-1 (MKP-1), a member of the immediate-early response gene product functioning as a dual specificity phosphase to reverse MAPK [40,41,42,43,44], by resveratrol. Previously, it has been reported that MKP-1 is rapidly induced in rat carotid arterial wall following balloon catheter injury [45]. Since no noticeable change in active ERK1/2 occurred at the 6 h or 2 day time points, where morphological changes clearly became visible, following treatment with resveratrol, it seems unlikely that the activation of MAPK is directly linked to the observed morphologic changes.
Responses of cultured endothelial cells to arterial shear stress
Another significant contribution of the present research is the demonstration that resveratrol promoted a greater adherence of BPAEC to the cultured vehicle in vitro. In vivo, such a cellular property could make endothelial cells less likely to dislodge to become part of a growing thrombotic plug. Restricted detachment could also imply that there is less degeneration of the endothelial cell monolayer, which, in turn, would reduce the exposure of the underlying subendothelial matrix components, thereby making platelet adhesion and aggregation less likely. The mechanism(s) responsible for the resveratrol-induced cellular properties remain to be further investigated. One possibility centers on modulation of the number of focal contact adhesion sites, and/or the effective redistribution of the focal contacts, as well as the increased production of the EC-specific integrin complex, by resveratrol, all of which could contribute to the cardioprotective mechanism of this polyphenol. However, it should be emphasized that these experiments involved growing EC on plastic, not on layers of subendothelial matrix components such as collagen or fibrinogen. The latter experimental format simulating a more physiologically relevant subendothelial matrices may yield vastly different results. These possibilities warrant further investigation.
Induction of eNOS and modulation of EC growth by resveratrol
The endothelial cell lining of the blood vessel is extremely sensitive to damage from reactive oxygen species (ROS), the results of which are losses of both microvascular metabolic function and barrier properties [46]. To minimize such oxidant damage, cells rely on the production of nitric oxide (NO) by the enzyme nitric oxide synthase (eNOS). Biological functions attributed to NO include vasodilation [47], inhibition of platelet adhesion and aggregation [48], reduction of expression of adhesion molecules and chemokines [49,50,51,52], and suppression of cell growth and migration [53,54]. Therefore, we investigated whether resveratrol treatment could lead to increased eNOS expression in endothelial cells. We determined that resveratrol treatment did induce eNOS expression at all time points tested (6 h, 2 days, and 4 days). These results agreed with a previous report from this laboratory [21]. The peak expression of eNOS occurred at 2 days in 100 μM resveratrol-treated cells. These findings could mean that dietary resveratrol is capable of providing a gradual yet sustained increase in NO. The ability of resveratrol to induce S/G2 growth arrest [20,21] suggests an additional mechanism for cells to rapidly and efficiently repair any endothelial cell damage. Overall, the collective effect of resveratrol would be to decrease endothelial injury and exposure of the subendothelial matrix, which would lessen the probability of formation of atherosclerotic plaques and the development of CHD [53,54,55].
Conclusions
Our studies suggest that resveratrol interacts with endothelial cells in vitro to elicit morphological and functional changes. Specifically, resveratrol induced an elongated shape, interspersed with long, tortuous projections, in cultured BPAEC. Treated cells resisted detachment from the plastic coverslips under simulated arterial shear stress conditions. These results, combined with data from our previous studies, provide additional support for the notion that resveratrol acts as an anti-atherosclerotic agent. Whether these findings can be extrapolated to humans require further study.
Materials and Methods
Stock solution (12.5 mM) of resveratrol (Sigma Chemical Co.) was prepared in DMSO and stored at -20°C. For treatment, the resveratrol was diluted in RPMI 1640 and added to cultures to give the desired final concentrations. Untreated cultures received the same amount of the carrier solvent (0.2% DMSO). The rhodamine-conjugated phalloidin, used to detect F-actin, was purchased from Molecular Probes, Eugene, OR, and stored at 4°C in methanol. Before use, the solvent methanol was evaporated using N2 gas and the F-actin probe was resolubilized in TBS (Tris-buffered saline).
