

Abstract
The nuclear vitamin D receptor (VDR) binds 1,25-dihydroxyvitamin D3 (1,25D), its high affinity renal endocrine ligand, to signal intestinal calcium and phosphate absorption plus bone remodeling, generating a mineralized skeleton free of rickets/osteomalacia with a reduced risk of osteoporotic fractures. 1,25D/VDR signaling regulates the expression of TRPV6, BGP, SPP1, LRP5, RANKL and OPG, while achieving feedback control of mineral ions to prevent age-related ectopic calcification by governing CYP24A1, PTH, FGF23, PHEX, and klotho transcription. Vitamin D also elicits numerous intracrine actions when circulating 25-hydroxyvitamin D3, the metabolite reflecting vitamin D status, is converted to 1,25D locally by extrarenal CYP27B1, and binds VDR to promote immunoregulation, antimicrobial defense, xenobiotic detoxification, anti-inflammatory/anticancer actions and cardiovascular benefits. VDR also affects Wnt signaling through direct interaction with β-catenin, ligand-dependently blunting β-catenin mediated transcription in colon cancer cells to attenuate growth, while potentiating β-catenin signaling via VDR ligand-independent mechanisms in osteoblasts and keratinocytes to function osteogenically and as a pro-hair cycling receptor, respectively. Finally, VDR also drives the mammalian hair cycle in conjunction with the hairless corepressor by repressing SOSTDC1, S100A8/S100A9, and PTHrP. Hair provides a shield against UV-induced skin damage and cancer in terrestrial mammals, illuminating another function of VDR that facilitates healthful aging.
Keywords: Vitamin D receptor; 1,25-dihydroxyvitamin D3; Calcium metabolism; Phosphate metabolism; Fibroblast growth factor 23; Klotho; CYP24A1; Osteocalcin (BGP); Osteopontin (SSP1); LRP5; TRPV6; RANKL; OPG; β-catenin; Hairless; S100A8; SOSTDC1
1. Introduction
The nuclear vitamin D receptor (VDR) is a member of the thyroid hormone and retinoic acid receptor subfamily of nuclear hormone receptors that heterodimerizes with retinoid X receptor (RXR) isoforms to regulate the expression of genes encoding factors which, in a variety of cell types, control functions such as proliferation, differentiation, metabolism, ion transport, apoptosis, etc. [1]. Evolutionarily, VDR is most closely related to the pregnane X receptor (PXR) that triggers xenobiotic detoxification and to the farnesoid X receptor (FXR) which governs bile acid metabolism [1]. In fact, the ancient function of VDR in chordates is thought to be that of detoxification [2, 3], a property that apparently has been retained in extant mammals as evidenced by the ability of VDR to bind the carcinogenic secondary bile acid, lithocholic acid (LCA), with low affinity and signal its detoxification in colon via induction of CYP3A4 and SULT2 [4, 5].
The modern vitamin D endocrine system, in which the high affinity hormonal ligand for VDR, 1,25-dihydroxyvitamin D3 (1,25D), is generated in the kidney according to the calcium and phosphorus needs of the animal, likely evolved when terrestrial animals were forced into locomotion facilitated by a mineralized skeleton in order to seek calcium, considering its limited availability on land compared to in the sea. Thus, under the control of PTH, which signals low calcium, the renal 1α-OHase (CYP27B1) catalyzes the production of 1,25D, the vitamin D hormone that binds VDR to induce small intestinal calcium absorption with the aid of TRPV6 and other calcium translocation gene products [6]. Accordingly, the predominant phenotype of VDR null mice is that of post-weaning rickets, rectifiable with a high calcium/lactose/phosphate rescue diet [7]. However, a second feature of the VDR null mouse, namely alopecia, is not ameliorated by the rescue diet, and seems to be independent of the vitamin D ligand [8]. Thus, a second major function of VDR is driving the mammalian hair cycle, a phenomenon which protects terrestrial animals seeking calcium from damaging UV exposure and co-evolved with the renal VDR ligand and a mineralized skeleton [9].
VDR regulates the expression of many genes beyond those involved in calcium absorption and the mammalian hair cycle. Based upon numerous studies, including cDNA microarray analysis of mRNAs, as many as 500-1000 genes may be modulated by VDR ligands [10]. Many of these effects on gene expression are primary in that they involve direct VDR-RXR binding to vitamin D responsive elements (VDREs) in or near the genes in question (i.e., within approximately 100 kb either 5′ or 3′ of the transcription start site), leading to the concept of a “vitamin D receptor cistrome” analogous to the estrogen receptor α cistrome mapped by Brown and colleagues [11]. Coupled with the knowledge that although VDR is most highly expressed in small intestine, colon, kidney, bone and skin, the receptor is present in reasonable copy numbers in many other tissues and cell types including brain, the vascular system, numerous endocrine organs, the immune system, muscle, etc. [12], plus the existence of many extrarenal sites of CYP27B1 expression [13] to catalyze local 1,25D production, it is reasonable to assume that many of the recently discovered health benefits of vitamin D beyond bone can be explained by intracrine 1,25D actions fed by the circulating 25(OH)D precursor. These effects of 1,25D are likely redundant with other biological modifiers, and their associated pathologies are subtle at best in VDR null mice, but they could be biologically significant in times of stress and especially in aging. The purpose of the present communication is to identify the key genes regulated by VDR for which their products mediate calcium and phosphate transport, bone remodeling, and other traditional VDR actions, as well as to unveil novel genes that mediate the other health benefits of vitamin D. We will also develop the thesis that many of the genes governed by VDR encode proteins which participate in processes that allow one to age well with lowered risk of chronic diseases which cause premature death. In this vein, we propose that VDR actions in effect constitute a biochemical “Fountain of Youth” to preserve health into old age. This hypothesis may seem counterintuitive in that vitamin D is produced in skin by sunlight, UV wavelengths of which are considered to promote aging, suppress the immune system, and cause skin cancer. Moreover, vitamin D facilitates calcium and phosphate absorption and retention which lead to arteriosclerosis and other disorders characterized by ectopic calcification. However, 1,25D and its receptor also possess the ability to modulate expression of genes which counterregulate calcium and phosphate to prevent ectopic calcification. Finally, the sunshine vitamin, via VDR, has the capacity to lower the risk of skin cancer by reversing the deleterious effects of UV light that produce it in skin. And VDR itself promotes hair growth and replenishment to shield against UV, possibly the only incidence in biology of independent actions of a receptor and its ligand to prevent a pathology resulting from conditions that promote synthesis of that same ligand.
2. Materials and methods
See Figure Legends for methods
3. Results and discussion
3.1 Mechanism of induction by 1,25D-VDR
Control of the murine osteopontin gene (SPP1) by 1,25D, originally reported by Noda et al. [14], exemplifies the prototypical mechanism whereby 1,25D-VDR directly upregulates the expression of target genes. The murine SPP1 promoter possesses the strongest single natural VDRE yet reported, residing at -757 bp. Fig. 1A illustrates this VDRE, a perfect direct repeat with a spacer of three nucleotides, GGTTCAcgaGGTTCA, in the context of the natural SPP1 promoter fused to a GH reporter gene. We altered three base pairs in the 3′-half element, where VDR binds to the DNA [15], to generate a TATCCA mutant half-element in the context of the promoter, leaving the 5′-half element, where RXR binds, unaltered and tested the effect of this modification on SPP1 induction by 1,25D. As is evident from the results in Fig. 1A, the robust 7.3-fold stimulation of transcription by 1,25D with the native promoter is completely abrogated by altering the 3′-half element of the VDRE. These data indicate that 1,25D-VDR-RXR must contact a native VDRE to induce the expression of genes such as SPP1, a conclusion that is confirmed by chromatin immunoprecipitation (ChIP) results showing that VDR associates with the murine SPP1 VDRE at -757 bp in a 1,25D ligand-dependent manner in intact MC3T3-E1 cells [16]. Interestingly, mutation of the 3′ half-element in SPP1 actually transforms 1,25D-VDR into a repressor, with a significant 60% reduction in transactivation (Fig. 1A). This suggests that, when bound to this mutated half-element, VDR assumes a repressive conformation, likely recruiting corepressors instead of coactivators. The reverse of this experiment was performed by Koszewski et al. [17], who transformed a negative VDRE in the chicken PTH gene into a positive, also by altering base pairs in the 3′-half element. Thus, both primary induction and repression of target genes by 1,25D consist of direct binding of VDR to VDREs, recruiting coactivators and corepressors, respectively. No intermediary protein transcription factors are involved in this mechanism, as it is insensitive to inhibition by cycloheximide, an inhibitor of new protein synthesis (Fig. 1B), again consistent with a major mechanism of 1,25D-VDR action being that of primary transcriptional regulation.
Fig. 1.
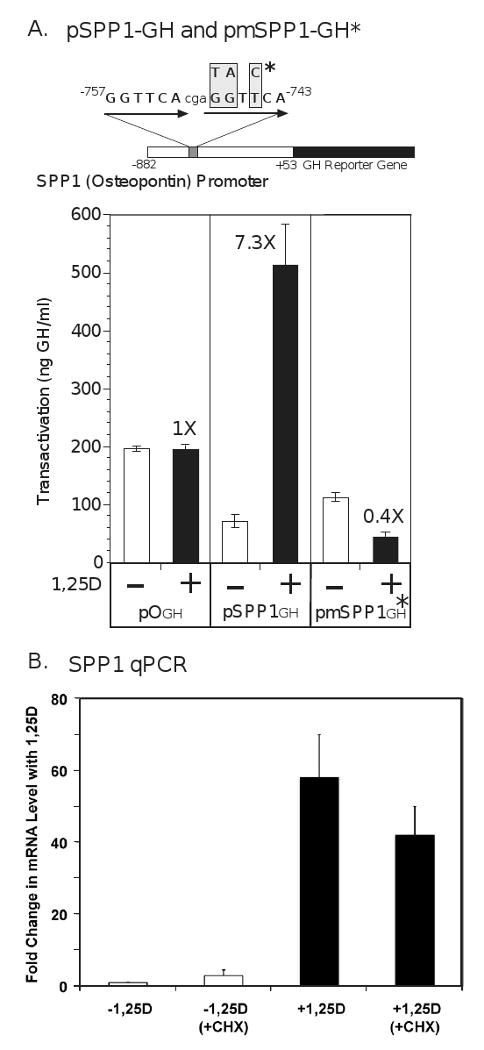
Osteopontin (SPP1) expression is induced by 1,25-VDR via a primary transcriptional mechanism involving a perfect direct repeat-3 (DR3) VDRE at -757 bp in the proximal promoter of the murine gene. (A) The mouse SPP1 proximal promoter obtained from Dr. David Dempster [94] was linked to the growth hormone (GH) reporter gene. ROS 17/2.8 rat osteosarcoma cells were cotransfected with an expression vector (1 μg) containing either the wild type version of the mouse osteopontin promoter (pSPP1GH) or its mutated VDRE version (pmSPP1GH*). The insertless vector p0GH was included as a control. Cells were treated at 12 hours post-transfection with either ethanol vehicle (-) or 1,25D at a concentration of 10 nM (+), and cell media assayed by RIA for growth hormone at 48 hours post-transfection. The data depicted represent the means (±SD) of two independent experiments with n=3 for each assay. The numbers at the top of the dark colored bars indicate the fold-induction of transcriptional activity following treatment with ligand. (B) UMR-106 rat osteosarcoma cells were treated with 100 nM 1,25D or ethanol vehicle for 24 hours in the presence (+CHX) or absence of 10 μM cycloheximide added 30 min prior to the 1,25D. All groups received both ethanol (for 1,25D) and DMSO (for cycloheximide) vehicles. RNA was isolated and reverse transcribed into cDNA that was assayed by real time PCR using custom primers against the coding region of SPP1, as described previously [16]. All values are normalized against the unregulated GAPDH control cDNA and are expressed as mean fold change ±SEM for three independent experiments, each performed in triplicate.
3.2 Genes for which expression is regulated by 1,25D
1,25D-VDR controls the expression of at least eleven genes (SPP1, TRPV6, LRP5, BGP, RANKL, OPG, CYP24A1, PTH, FGF23, PHEX, and klotho) which encode bone and mineral homeostasis effectors that also facilitate aging well. The first, osteopontin or SPP1, which is dramatically induced by 1,25D in osteoblast-like cells (Fig. 1B), was initially named for its positive actions in bone, which include inducing ossification, especially in response to mechano-stress/fracture, and remodeling [18], both of which strengthen the skeleton to promote longevity and reduce fracture risk. Moreover, SPP1 facilitates angiogenesis and osteoclast accumulation for the resorption of ectopic bone; in fact, SPP1 is an inducible inhibitor of vascular calcification and associated disease [19]. Other benefits of SPP1 are the effecting of immune homeostasis and the promotion of myelination [20]. However, there is a down-side to induction of osteopontin in that it is proinflammatory and possesses promalignancy/prometastasis [21] properties. Fortunately, these potentially deleterious effects likely are counterbalanced by numerous other 1,25D-induced gene products which are anti-inflammatory and anticancer as discussed below.
Intestinal calcium absorption is mediated, in part, by 1,25D-VDR induction of TRPV6, which is illustrated for human Caco-2 cells in Fig. 2 (inset at upper right). This is a direct action of VDR, because the VDRE at -2.1 kb as well as other VDREs in the human TRPV6 gene are occupied by the liganded receptor complex in intact intestinal cells as assessed by ChIP experiments [16, 22]. TRPV6 represents a key calcium channel gene product that supplies dietary calcium via transport to build the mineralized skeleton and thereby delay the inevitable calcium leaching from bone in senile osteoporosis. Accordingly, TRPV6 null mice have 60% decreased intestinal calcium absorption, decreased bone mineral density (BMD) and, strikingly, 20% of animals exhibit alopecia and dermatitis [23] similar to VDR knockout mice [24]. Since the skin phenotype in VDR null mice is not ameliorated by the high calcium rescue diet [7], we speculate that TRPV6 may mediate calcium entry into keratinocytes to elicit differentiation and hair cycling. Because calcium is protective against colon cancer [25], while hair plus a full stratum corneum reduce UV-induced skin damage and cancer, VDR-induced TRPV6 could function in colon and skin to lower the risk of neoplasia in these two epithelial cell types.
Fig. 2.
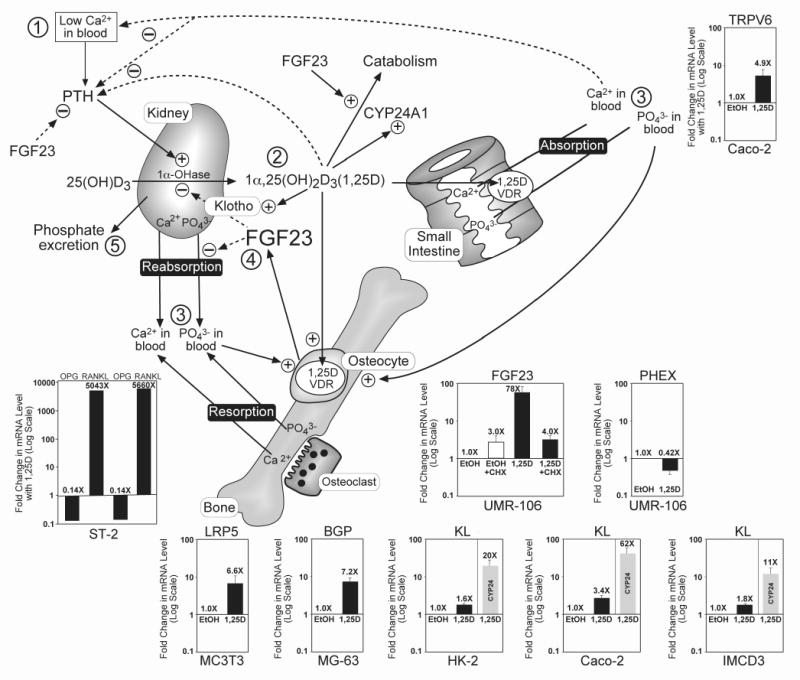
1,25D-VDR regulates the expression of many genes whose products effect bone mineral homeostasis and the integrity of the endoskeleton, as well as promote healthful aging. Insets show the induction or repression of each gene listed at the top, with the cell line denoted at the bottom of the inset. KL=klotho, a recognized longevity gene [51]. All mRNAs were quantitated via real time PCR as described previously [16], 24 hours after treatment of cells with 100 nM 1,25D or ethanol vehicle and expressed as fold-change (induction or repression) with 1,25D, plotted on a log scale with each value representing the mean ±SEM of at least three experiments each carried out in triplicate; an exception to the latter is ST2 mouse stromal cells, where RANKL and OPG mRNAs were measured in duplicate in the two experiments depicted in the inset and yielded essentially identical (0.14X) repression of OPG and over 5,000-fold induction of RANKL by 1,25D. Other cells utilized included Caco-2 human colon cancer, UMR-106 rat osteosarcoma, MG-63 human osteosarcoma, MC3T3 E1 mouse osteoblast-like, HK-2 human renal proximal tubule and mouse IMCD3 intermedullary collecting duct cells. The cycloheximide (CHX) protocol in the case of FGF23 regulation in UMR-106 cells was performed as described for SPP1 in Fig. 1B. Induction by 1,25D of LRP5, BGP and RANKL, plus repression of OPG, is discussed in the text as events that strengthen the skeleton via anabolic actions, extracellular matrix protein production and remodeling. The circled numbers in the schematic model portion of the figure represent the sequence of physiologic adjustments and feedback regulations of mineral metabolism after a reduction in blood calcium (➀) as follows: PTH-mediated enhancement of CYP27B1 (1α-OHase) and consequent rise in 1,25D hormone (➁); 1,25D-VDR action to cause calcium and phosphate absorption, reabsorption and resorption (➂) to raise both calcium and phosphate in blood; calcium and 1,25D negatively feedback (dashed lines) on PTH to close the calcemic loop, whereas 1,25D and phosphate cause osteocyte elaboration of FGF23 (➃), and 1,25D induces klotho to reduce 1,25D by inhibiting its production catalyzed by the 1α-OHase and inducing the CYP24A1 detoxification enzyme to accelerate its degradation; FGF23 also inhibits Npt2a/Npt2c-facilitated phosphate reabsorption to effect phosphate excretion (➄), protecting against hyperphosphatemia and ectopic calcification.