Cell culture and treatment with resveratrol
The BPAEC isolated from the distal main intrapulmonary artery of calf lungs were cultured in minimal essential medium (MEM) supplemented with 15% fetal bovine serum and containing D-valine as previously described [56]. Plastic coverslips on which the cells were cultured were sterilized by washing in ethanol, followed by rinsing three times in MEM and 15 min exposure to UV radiation. The coverslips were placed in 6-well tissue culture plates, and 2 ml of the cells, at an initial density of 1 × 105 cells/ml, were added to each of the wells. Twenty-four h after seeding, the coverslips were treated with 0,10, 25, 50, and 100 μM resveratrol and maintained for 2 days at 37°C until they were fixed and stained for confocal microscopy.
Preparation and staining of slides
Coverslips containing adhered BPAEC were washed for 5 min in TBS, fixed with 5% glutaraldehyde in 0.1 M cacodylate (pH 7.4) for 15 min, washed again for 5 min in TBS, then stored in 0.1 M cacodylate/7% sucrose at 4°C until staining. Cells were permeabilized with 0.1% Triton X-100/TBS for 3 min, washed twice with TBS, blocked twice for 3 min with 0.1% bovine serum albumin in TBS, and incubated with rhodamine-conjugated phalloidin for 20 min. Routinely, 5 μl of rhodamine-phalloidin, dissolved in 200 μl TBS, was used for each coverslip stained. The coverslips were wash with 0.1% BSA/TBS for 3 min, then mounted in an inverted position onto TBS-containing beveled glass slides and sealed with clear nail polish.
Perfusion
A parallel plate perfusion chamber as described previously [57] was used to generate simulated arterial flow conditions. The chamber consisted of three pieces: two rectangular slabs and a central knob with a depression that fits 18 by 21 mm coverslips. The depression containing the coverslip was placed perpendicularly to the flow of perfusate. A laminar arterial shear rate of 650 s-1 was achieved based on the dimensions of the flow chamber slit and a constant flow rate of 10 ml/min. After preincubation at 37°C, minimal medium was drawn directly by an eight roller peristaltic pump through the perfusion chamber containing the BPAEC-covered coverslip positioned 70 mm from the inlet valve at a rate of 10 ml/min for either 2 min or 5 min. A recirculating system was employed to recycle the medium after passing through the chamber.
Confocal microscopy
Prepared slides containing adhered BPAEC were examined using a BioRad confocal microscope. The cells were imaged following excitation of rhodamine-phalloidin with an argon laser beam at a wavelength of 554 nm using either an inverted- or an uprighted-staged microscope equipped with either a 10X or a 20X oil-free objective and an epifluorescent illumination attachment. Cells viewed in representative field screen captures were counted manually for cell morphology and classified as either normal or elongated.
Inhibitor study
Passages 4-5 BPAEC were seeded on plastic coverslips as described above. Inhibitor concentrations were the same as in [28]. Stock solutions were prepared by dissolving the inhibitors in DMSO. The inhibitors used and their stock solutions were as follows: quin2-AM (Sigma, 2 mM), herbimycin A (GibcoBRL, 0.175 mM), chelerythrine (Sigma, 0.4 mM), cytochalasin D (Sigma, 8 μM), nocodazole (Sigma, 0.66 mM). Quin2-AM, herbimycin A, chelerythrine, and cytochalasin D were stored at -20°C, and nocodazole was stored at 4°C. The stock solutions were diluted 1:200 in MEM to give the following final concentrations: quin2-AM (10 μM), herbimycin A (875 nM), chelerythrine (2 μM), cytochalasin D (40 nM), nocodazole (3.3 μM). Inhibitors were applied 24 h prior to the addition of resveratrol, except for quin2-AM, which was applied 1 h prior to resveratrol treatment. Since cells treated with various inhibitors grew less well, compared to controls, treatment with 100 μM resveratrol was reduced to approximately 40 h. Fixing and staining of cells for confocal microscopy was as described above.