As illustrated in Fig. 2 (inset at lower left), 1,25D significantly induces LRP5, a gene product that promotes osteoblastogenesis via canonical Wnt signaling, and is thereby anabolic to bone [26]. This action of 1,25D is VDR-mediated directly through association with VDREs in the murine LRP5 gene [27], including one in the first intron at +656 bp in relation to the transcription start site which is occupied by liganded VDR as monitored by ChIP assay [16]. Interestingly, LRP5 is not only beneficial to bone through an anabolic action in osteoblasts, but we speculate that induction of LRP5 by 1,25D-VDR in duodenal enterochromaffin cells suppresses gut serotonin, a neurotransmitter which Karsenty and colleagues [28] have recently demonstrated is deleterious to bone and must be kept in check via an LRP5-triggered silencing mechanism. This is in contrast to central serotonin, which is downstream of leptin signaling and indirectly preserves bone via inhibition of sympathetic nervous system tone and β-adrenergic agonist release [29].
Osteocalcin (BGP) is another gene classically induced by 1,25D in osteoblasts, particularly in the rat [30-32] and the human [33]. The primary VDRE in the rat BGP gene is located at -456 bp, and is conserved in the human BGP promoter. Fig. 2 (inset) illustrates the induction of BGP by 1,25D in human osteoblast-like osteosarcoma cells (MG-63), and the proximal 5′ VDRE in the human gene is occupied by liganded VDR in these cells as demonstrated by ChIP assay [16]. Recently, utilizing BGP null animals, it has been shown that normal osteocalcin expression is important for robust, fracture-resistant bones [34]. Extracellular BGP also is thought to bind calcified bone matrix where it functions as a chemotactic agent for osteoclasts, perhaps strengthening the skeleton via enhanced remodeling cycles. Finally, osteocalcin has been identified by Karsenty and coworkers [35] as a bone-secreted hormone that both improves insulin release from pancreatic β-cells and increases insulin metabolic responsiveness in target tissues. Thus, vitamin D-induced bone osteocalcin, by supporting insulin release and action, could be considered an important adjunct in insuring glucose control to delay or lower the risk of advanced glycosylation end product formation (“AGEing”) characteristic of uncontrolled diabetes mellitus which elicits retinopathy, neuropathy, and vascular disease.
RANKL constitutes one of the most dramatically 1,25D-upregulated bone genes, the product of which effects 1,25D-VDR mediated bone resorption through osteoclastogenesis. Pike and associates [36] have proven that RANKL expression is controlled by VDR through multiple VDREs, the most distal of which is 76 kb 5′ of the transcriptional start site in the mouse gene. This distal region, which is conserved among other mammals, also mediates the effects on RANKL expression of PTH, prostaglandins, and other bone resorbing agents [36]. Fig. 2 (inset at lower left) reveals that RANKL is induced over 5000-fold by 1,25D in mouse ST-2 stromal cells in culture. OPG, the soluble decoy receptor for RANKL that tempers its activity, is simultaneously repressed by 86% (Fig. 2 inset at lower left) to amplify the bioeffect of displayed (or secreted) RANKL. Thus, like PTH, 1,25D is a potent bone-resorbing, hypercalcemic hormone and, although chronic excess of either hormone elicits a severe osteopenic pathology, physiologic bone remodeling can be argued to strengthen the skeleton. In other words, like a well-mineralized bone, an appropriately remodeled bone is a healthy bone, less susceptible to fractures and the eventual ravages of senile osteoporosis.
3.3 Feedback control of the calcemic, phosphatemic, and bone resorbing actions of 1,25D
The 1,25D-modulated genes enumerated in the previous section are all calcemic, phosphatemic, or affect bone remodeling. To govern these 1,25D-induced phenomena, there exists a separate class of feedback regulatory genes which curb the mineralotropic and osteotrophic actions of 1,25D. Control of these genes by 1,25D-VDR delimits bone mineralization to the defined endoskeleton, prevents ectopic calcification elicited by excesses of either calcium or phosphate, reduces age-related vascular pathology and atherosclerosis, protects against muscle and skin atrophy as well as respiratory failure, and generally prevents premature aging and lengthens lifespan [37]. Many of these pathologies are also the consequence of hypervitaminosis D [37]. Thus, excess vitamin D and its actions actually reduce lifespan, meaning that the level of 1,25D as well as the sequelae of its effects through VDR must be “detoxified” and sustained in an optimal range to maintain healthful aging.
The vitamin D 24-hydroxylase, CYP24A1, is one of the most strongly 1,25D-induced genes in all cells expressing VDR. For example, Fig. 2 insets (gray bars) illustrate CYP24A1 induction by 1,25D in human proximal tubule cells, HK-2 (20-fold), and human colon cancer, Caco-2 cells (62-fold). The endocrine and intracrine effects of 1,25D are curtailed by CYP24A1 catalyzed catabolism of 1,25D, providing an “off” signal once the hormone has executed its physiologic modulation of gene expression. Mice with ablation of the CYP24A1 gene die early because of 1,25D excess, and those that survive by compensatory adaptation lack endochondral bone formation caused by excess 1,25D [38]. Therefore, as summarized schematically in Fig. 2, 1,25D induces CYP24A1 to feedback attenuate its actions through catabolism to inactive vitamin D metabolites.
PTH is the major tropic hormone that stimulates the renal 1α-OHase, CYP27B1, to produce the 1,25D hormone, primarily under low calcium conditions when the calcium-sensing receptor in the parathyroid glands recognizes hypocalcemia and signals the synthesis and release of PTH. In a short feedback loop pictured in Fig. 2, 1,25D-VDR feedback represses PTH gene expression to close the endocrine loop; PTH repression by 1,25D is well documented [39], and a negative VDRE in the chicken PTH gene has been identified [17]. Calcium, mobilized from bone via resorption, or derived from 1,25D-enhanced intestinal absorption, is a second negative feedback regulator of PTH elaboration (Fig. 2). Ultimately, the combination of 1,25D-VDR and calcium close the endocrine loop by suppressing PTH and eliminating its potentiation of CYP27B1, thereby reducing 1,25D production. Normally, through its phosphaturic action, PTH would eliminate any excess, unneeded phosphate mobilized by 1,25D-VDR in the process of correcting hypocalcemia. However, because PTH is so quickly and effectively suppressed in this situation, there is a need for a second phosphaturic hormone to enter the scene.
FGF23 is the newly recognized phosphaturic peptide hormone which functions initially in concert with PTH, and chronically when PTH is suppressed by calcium and 1,25D. In fact, FGF23 directly represses PTH (Fig. 2) [40] to eliminate the activation of CYP27B1 by PTH, while at the same time taking over from PTH the role of phosphate elimination. Like PTH, FGF23 inhibits renal Npt2a and Npt2c to elicit phosphaturia. Yet oppositely to PTH, FGF23 is upregulated by 1,25D as shown in Fig. 2 (inset) for the case of rat UMR-106, osteocyte-like cells of the osteoblast lineage. This striking 78-fold induction of FGF23 by 1,25D is, however, blocked by cycloheximide treatment of the cells (Fig. 2 inset), indicating that a newly synthesized intermediary transcription factor targeted by VDR is secondarily inducing FGF23. This is consistent with the lack of reported conserved VDREs in the FGF23 gene as well as the apparent inability of ChIP assays to identify liganded VDR association with the gene. Notably, FGF23 regulation is complex and multifactorial. For instance, PHEX, the gene encoding an endoproteinase which is loss-of-function mutated in X-linked hypophosphatemic rickets (XLH), acts to repress FGF23 expression [41], and excess FGF23 causes the hypophosphatemic pathology in XLH, tumor-induced osteomalacia (ectopic FGF23 secretion) [42], and autosomal dominant hypophosphatemic rickets (ADHR) [42], wherein FGF23 is altered to preclude proteolytic inactivation. As depicted in Fig. 2 (inset), 1,25D represses PHEX expression in UMR-106 osteocyte-like cells, which is in accordance with the induction of FGF23 in that the PHEX suppressor is removed to permit maximal induction of FGF23 by 1,25D. It is conceivable that the mechanism of FGF23 induction by 1,25D is, in part or entirely, a consequence of PHEX repression, yet the PHEX substrate which ultimately regulates FGF23 transcription is not known. Although dentin matrix acid phosphoprotein 1 (DMP1) is apparently not a PHEX substrate, loss of function mutations in DMP1 cause a phenotype identical to XLH, with excess FGF23 producing hypophosphatemia. This suggests that DMP1, like PHEX, normally represses FGF23 expression in osteocytes; although this is inconsistent with the recent observation [43] that 1,25D induces DMP1 in UMR-106 cells. Conversely, as illustrated in Fig. 2, hyperphosphatemia enhances FGF23 independently of 1,25D, rendering FGF23 the perfect phosphaturic counter-1,25D hormone because it inhibits both renal phosphate reabsorption and 1,25D biosynthesis (Fig. 2). FGF23 also induces CYP24A1 (Fig. 2) in kidney to further reduce ambient circulating 1,25D levels. Regardless of the mechanism whereby FGF23 is induced, this hormone allows osteocytes in bone to communicate with the kidney to govern vitamin D bioactivation and circulating phosphate, thereby preventing excess 1,25D function and ectopic calcification due to hyperphosphatemia. That FGF23 production in bone is absolutely dependent on 1,25D is proven by the extremely low levels of circulating FGF23 in VDR null mice [44], and the dramatic 80-fold increase in serum FGF23, emanating almost entirely from bone, when normal mice are treated with 1,25D [45]. The primary mission of FGF23 is phosphate excretion to protect against hyperphosphatemia and ectopic calcification, which not coincidentally are the two dominant characteristics of the FGF23 knockout mouse [46]. As predicted, FGF23 null mice also possess markedly elevated 1,25D in blood, and their phenotype is characterized by a shortened lifespan, skin atrophy, arteriosclerosis and chronic obstructive pulmonary disease, with complications from the latter usually having fatal consequences [46]. Thus, FGF23, a gene induced by 1,25D, could be considered a longevity gene. Interestingly, double knockouts of FGF23 with either VDR [47] or CYP27B1 [48] essentially rescue the FGF23 null phenotype in mice, indicating that the capability of FGF23 to function as a counter-regulatory hormone to 1,25D is the key to its health and longevity benefits. Thus, the physiologic activities of 1,25D provide for healthful aging and prolong life by lowering the risk of chronic disorders of old age, but in pharmacologic doses or pathological excess, 1,25D generates the phenotype of the FGF23 ablated mouse, including ectopic calcification, skin atrophy, osteoporosis, vascular disease and emphysema. This situation of 1,25D toxicity is analogous to the actions of the only other known toxic fat soluble vitamin, namely vitamin A and its active retinoic acid metabolite. In physiologic quantities, retinoids mediate epithelial cell differentiation and barrier formation, embryonic development, etc., yet pharmacologic excesses of vitamin A and retinoic acid yield epithelial cell pathologies such as gastroenteritis and exfoliation, and embryopathy, respectively. Whereas no feedback control exists in the vitamin A system, the endocrine “Rosetta Stone” that allows the body to keep vitamin D in check is FGF23 and its signaling.
FGF23 signals via renal FGFR1 and klotho coreceptors to promulgate phosphaturia and repress CYP27B1/induce CYP24A1 [49], functioning through phospho-ERK and resulting in induction of the early response gene Egr-1 [50]. Klotho is a bona fide longevity gene expressed primarily in the distal renal tubule which, when inactivated in mice, generates a phenotype identical to that of FGF23 null mice [51], including premature aging/decreased lifespan, elevated 1,25D, hyperphosphatemia, ectopic calcification, skin and muscle atrophy, osteoporosis and hearing loss. FGF23 downregulates its klotho coreceptor [52] but, as depicted in Fig. 2 (insets), 1,25D induces klotho in kidney and colon cancer cells. Upregulation of klotho by 1,25D is modest, but statistically significant, and consistent with potentiation of FGF23 signaling in kidney and perhaps in other cell types (e.g., vascular) where a secreted form of klotho is considered a potential beneficial hormone [53]. In one particularly striking study, klotho introduction via a viral vector prevented progression of spontaneous hypertension and renal damage in the SHR rat model [54]. We propose that many of the health and longevity benefits of 1,25D may be effected through VDR-mediated enhancement of klotho expression in kidney (Fig. 2) and perhaps other cell types.
3.4 Tissue selective interaction of VDR with Wnt signaling/β-catenin
Wnts consist of approximately 20 members of a secreted peptide signaling molecule family that act by binding to transmembrane Frizzled (Fz) and low density lipoprotein-related protein (LRP) coreceptors releasing intracellular Disheveled (Dsh) to inhibit the β-catenin degradation complex. The result is an accumulation of β-catenin which translocates into the nucleus and induces the transcription of target genes encoding proteins that function to alter cell proliferation, differentiation, polarity and tissue remodeling. β-catenin also functions extranuclearly to affect cell adhesion and motility. By inducing LRP5 (Fig. 2 inset), 1,25D-VDR potentiates Wnt signaling in select cell types, likely in bone to function anabolically via osteoblast proliferation [55], and in skin and hair follicles to promote organogenesis and hair cycling, respectively [56].
However, in the colon, β-catenin constitutes a proto-oncogene, with either mutational activation of its function [57] or loss-of-function mutation in its counterregulatory protein partner(s) [58] causing colon cancer. Calcium and 1,25D are known to lower the risk of colon cancer [25], a disease prevalent at higher latitudes where vitamin D production from sunlight exposure is limited. 1,25D protects against colon cancer by detoxifying LCA, the carcinogenic secondary bile acid [4] ligand for VDR, and by controlling the expression of genes which influence cell growth and differentiation such as TGFβR1, EGFR, etc. We show here (Fig. 3A) that VDR, in a 1,25D ligand-dependent manner, suppresses the transcriptional activity of β-catenin in HT-29 human colorectal carcinoma cells, likely via direct VDR-β-catenin protein interaction to divert transcriptional activity away from β-catenin target genes that promote proliferation and toward VDR target genes that encode factors stimulating cell differentiation and apoptosis [59]. Palmer and coworkers [60] have independently reached this same conclusion. Moreover, the interaction between VDR and β-catenin is significantly attenuated when VDR is instead liganded with carcinogenic LCA [59]. These observations provide further evidence that suggests a mechanism whereby 1,25D-VDR acts to prevent colon cancer while LCA may contribute to tumorigenesis.
Fig. 3.
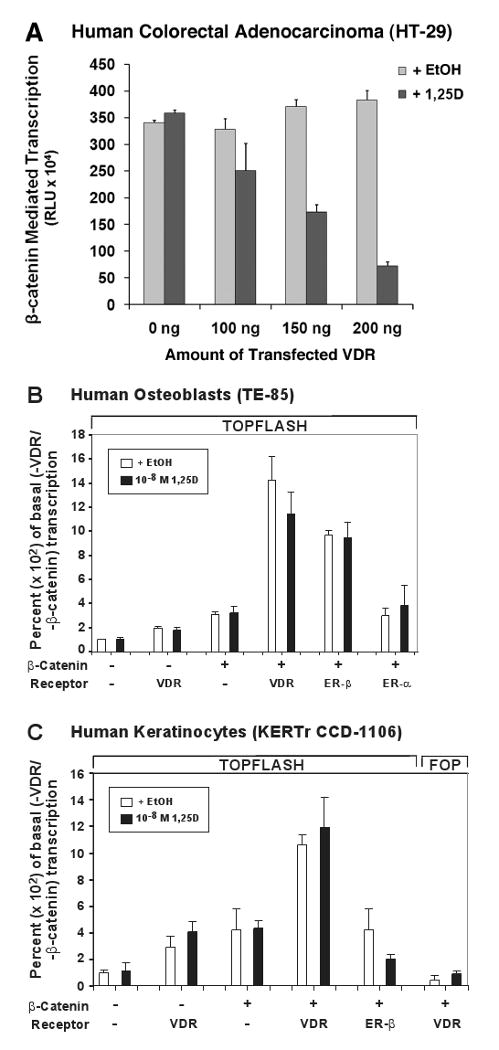
VDR modulates β-catenin signaling in a cell-specific manner to impact chemoprevention, bone remodeling, and hair cycling. (A) VDR transfection of human colorectal adenocarcinoma (HT-29) cells dose-responsively blunts β-catenin-mediated transcription in a 1,25D ligand-dependent fashion. β-catenin transcriptional activity was measured via TOPFLASH assay as described elsewhere [59], after treating human β-catenin-transfected HT-29 cells with 1 nM 1,25D for 24 hours. Error bars are ±SD, with n=3 for this representative experiment. VDR transfection of human osteoblast-like TE-85 (B), or human keratinocyte KERTr CCD-1106 (C) cells potentiates β-catenin transcriptional activity in the absence or presence of 10 nM 1,25D. TOPFLASH was assayed as described elsewhere [59], and FOP represents a loss of function mutant β-catenin responsive element construct that serves as a negative control.