Western blot analysis
Control and treated cells were lysed by repeated freeze-thaw cycles with buffer containing 10 mM HEPES (pH 7.5), 1.5 mM Mg(OAc)2, 1 mM dithiothreitol (DTT), 0.5% NP40, 5% glycerol, 0.5 mM phenylmethylsulfonyl fluoride, and 10 μg/ml each of the protease inhibitors aprotinin, pepstatin, and leupeptin. Cell-free extracts were obtained by centrifugation in a microcentrifuge. Lysates (7-10 μg) from control and treated cells were separated on 10% SDS-PAGE. The separated proteins were transferred to nitrocellulose membranes, and the membranes were incubated with the respective primary and secondary antibodies. The monoclonal primary antibodies used and their dilutions were as follows: Phosphorylated, active ERK1/2-P (RBI/Sigma, 1:1000), total ERK1/2 (RBI/Sigma, 1:1000), eIF4E (Santa Cruz, 1:250), eNOS (Sigma 1:1000). Membranes were probed with alkaline phosphatase-conjugated IgG (Santa Cruz, 1:1500) or horseradish peroxidase-conjugated IgG (Santa Cruz, 1:2000). Specific immunoreactive bands were identified by color reaction or enhanced chemiluminescence, respectively.
Acknowledgments
Acknowledgements
Supported in part by the Vivien Wu-Au Memorial Cancer Research Fund and an unrestricted grant from the Philip Morris Company to J.M.W., and by grant 9816832 from the National Science Foundation to K.M.L.
Contributor Information
Jed L Bruder, Email: Jed_bruder@nymc.edu.
Tze-chen Hsieh, Email: Tze-chen_Hsiech@nymc.edu.
Kenneth M Lerea, Email: Ken_Lerea@nymc.edu.
Susan C Olson, Email: Susan_Olson@nymc.edu.
Joseph M Wu, Email: Joseph_Wu@nymc.edu.
References
- Hunink MG, Goldman L, Tosteson AN, Mittleman MA, Goldman PA, Williams LW, Tsevat J, Weinstein MC. The recent decline in mortality from coronary heart disease, 1980-1990. J Am Med Assoc. 1977;277:535–542. doi: 10.1001/jama.277.7.535. [DOI] [PubMed] [Google Scholar]
- Strong JP, Malcom GT, McMahan CA, Tracey RE, Newman WP, III, Herderick EE, Cornhill JF. Prevalence and extent of atherosclerosis in adolescents and young adults. Implications for prevention from the pathobiological determinants of atherosclerosis in youth study. J Am Med Assoc. 1999;281:727–735. doi: 10.1001/jama.281.8.727. [DOI] [PubMed] [Google Scholar]
- Gutstein WH. Vasopasm, vascular injury, and atherogenesis: A perspective. Hum Pathol. 1999;30:365–371. doi: 10.1016/s0046-8177(99)90109-0. [DOI] [PubMed] [Google Scholar]
- Bronte-Stewart B. The role of dietary fats in ischemic heart disease. Br Med Bull. 1958;34:243–251. doi: 10.1093/oxfordjournals.bmb.a069691. [DOI] [PubMed] [Google Scholar]
- Ducimetiere P, Richard JL, Cambien R, Rakotovas R, Claude JR. Coronary heart disease in middle-aged Frenchmen. Comparison between Paris prospective study, Seven Counties and Pooling project. Lancet. 1980;1:1346–1350. doi: 10.1016/S0140-6736(80)91796-1. [DOI] [PubMed] [Google Scholar]
- Renaud S, De Lorgeril M. The French paradox: dietary factors and cigarette smoking-related health risks. Ann NY Acad Sci. 1993; 686:299–309. doi: 10.1111/j.1749-6632.1993.tb39191.x. [DOI] [PubMed] [Google Scholar]
- Renaud S, De Lorgeril M. Wine, alcohol, platelets and the French paradox for coronary heart disease. Lancet. 1992;339:1523–1526. doi: 10.1016/0140-6736(92)91277-F. [DOI] [PubMed] [Google Scholar]
- Seigneur M, Bonnet J, Dorian B, Benchimol D, Drouillet D, Gouverneur G, Larrue J, Crockett R, Boiseau M, Gayon R, Bricaus H. Effect of consumption of alcohol, wine wine, and red wine on platelet function and serum lipids. J Appl Cardiol. 1990;5:215–222. [Google Scholar]