The relationship between 1,25D-VDR and β-catenin is interestingly different in bone. As illustrated in Fig. 3B, VDR, as well as ERβ but not ERα, significantly potentiates β-catenin-directed transcription in human osteoblast-like TE-85 osteosarcoma cells, and this effect is ligand independent. By potentiating β-catenin signaling in osteoblasts, VDR is apparently anabolic to bone via enhanced Wnt signaling. This action is consistent with the osteoporotic phenotype occurring in LRP5 knockout [61] mice, and the fact that the most promising new bone anabolic drug potentially to treat osteoporosis is a sclerosterin-1 (SOST-1)-immunoneutralizing monoclonal antibody [62] which promotes Wnt/β-catenin signaling by removing the soluble, extracellular SOST-1 Wnt antagonist. Interestingly, osteosarcoma may be caused by a silencing of WIF, another Wnt/β-catenin antagonist [63], revealing that the downside of inducing β-catenin signaling for osteogenic purposes could be the development of osteosarcoma. Nevertheless, the results in Fig. 3A and 3B illustrate that VDR can either blunt or amplify β-catenin transcriptional action depending on the cell type. These observations shed considerable light on the antiproliferative action of the receptor in colon [64] and the osteogenic function of VDR in bone osteoblasts [65].
Similar to the situation in bone cells, VDR ligand-independently upregulates β-catenin transcriptional signaling in keratinocytes (Fig. 3C), an effect which is not shared with ERβ in keratinocytes as it is in osteoblasts. β-catenin is absolutely required in keratinocytes [66], as is VDR [67], to permit mammalian hair cycling, and the ligand-independent action of VDR to mediate the hair cycle is thought to involve Wnt signaling in skin stem cells [67, 68]. Thus, mammalian hair cycling, which does not require a VDR ligand, appears to be driven by unliganded VDR (or VDR liganded with a non-vitamin D lipid) through modulation of canonical Wnt signaling, with β-catenin as a key player. All of the effects of VDR on β-catenin can be considered “Fountain of Youth” actions because a lowered risk of colon cancer, enhanced bone volume, and competent skin protected by hair all lead to more healthful aging.
3.5 Multifaceted mechanism whereby VDR drives the mammalian hair cycle
As illustrated schematically in Fig. 4C, the regulation of mammalian hair cycling is complex, consisting of the convergence of two signaling pathways, BMP and Wnt. Starting at the upper left of Fig. 4C, Noggin from the dermal papilla initially antagonizes BMP4 signaling in bulb (or bulge) keratinocytes, allowing for the accumulation of Lef1/TCF (❶ in Fig. 4C), a transcriptional coactivator which targets genes via DNA-binding partners such as β-catenin. Cessation of Noggin signaling reinstates BMP signal transduction via SMADs (❷ in Fig. 4C) provided that the Wnt modulator in surface ectoderm (Wise or SOSTDC1), which antagonizes both Wnt and BMP pathways [69], has also been repressed. Wnt ligand, e.g., Wnt 10b, signaling and resulting accumulation of β-catenin facilitates cooperation with Lef1/TCF to induce the genes encoding factors, such as Shh, that trigger the hair cycle to transition from telogen (resting) to anagen (growth).
Fig. 4.
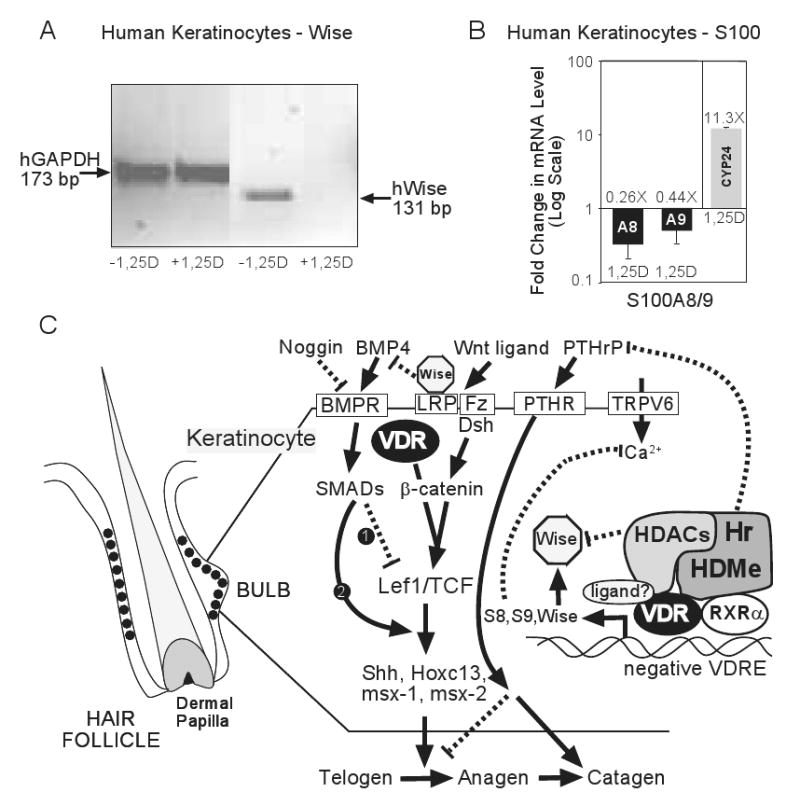
VDR action in keratinocytes to sustain the mammalian hair cycle. (A) Repression of human Wise (SOSTDC1) gene expression by 1,25D in human keratinocytes (KERTr CCD-1106). Cultured cells were incubated with or without 10 nM 1,25D for 18 h. Levels of mRNA expression were evaluated by RT-PCR. Total RNA was isolated, reverse transcribed into cDNA, and the products analyzed via gel electrophoresis with primers designed to generate 131 bp and 173 bp respective coding region products from Wise and control GAPDH. (B) S100A8 (A8) and S100A9 (A9) genes are rapidly (3 h) repressed in KERTr CCD-1106 cells by 1,25D (10 nM) treatment in the face of an 11.3-fold induction of CYP24A1. RNA was isolated, reverse transcribed into cDNA, and analyzed by real time PCR using GAPDH as an uncontrolled normalizing factor. Error bars are ±SD with n=9. (C) Model for control of the mammalian hair cycle and the proposed role of VDR, RXRα, and Hr. See text for an explanation of the model.
However, we propose that VDR also functions in keratinocytes to drive the hair cycle by controlling gene expression. This proposal is based upon gene ablation studies which indicate that VDR, its RXRα [70] DNA binding heteropartner, as well as the coactivator DRIP205 [71] and the corepressor, hairless (Hr) [72], all are required for normal hair cycling. Another candidate gene is the calcium channel TRPV6, which not only is required for hair cycling [23], but also is induced by 1,25D-VDR (Fig. 2 inset). Finally, intracellular calcium, which can be enhanced by TRPV6 activity, is itself a trigger for keratinocyte differentiation [73].
The most important role of VDR in controlling the hair cycle, however, appears to be in repression of key target genes. This conclusion is based upon the observation that knockout of Hr, a VDR co-repressor which colocalizes with VDR in the outer root sheath of the hair follicle [74], produces a phenocopy of the VDR-null mouse with respect to alopecia, but does not affect bone and mineral metabolism [75]. At least one of the molecular functions of Hr is recruitment of histone deacetylases (HDACs) to reconform chromatin to a repressive architecture [76]; another proposed function of Hr is to catalyze histone demethylation (HDMe) to further attenuate transcription [77].
What genes are targeted by the VDR-RXRα-Hr complex for repression? Thompson and coworkers [68] have defined Wise (SOSTDC1) as a gene overexpressed in keratinocytes from Hr-null mice, and Kato and colleagues [78] characterized S100A8 as a gene overexpressed in VDR-null keratinocytes. To determine the effect of activated VDR on the expression of SOSTDC1 and S100A8, we examined their mRNA levels by PCR in human keratinocytes treated with 1,25D. Fig. 4A reveals that, as monitored by reverse transcriptase-PCR, Wise/SOSTDC1 is strikingly repressed after 18 hours of 1,25D treatment of keratinocytes. We verified that SOSTDC1 is suppressed by 1,25D utilizing independent cDNA microarray analysis of human cells (data not shown). Because Wise not only antagonizes the Wnt pathway by binding to LRP, but also inhibits the BMP pathway through neutralization of BMP4 [69], repression of Wise by VDR could constitute a major event in initiating the mammalian hair cycle (Fig. 4C). Similarly, Fig. 4B shows that 1,25D rapidly represses S100A8 and its obligatory S100A9 heteropartner in calcium binding. This inhibition of S100A8/A9 expression by 1,25D-VDR is in stark contrast to a CYP24A1 induction of 11.3-fold (Fig. 4B) in keratinocytes, as well as to the induction of S100A8/A9 observed in HL-60 cells when differentiated by 1,25D along the macrophage lineage [79]. Thus, 1,25D regulates S100A8/A9 expression differentially in a cell selective fashion. The S100A8 protein is a general biomarker for inflammation and malignancy [80], but we propose that its role in keratinocytes is to dampen intracellular calcium oscillations required for skin differentiation and hair cycling (Fig. 4C). Therefore, repressing S100A8/A9 could conceivably restore the cellular calcium gradient which appears to be the ultimate messenger that controls keratinocyte function. One more gene repressed by 1,25D, namely PTHrP [81], is already known to encode a suppressor of the telogen to anagen transition in the hair follicle, as well as promote entry into catagen [82], providing yet another VDR-RXRα-Hr repressed gene target that participates in hair cycle control (Fig. 4C). In summary, VDR appears to usher the regeneration of hair, an obvious shield that protects skin and facilitates healthful aging, via both protein-protein and protein-DNA interactions that potentiate Wnt, BMP and calcium signaling.
4. Conclusion
In the present communication we have highlighted numerous genes for which the expression is modulated by 1,25D-VDR, and theorized that through physiological regulation of the transcription of these and other genes, the nuclear VDR can be characterized as an “age-well” receptor. These genes fall into four general classes. The first group is comprised of genes for which the products homeostatically control bone and mineral metabolism, as well as the integrity of the endoskeleton. By inducing TRPV6/TRPV5 and Npt2b/Npt2c, VDR signaling supplies respective calcium and phosphorus minerals via absorption/reabsorption to generate the fully mineralized endoskeleton. By repressing PTH and PHEX, and inducing the FGF23/klotho system, plus upregulating CYP24A1, VDR prevents the production of excess 1,25D hormone and protects against ectopic calcification arising from hypercalcemia and/or hyperphosphatemia. Through regulation of BGP, SPP1, LRP5, RANKL and OPG, VDR ensures the formation of high volume, fracture-resistant bone with connectivity that is modeled for strength via osteocyte mechanosensing endocrine cells in the skeleton. Klotho can be considered to be in a class by itself because it has been shown to represent a longevity gene, with actions that include but are not limited to control of phosphate metabolism [53]. The third group is composed of genes encoding factors impacting Wnt signaling that effects organogenesis (especially skin and bone) and hair growth, including LRP5 (also in group 1), SOSTDC1, Dkk4, DkkL1, S100A8/S100A9 and PTHrP. Control of this set of genes by VDR guarantees the proper formation of epithelial cell barriers, especially the cornified epithelium of the epidermis, to guard against invasion by microorganisms and elicits hair growth and cycling to shield against the age-related damages from UV irradiation. The fourth group of VDR-regulated genes are unrelated to skin, bone and mineral metabolism, and are not a focus of the present investigation, but regulation of their expression by vitamin D is well documented to facilitate healthful aging by delaying or eliminating a host of diseases. For example, 1,25D-VDR induces cathelicidin [83] as a part of activating the innate immune system to fight infection, and represses IL-17 [84] to temper the adaptive immune system and lower the risk of autoimmune disorders such as type I diabetes mellitus, multiple sclerosis, lupus and rheumatoid arthritis. Liganded VDR also functions as a detoxification nuclear receptor by inducing CYP3A4 [4] and SULT2 [5] to eliminate toxic xenobiotics that might affect the gastrointestinal tract, skin and other tissues. High vitamin D concentrations are associated with longer leukocyte telomere length, a parameter which decreases with each cell cycle and increased inflammation, highlighting the potential beneficial effects of 1,25D-VDR on aging and age-related diseases [85]. 1,25D-VDR is anti-inflammatory by blunting NFkB [86] and COX2 [87] and, because inflammation is considered a common denominator in maladies such as heart disease and stroke, as well as cancer, the inflammation-reducing actions of VDR could be crucial in the healthy aging property of the vitamin D receptor. VDR likely also reduces risk for many cancers by inducing the p53 [88] and p21 [88] tumor suppressors, as well as DNA repair enzymes in skin [10]. Thus, it is no surprise that the VDR null mouse is supersensitive to DMBA-induced skin cancer [89] as well as UV light-induced skin malignancy [90]. VDR knockout mice exhibit enhanced colonic proliferation [64] plus amplified mammary gland ductal extension, end buds and density [91], indicating that the fundamental actions of VDR to promote cell differentiation and apoptosis [59] play an important role in reducing the risk of age-related epithelial cell cancers such as those of the colon and breast. Finally, 1,25D-VDR induces FOXO3 [92], an important molecular player in preventing oxidative damage, a leading candidate for the cause of aging [93]. Clearly, through control of key genes, VDR indeed feeds the “Fountain of Youth” and allows one to age well by delaying fractures, ectopic calcification, oxidative damage, infections, autoimmunity, inflammation, pain, cardiovascular disease, and malignancy.
Acknowledgments
This work was supported by National Institutes of Health grants to M.R.H. (DK33351 and DK063930).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Whitfield GK, Jurutka PW, Haussler CA, Hsieh JC, Barthel TK, Jacobs ET, Encinas Dominguez C, Thatcher ML, Haussler MR. Nuclear vitamin D receptor: structure-function, molecular control of gene transcription, and novel bioactions. In: Feldman D, Pike JW, Glorieux FH, editors. Vitamin D. Elsevier Academic Press; Oxford, UK: 2005. pp. 219–261. [Google Scholar]
- 2.Whitfield GK, Dang HTL, Schluter SF, Bernstein RM, Bunag T, Manzon LA, Hsieh G, Dominguez CE, Youson JH, Haussler MR, Marchalonis JJ. Cloning of a functional vitamin D receptor from the lamprey (Petromyzon marinus), an ancient vertebrate lacking a calcified skeleton and teeth. Endocrinology. 2003;144(6):2704–2716. doi: 10.1210/en.2002-221101. [DOI] [PubMed] [Google Scholar]
- 3.Nehring JA, Zierold C, DeLuca HF. Lithocholic acid can carry out in vivo functions of vitamin D. Proc Natl Acad Sci U S A. 2007;104(24):10006–10009. doi: 10.1073/pnas.0703512104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 5.Echchgadda I, Song CS, Roy AK, Chatterjee B. Dehydroepiandrosterone sulfotransferase is a target for transcriptional induction by the vitamin D receptor. Mol Pharmacol. 2004;65(3):720–729. doi: 10.1124/mol.65.3.720. [DOI] [PubMed] [Google Scholar]
- 6.Christakos S, Dhawan P, Benn B, Porta A, Hediger M, Oh GT, Jeung EB, Zhong Y, Ajibade D, Dhawan K, Joshi S. Vitamin D: molecular mechanism of action. Ann N Y Acad Sci. 2007;1116:340–348. doi: 10.1196/annals.1402.070. [DOI] [PubMed] [Google Scholar]
- 7.Amling M, Priemel M, Holzmann T, Chapin K, Rueger JM, Baron R, Demay MB. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140(11):4982–4987. doi: 10.1210/endo.140.11.7110. [DOI] [PubMed] [Google Scholar]
- 8.Dardenne O, Prud'homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D3-1α-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142(7):3135–3141. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- 9.Haussler MR, Haussler CA, Bartik L, Whitfield GK, Hsieh JC, Slater S, Jurutka PW. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutr Rev. 2008;66(10 Suppl 2):S98–112. doi: 10.1111/j.1753-4887.2008.00093.x. [DOI] [PubMed] [Google Scholar]
- 10.Lin R, Nagai Y, Sladek R, Bastien Y, Ho J, Petrecca K, Sotiropoulou G, Diamandis EP, Hudson TJ, White JH. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 analog EB1089 signaling on cell proliferation, differentiation, and immune system regulation. Mol Endocrinol. 2002;16(6):1243–1256. doi: 10.1210/mend.16.6.0874. [DOI] [PubMed] [Google Scholar]
- 11.Lupien M, Eeckhoute J, Meyer CA, Krum SA, Rhodes DR, Liu XS, Brown M. Coactivator function defines the active estrogen receptor alpha cistrome. Mol Cell Biol. 2009;29(12):3413–3423. doi: 10.1128/MCB.00020-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH, Dominguez CE, Jurutka PW. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13(3):325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 13.Bikle D. Extrarenal synthesis of 1,25-dihydroxyvitamin D and its health implications. Clin Rev Bone Miner Metab. 2009;7(2):114–125. [Google Scholar]
- 14.Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF, Denhardt DT. Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (Spp-1 or osteopontin) gene expression. Proc Natl Acad Sci U S A. 1990;87:9995–9999. doi: 10.1073/pnas.87.24.9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin CH, Kerner SA, Hong MH, Pike JW. Transcriptional activation and dimerization functions in the human vitamin D receptor. Mol Endocrinol. 1996;10:945–957. doi: 10.1210/mend.10.8.8843411. [DOI] [PubMed] [Google Scholar]
- 16.Barthel TK, Mathern DR, Whitfield GK, Haussler CA, Hopper HAt, Hsieh JC, Slater SA, Hsieh G, Kaczmarska M, Jurutka PW, Kolek OI, Ghishan FK, Haussler MR. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J Steroid Biochem Mol Biol. 2007;103(3-5):381–388. doi: 10.1016/j.jsbmb.2006.12.054. [DOI] [PubMed] [Google Scholar]
- 17.Koszewski NJ, Ashok S, Russell J. Turning a negative into a positive: vitamin D receptor interactions with the avian parathyroid hormone response element. Molecular Endocrinology. 1999;13(3):455–465. doi: 10.1210/mend.13.3.0249. [DOI] [PubMed] [Google Scholar]