- Klatsky AL, Armstrong MA, Friedman GD. Relations of alcoholic beverage use to subsequent coronary artery disease hospitalization. Am J Cardiol. 1986;58:710–714. doi: 10.1016/0002-9149(86)90342-5. [DOI] [PubMed] [Google Scholar]
- Siscovick DS, Weiss NS, Fox N. Moderate alcohol consumption and primary cardiac arrest. Am J Epidemiol. 1986;123:499–503. doi: 10.1093/oxfordjournals.aje.a114265. [DOI] [PubMed] [Google Scholar]
- Scragg R, Stewart A, Jackson R, Beaglehole R. Alcohol and exercise in myocardial infarction and sudden coronary death in men and women. Am J Epidemiol. 1987;126:77–85. doi: 10.1093/oxfordjournals.aje.a114664. [DOI] [PubMed] [Google Scholar]
- Klatsky AL, Armstrong MA, Friedman GD. Risk of cardiovascular mortality in alcohol drinkers, ex-drinkers and nondrinkers. Am J Cardiol. 1990;66:1237–1242. doi: 10.1016/0002-9149(90)91107-h. [DOI] [PubMed] [Google Scholar]
- Jackson R, Scragg R, Beaglehole R. Alcohol consumption and risk of coronary heart disease. Br Med J. 1991;303:211–216. doi: 10.1136/bmj.303.6796.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klatsky AL, Armstrong MA, Friedman GD. Alcohol and mortality. Ann Intern Med. 1992;117:646–654. doi: 10.7326/0003-4819-117-8-646. [DOI] [PubMed] [Google Scholar]
- Klurfield DM, Kritchevsky D. Differential effects of alcoholic beverages on experimental atherosclerosis in rabbits. Exp Mol Pathol. 1981;34:62–71. doi: 10.1016/0014-4800(81)90036-8. [DOI] [PubMed] [Google Scholar]
- Demrow HS, Slane BS, Folts JD. Administration of wine and grape juice inhibits in vivo platelet activity and thrombosis in stenosed canine coronary arteries. Circulation. 1995;91:1182–1188. doi: 10.1161/01.cir.91.4.1182. [DOI] [PubMed] [Google Scholar]
- Meyer AS, Yi OS, Pearson DA, Waterhouse AL, Frankel EN. Inhibition of human low-density lipoprotein oxidation in relation to composition of phenolic antioxidants in grapes (Vitis vinifera). J Agric Food Chem. 1997;45:1638–1643. doi: 10.1021/jf960721a10.1021/jf960721a. [DOI] [Google Scholar]
- Siemann EH, Creasy LL. Concentration of the phytoalexin resveratrol in wine. Am J Enol Vitic. 1992;43:49–52. [Google Scholar]
- Zou JG, Huang YZ, Chen Q, Wei EH, Cao KJ, Wu JM. Effect of resveratrol on oxidative modification of human low density lipoprotein. Chinese Med J. 2000;113:99–102. [PubMed] [Google Scholar]
- Zou JG, Huang YZ, Chen Q, Wang N, Cao KJ, Hsieh TC, Wu JM. Suppression of mitogenesis and regulation of cell cycle traverse by resveratrol in cultured smooth muscle cells. Int J Oncol. 1999;15:647–651. doi: 10.3892/ijo.15.4.647. [DOI] [PubMed] [Google Scholar]
- Hsieh TC, Juan G, Darzynkiewicz Z, Wu JM. Resveratrol increases nitric oxide synthase, induces accumulation of p53 and p21 (WAF1/CiP1), and suppresses cultured bovine pulmonary artery endothelial cell proliferation by perturbing progression through S and G2. Canc Res. 1999;59:2596–2601. [PubMed] [Google Scholar]
- Pace-Asciak CR, Hahn S, Diamandis EP, Soleas G, Goldberg DM. The red wine phenolics trans-resveratrol and quercetin block human platelet aggregation and eicosanoid synthesis: Implications for protection against coronary heart disease. Clin Chim Acta. 1995;235:207–219. doi: 10.1016/0009-8981(95)06045-1. [DOI] [PubMed] [Google Scholar]
- Bertelli AA, Giovannini L, De Caterina R, Bernini W, Migliori M, Fregoni M, Bavaresco L, Bertelli A. Antiplatelet activity of cis-resveratrol. Drugs Exptl Clin Res. 1996;222:61–63. [PubMed] [Google Scholar]