- 18.Noda M, Nagao M, Hanyu R, Miyai K, Ezura Y. Bone fracture and the healing mechanisms. Molecular bases of fracture healing. Clin Calcium. 2009;19(5):634–640. [PubMed] [Google Scholar]
- 19.Weissen-Plenz G, Nitschke Y, Rutsch F. Mechanisms of arterial calcification: spotlight on the inhibitors. Adv Clin Chem. 2008;46:263–293. [PubMed] [Google Scholar]
- 20.Wang KX, Denhardt DT. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008;19(5-6):333–345. doi: 10.1016/j.cytogfr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Ramaiah SK, Rittling S. Pathophysiological role of osteopontin in hepatic inflammation, toxicity, and cancer. Toxicol Sci. 2008;103(1):4–13. doi: 10.1093/toxsci/kfm246. [DOI] [PubMed] [Google Scholar]
- 22.Meyer MB, Watanuki M, Kim S, Shevde NK, Pike JW. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol Endocrinol. 2006;20(6):1447–1461. doi: 10.1210/me.2006-0031. [DOI] [PubMed] [Google Scholar]
- 23.Bianco SD, Peng JB, Takanaga H, Suzuki Y, Crescenzi A, Kos CH, Zhuang L, Freeman MR, Gouveia CH, Wu J, Luo H, Mauro T, Brown EM, Hediger MA. Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J Bone Miner Res. 2007;22(2):274–285. doi: 10.1359/jbmr.061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94(18):9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garland C, Shekelle RB, Barrett-Connor E, Criqui MH, Rossof AH, Paul O. Dietary vitamin D and calcium and risk of colorectal cancer: a 19-year prospective study in men. Lancet. 1985;1(8424):307–309. doi: 10.1016/s0140-6736(85)91082-7. [DOI] [PubMed] [Google Scholar]
- 26.Milat F, Ng KW. Is Wnt signalling the final common pathway leading to bone formation? Mol Cell Endocrinol. 2009;310(1-2):52–62. doi: 10.1016/j.mce.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Fretz JA, Zella LA, Kim S, Shevde NK, Pike JW. 1,25-Dihydroxyvitamin D3 regulates the expression of low-density lipoprotein receptor-related protein 5 via deoxyribonucleic acid sequence elements located downstream of the start site of transcription. Mol Endocrinol. 2006;20(9):2215–2230. doi: 10.1210/me.2006-0102. [DOI] [PubMed] [Google Scholar]
- 28.Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135(5):825–837. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, Confavreux C, Klemenhagen KC, Tanaka KF, Gingrich JA, Guo XE, Tecott LH, Mann JJ, Hen R, Horvath TL, Karsenty G. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138(5):976–989. doi: 10.1016/j.cell.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markose ER, Stein JL, Stein GS, Lian JB. Vitamin D-mediated modifications in protein-DNA interactions at two promoter elements of the osteocalcin gene. Proc Natl Acad Sci U S A. 1990;87:1701–1705. doi: 10.1073/pnas.87.5.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeMay MB, Gerardi JM, DeLuca HF, Kronenberg HM. DNA sequences in the rat osteocalcin gene that bind the 1,25-dihydroxyvitamin D3 receptor and confer responsiveness to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1990;87:369–373. doi: 10.1073/pnas.87.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terpening CM, Haussler CA, Jurutka PW, Galligan MA, Komm BS, Haussler MR. The vitamin D-responsive element in the rat bone Gla protein gene is an imperfect direct repeat that cooperates with other cis-elements in 1,25-dihydroxyvitamin D3-mediated transcriptional activation. Mol Endocrinol. 1991;5(3):373–385. doi: 10.1210/mend-5-3-373. [DOI] [PubMed] [Google Scholar]
- 33.Ozono K, Liao J, Kerner SA, Scott RA, Pike JW. The vitamin D-responsive element in the human osteocalcin gene: association with a nuclear proto-oncogene enhancer. J Biol Chem. 1990;265:21881–21888. [PubMed] [Google Scholar]
- 34.Poundarik A, Gambino C, Gundberg C, Vashishth D. Osteocalcin - a determinant of bone toughness. J Bone Miner Res. 2009;24(Suppl 1) Available at http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=51d54e88b-f79d-47e52-a15b-134f130c157b152e.
- 35.Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S, Yamazaki M, Shevde NK, Pike JW. Transcriptional control of receptor activator of nuclear factor-kappaB ligand by the protein kinase A activator forskolin and the transmembrane glycoprotein 130-activating cytokine, oncostatin M, is exerted through multiple distal enhancers. Mol Endocrinol. 2007;21(1):197–214. doi: 10.1210/me.2006-0315. [DOI] [PubMed] [Google Scholar]
- 37.Keisala T, Minasyan A, Lou YR, Zou J, Kalueff AV, Pyykko I, Tuohimaa P. Premature aging in vitamin D receptor mutant mice. J Steroid Biochem Mol Biol. 2009;115(3-5):91–97. doi: 10.1016/j.jsbmb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Masuda S, Byford V, Arabian A, Sakai Y, Demay MB, St-Arnaud R, Jones G. Altered pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology. 2005;146(2):825–834. doi: 10.1210/en.2004-1116. [DOI] [PubMed] [Google Scholar]
- 39.Silver J, Naveh-Many T, Mayer H, Schmelzer HJ, Popovtzer MM. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. J Clin Invest. 1986;78(5):1296–1301. doi: 10.1172/JCI112714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukagawa M, Kazama JJ. FGF23: its role in renal bone disease. Pediatr Nephrol. 2006;21(12):1802–1806. doi: 10.1007/s00467-006-0230-3. [DOI] [PubMed] [Google Scholar]
- 41.Yuan B, Takaiwa M, Clemens TL, Feng JQ, Kumar R, Rowe PS, Xie Y, Drezner MK. Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest. 2008;118(2):722–734. doi: 10.1172/JCI32702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukumoto S. Fibroblast growth factor (FGF) 23 works as a phosphate-regulating hormone and is involved in the pathogenesis of several disorders of phosphate metabolism. Rinsho Byori. 2007;55(6):555–559. [PubMed] [Google Scholar]
- 43.Farrow EG, Davis SI, Ward LM, Summers LJ, Bubbear JS, Keen R, Stamp TC, Baker LR, Bonewald LF, White KE. Molecular analysis of DMP1 mutants causing autosomal recessive hypophosphatemic rickets. Bone. 2009;44(2):287–294. doi: 10.1016/j.bone.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005;36(6):971–977. doi: 10.1016/j.bone.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Kolek OI, Hines ER, Jones MD, Lesueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1{alpha},25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289(6):G1036–G1042. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- 46.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hesse M, Frohlich LF, Zeitz U, Lanske B, Erben RG. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 2007;26(2):75–84. doi: 10.1016/j.matbio.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Renkema KY, Alexander RT, Bindels RJ, Hoenderop JG. Calcium and phosphate homeostasis: concerted interplay of new regulators. Ann Med. 2008;40(2):82–91. doi: 10.1080/07853890701689645. [DOI] [PubMed] [Google Scholar]
- 49.Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5(11):611–619. doi: 10.1038/nrendo.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukuda T, Kanomata K, Nojima J, Urakawa I, Suzawa T, Imada M, Kukita A, Kamijo R, Yamashita T, Katagiri T. FGF23 induces expression of two isoforms of NAB2, which are corepressors of Egr-1. Biochem Biophys Res Commun. 2007;353(1):147–151. doi: 10.1016/j.bbrc.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 51.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, ShirakiI-ida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 52.Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118(12):3820–3828. doi: 10.1172/JCI36479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuro-o M. Klotho and aging. Biochim Biophys Acta. 2009;1790(10):1049–1058. doi: 10.1016/j.bbagen.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54(4):810–817. doi: 10.1161/HYPERTENSIONAHA.109.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urano T, Shiraki M, Usui T, Sasaki N, Ouchi Y, Inoue S. A1330V variant of the low-density lipoprotein receptor-related protein 5 (LRP5) gene decreases Wnt signaling and affects the total body bone mineral density in Japanese women. Endocr J. 2009;56(4):625–631. doi: 10.1507/endocrj.k09e-133. [DOI] [PubMed] [Google Scholar]
- 56.Widelitz RB. Wnt signaling in skin organogenesis. Organogenesis. 2008;4(2):123–133. doi: 10.4161/org.4.2.5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin T, George Fantus I, Sun J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta-catenin. Cell Signal. 2008;20(10):1697–1704. doi: 10.1016/j.cellsig.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 58.Kohler EM, Chandra SH, Behrens J, Schneikert J. Beta-catenin degradation mediated by the CID domain of APC provides a model for the selection of APC mutations in colorectal, desmoid and duodenal tumours. Hum Mol Genet. 2009;18(2):213–226. doi: 10.1093/hmg/ddn338. [DOI] [PubMed] [Google Scholar]