- Rotondo S, Totilio D, Cerletti C, deGaetano G. Red wine, aspirin and platelet function. Thrombosis Haemostasis. 1996;76:818–819. [PubMed] [Google Scholar]
- Diaz MN, Frei B, Vita JA, Keaney JF., Jr Antioxidants and atherosclerotic heart disease. New Engl J Med. 1997;337:408–416. doi: 10.1056/NEJM199708073370607. [DOI] [PubMed] [Google Scholar]
- Lichtenstein AH. Soy protein, isoflavones and cardiovascular disease risk. J Nutr. 1999;128:1589–1592. doi: 10.1093/jn/128.10.1589. [DOI] [PubMed] [Google Scholar]
- Heller R, Munscher-Paulig F, Grabner R, Till U. L-ascorbic acid potentiatesx nitric oxide synthesis in endothelial cells. J Biol Chem. 1999;274:8254–8260. doi: 10.1074/jbc.274.12.8254. [DOI] [PubMed] [Google Scholar]
- Malek AM, Izumo S. Mechanism of endothelial cell shape change and cytoskeletal remodeling in response to fluid shear stress. J Cell Sci. 1996;109:713–726. doi: 10.1242/jcs.109.4.713. [DOI] [PubMed] [Google Scholar]
- Galbraith CG, Skalak R, Chien S. Shear stress induces spatial reorganization of the endothelial cell cytoskeleton. Cell Motility and the Cytoskeleton. 1998;40:317–330. doi: 10.1002/(SICI)1097-0169(1998)40:4<317::AID-CM1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Berk BC. Mitogen-activated protein kinase (ERK1/2) activation by shear stress and adhesion in endothelial cells. Essential role for herbimycin-sensitive kinase. J Clin Invest. 1996;98:2623–2631. doi: 10.1172/JCI119083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo H, Sipos K, Go YM, Law R, Rong J, McDonald JM. Differential effect of shear stress on extracellular signal-regulated kinase and N-terminal Jun kinase in endothelial cells. Gi2 and Gβ/γ-dependent signaling pathways. J Biol Chem. 1997;272:1395–1401. doi: 10.1074/jbc.272.2.1395. [DOI] [PubMed] [Google Scholar]
- Tertov VV, Orekhov AN, Repin VS, Smirnov VN. Dibutyryl cyclic AMP decreases proliferative activity and the cholesteryl ester content in cultured cells of atherosclerotic human aorta. Biochem Biophys Res Commun. 1982;109:1228–1233. doi: 10.1016/0006-291x(82)91908-8. [DOI] [PubMed] [Google Scholar]
- Antonov AS, Lukashev ME, Romanov YA, Tkachuk VA, Repin VS, Smirnov VN. Morphological alterations in endothelial cells from human aorta and umbilical vein induced by forskolin and phorbol 12-myristate 13-acetate: a synergistic action of adenyl cyclase and protein kinase C activators. Proc Natl Acad Sci USA. 1986;83:9704–9708. doi: 10.1073/pnas.83.24.9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunhut SJ, Palomares M. Modulation of endothelial cell shape and growth by retinoids. Microvasc Res. 1991;41:47–62. doi: 10.1016/0026-2862(91)90007-x. [DOI] [PubMed] [Google Scholar]
- Huonker M, Halle M, Keul J. Structural and functional adaptations of the cardiovascular system by training. Int J Sports Med. 1996;17 (Suppl. 3):S164–S172. doi: 10.1055/s-2007-972919. [DOI] [PubMed] [Google Scholar]
- Niebauer J, Cooke JP. Cardiovascular effects of exercise: role of endothelial shear stress. J Am Coll Cardiol. 1996;28:1652–1660. doi: 10.1016/S0735-1097(96)00393-2. [DOI] [PubMed] [Google Scholar]
- Sato M, Ohshima N. Flow-induced changes in shape and cytoskeletal structure of vascular endothelial cells. Biorheology. 1994;31:143–153. doi: 10.3233/bir-1994-31202. [DOI] [PubMed] [Google Scholar]
- Guzman RJ, Abe K, Zarins CK. Flow-induced arterial enlargement is inhibited by suppression of nitric oxide synthase activity in vivo. Surgery. 1997;122:273–279. doi: 10.1016/s0039-6060(97)90018-0. [DOI] [PubMed] [Google Scholar]
- Barakat AI. Responsiveness of vascular endothelium to shear stress: potential role of ion channels and cellular cytoskeleton (review). Int J Mol Med. 1999;4:323–332. doi: 10.3892/ijmm.4.4.323. [DOI] [PubMed] [Google Scholar]
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylate MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- Bokemeyer D, Sorokin A, Yan M, Ahn NG, Templeton DJ, Dunn MJ. Induction of mitogen-activated protein kinase phosphatase 1 by the stress-activated protein kinase signaling pathway but not by extracellular signal-regulated kinase in fibroblasts. J Biol Chem. 1996;271:639–642. doi: 10.1074/jbc.271.2.639. [DOI] [PubMed] [Google Scholar]
- Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U-937 cells. J Biol Chem. 1997;272:16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- Shapiro PS, Ahn NG. Feedback regulation of Raf-1 and mitogen-activated protein kinase (MAP) kinase kinases 1 and 2 by MAP kinase phosphatase-1 (MKP-1). J Biol Chem. 1998;273:1788–1793. doi: 10.1074/jbc.273.3.1788. [DOI] [PubMed] [Google Scholar]
- Lee HY, Sueoka N, Hong WK, Mangelsdorf DJ, Claret FX, Kurie JM. All-trans-retinoic acid inhibits Jun N-terminal kinase by increasing dual-specificity phosphatase activity. Mol Cell Biol. 1999;19:1973–1980. doi: 10.1128/mcb.19.3.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama H, Olson NE, Dastvan FF, Reidy MA. Cell replication in the arterial wall: activation of signaling pathway following in vivo injury. Circ Res. 1998;82:713–721. doi: 10.1161/01.res.82.6.713. [DOI] [PubMed] [Google Scholar]
- Kuo PC, Schroeder PA. The emerging multifaceted roles of nitric oxide. Ann Surg. 1995;221:220–235. doi: 10.1097/00000658-199503000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2:1057–1058. doi: 10.1016/S0140-6736(87)91481-4. [DOI] [PubMed] [Google Scholar]
- Bath PM, Hassall DG, Gladwin AM, Palmer RM, Martin JF. Nitric oxide and prostacyclin. Divergence of inhibitory effects on monocyte chemotaxis and adhesion to endothelium in vitro. Arterioscler Thromb. 1991;11:254–260. doi: 10.1161/01.atv.11.2.254. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier TW, Scalia R, Murohara T, Guo JP, Lefer AM. Nitric oxide protects against leukecyte-endothelium interactions in the early stages of hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1995;15:1652–1659. doi: 10.1161/01.atv.15.10.1652. [DOI] [PubMed] [Google Scholar]
- Zeiher AM, Fisslthaler B, Schray Utz B, Busse R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ Res. 1995;76:980–986. doi: 10.1161/01.res.76.6.980. [DOI] [PubMed] [Google Scholar]
- Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res. 1996;78:225–230. doi: 10.1161/01.res.78.2.225. [DOI] [PubMed] [Google Scholar]
- Lloyd Jones DM, Bloch KD. The vascular biology of nitric oxide and its role in atherogenesis. Annu Rev Med. 1996;47:365–375. doi: 10.1146/annurev.med.47.1.365. [DOI] [PubMed] [Google Scholar]
- Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- Olson S, Dowds TA, Pino PA, Barry MT, Burke-Wolin T. ANG II stimulates endothelial nitric oxide synthase expression in bovine pulmonary artery endothelium. Am J Physiol. 1997;273:L315–L321. doi: 10.1152/ajplung.1997.273.2.L315. [DOI] [PubMed] [Google Scholar]
- Sakariassen KS, Aarts PA, de Groot PG, Houdijk WP, Sixma JJ. A perfusion chamber developed to investigate platelet interaction in flowing blood with human vessel wall cells, their extracellular matrix, and purified components. J Lab Clin Med. 1983;102:522–535. [PubMed] [Google Scholar]