- 59.Egan JB, Thompson PA, Vitanov MV, Bartik L, Jacobs ET, Haussler MR, Gerner EW, Jurutka PW. Vitamin D receptor ligands, adenomatous polyposis coli, and the vitamin D receptor FokI polymorphism collectively modulate beta-catenin activity in colon cancer cells. Mol Carcinog. 2009 doi: 10.1002/mc.20603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palmer HG, Gonzalez-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, Quintanilla M, Cano A, de Herreros AG, Lafarga M, Munoz A. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154(2):369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA, 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Chan L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157(2):303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shahnazari M, Yao W, Corr M, Lane NE. Targeting the Wnt signaling pathway to augment bone formation. Curr Osteoporos Rep. 2008;6(4):142–148. doi: 10.1007/s11914-008-0025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kansara M, Tsang M, Kodjabachian L, Sims NA, Trivett MK, Ehrich M, Dobrovic A, Slavin J, Choong PF, Simmons PJ, Dawid IB, Thomas DM. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest. 2009;119(4):837–851. doi: 10.1172/JCI37175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kallay E, Pietschmann P, Toyokuni S, Bajna E, Hahn P, Mazzucco K, Bieglmayer C, Kato S, Cross HS. Characterization of a vitamin D receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis. 2001;22(9):1429–1435. doi: 10.1093/carcin/22.9.1429. [DOI] [PubMed] [Google Scholar]
- 65.Misof BM, Roschger P, Tesch W, Baldock PA, Valenta A, Messmer P, Eisman JA, Boskey AL, Gardiner EM, Fratzl P, Klaushofer K. Targeted overexpression of vitamin D receptor in osteoblasts increases calcium concentration without affecting structural properties of bone mineral crystals. Calcif Tissue Int. 2003;73(3):251–257. doi: 10.1007/s00223-002-2139-6. [DOI] [PubMed] [Google Scholar]
- 66.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105(4):533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 67.Cianferotti L, Cox M, Skorija K, Demay MB. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc Natl Acad Sci U S A. 2007;104(22):9428–9433. doi: 10.1073/pnas.0702884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beaudoin GM, 3rd, Sisk JM, Coulombe PA, Thompson CC. Hairless triggers reactivation of hair growth by promoting Wnt signaling. Proc Natl Acad Sci U S A. 2005;102(41):14653–14658. doi: 10.1073/pnas.0507609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lintern KB, Guidato S, Rowe A, Saldanha JW, Itasaki N. Characterization of wise protein and its molecular mechanism to interact with both Wnt and BMP signals. J Biol Chem. 2009;284(34):23159–23168. doi: 10.1074/jbc.M109.025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li M, Indra AK, Warot X, Brocard J, Messaddeq N, Kato S, Metzger D, Chambon P. Skin abnormalities generated by temporally controlled RXRalpha mutations in mouse epidermis. Nature. 2000;407(6804):633–636. doi: 10.1038/35036595. [DOI] [PubMed] [Google Scholar]
- 71.Bikle D, Schauber J, Gallo R, Elias P, Uchida Y, Oda Y. Abstracts from the 14th Workshop on Vitamin D. Brugge, Belgium: 2009. Differential regulation of epidermal function by VDR coactivators; p. 134. October 4-8, 2009. [Google Scholar]
- 72.Thompson CC, Sisk JM, Beaudoin GM., 3rd Hairless and Wnt signaling: allies in epithelial stem cell differentiation. Cell Cycle. 2006;5(17):1913–1917. doi: 10.4161/cc.5.17.3189. [DOI] [PubMed] [Google Scholar]
- 73.Bikle D. Nonclassic actions of vitamin D. J Clin Endocrinol Metab. 2009;94(1):26–34. doi: 10.1210/jc.2008-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsieh JC, Sisk JM, Jurutka PW, Haussler CA, Slater SA, Haussler MR, Thompson CC. Physical and functional interaction between the vitamin D receptor and hairless corepressor, two proteins required for hair cycling. J Biol Chem. 2003;278(40):38665–38674. doi: 10.1074/jbc.M304886200. [DOI] [PubMed] [Google Scholar]
- 75.Zarach JM, Beaudoin GM, 3rd, Coulombe PA, Thompson CC. The co-repressor hairless has a role in epithelial cell differentiation in the skin. Development. 2004;131(17):4189–4200. doi: 10.1242/dev.01303. [DOI] [PubMed] [Google Scholar]
- 76.Potter GB, Zarach JM, Sisk JM, Thompson CC. The thyroid hormone-regulated corepressor hairless associates with histone deacetylases in neonatal rat brain. Mol Endocrinol. 2002;16(11):2547–2560. doi: 10.1210/me.2002-0115. [DOI] [PubMed] [Google Scholar]
- 77.Hsieh JC, Slater S, Whitfield G, Dawson J, Hsieh G, Sheedy C, Haussler C, Haussler M. Analysis of hairless corepressor mutants to characterize molecular cooperation with the vitamin D receptor in promoting the mammalian hair cycle. J Cell Biochem Submitted for publication. 2010 doi: 10.1002/jcb.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamamoto Y, Memezawa A, Takagi K, Ochiai E, Shindo M, Kato S. Abstracts from the 14th Workshop on Vitamin D. Brugge, Belgium: 2009. A tissue-specific function by unliganded VDR; p. 66. October 4-8, 2009. [Google Scholar]
- 79.Suzuki T, Tazoe H, Taguchi K, Koyama Y, Ichikawa H, Hayakawa S, Munakata H, Isemura M. DNA microarray analysis of changes in gene expression induced by 1,25-dihydroxyvitamin D3 in human promyelocytic leukemia HL-60 cells. Biomed Res. 2006;27(3):99–109. doi: 10.2220/biomedres.27.99. [DOI] [PubMed] [Google Scholar]
- 80.Logsdon CD, Fuentes MK, Huang EH, Arumugam T. RAGE and RAGE ligands in cancer. Curr Mol Med. 2007;7(8):777–789. doi: 10.2174/156652407783220697. [DOI] [PubMed] [Google Scholar]
- 81.Falzon M. DNA sequences in the rat parathyroid hormone-related peptide gene responsible for 1,25-dihydroxyvitamin D3-mediated transcriptional repression. Mol Endocrinol. 1996;10:672–681. doi: 10.1210/mend.10.6.8776727. [DOI] [PubMed] [Google Scholar]
- 82.Cho YM, Woodard GL, Dunbar M, Gocken T, Jimenez JA, Foley J. Hair-cycle-dependent expression of parathyroid hormone-related protein and its type I receptor: evidence for regulation at the anagen to catagen transition. J Invest Dermatol. 2003;120(5):715–727. doi: 10.1046/j.1523-1747.2003.12147.x. [DOI] [PubMed] [Google Scholar]
- 83.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311(5768):1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 84.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Richards JB, Valdes AM, Gardner JP, Paximadas D, Kimura M, Nessa A, Lu X, Surdulescu GL, Swaminathan R, Spector TD, Aviv A. Higher serum vitamin D concentrations are associated with longer leukocyte telomere length in women. Am J of Clin Nutr. 2007;86(5):1420–1425. doi: 10.1093/ajcn/86.5.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cohen-Lahav M, Shany S, Tobvin D, Chaimovitz C, Douvdevani A. Vitamin D decreases NFkappaB activity by increasing IkappaBalpha levels. Nephrol Dial Transplant. 2006;21(4):889–897. doi: 10.1093/ndt/gfi254. [DOI] [PubMed] [Google Scholar]
- 87.Moreno J, Krishnan AV, Swami S, Nonn L, Peehl DM, Feldman D. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res. 2005;65(17):7917–7925. doi: 10.1158/0008-5472.CAN-05-1435. [DOI] [PubMed] [Google Scholar]
- 88.Audo I, Darjatmoko SR, Schlamp CL, Lokken JM, Lindstrom MJ, Albert DM, Nickells RW. Vitamin D analogues increase p53, p21, and apoptosis in a xenograft model of human retinoblastoma. Invest Ophthalmol Vis Sci. 2003;44(10):4192–4199. doi: 10.1167/iovs.02-1198. [DOI] [PubMed] [Google Scholar]
- 89.Zinser GM, Sundberg JP, Welsh J. Vitamin D3 receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002;23(12):2103–2109. doi: 10.1093/carcin/23.12.2103. [DOI] [PubMed] [Google Scholar]
- 90.Ellison TI, Smith MK, Gilliam AC, MacDonald PN. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol. 2008;128(10):2508–2517. doi: 10.1038/jid.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zinser G, Packman K, Welsh J. Vitamin D3 receptor ablation alters mammary gland morphogenesis. Development. 2002;129(13):3067–3076. doi: 10.1242/dev.129.13.3067. [DOI] [PubMed] [Google Scholar]
- 92.Eelen G, Gysemans C, Verlinden L, Gijsbers R, Beullens I, Van Camp M, Paik J, DePinho R, Bouillon R, Verstuyf A. Abstracts from the 14th Workshop on Vitamin D. Brugge, Belgium: 2009. Induction of FOXO3a by 1,25D in MC3T3E1 cells mediates resistance to oxidative stress; p. 60. October 4-8, 2009. [Google Scholar]
- 93.Lin M, Beal M. The oxidative damage theory of aging. Clinical Neuroscience Research. 2003;2:305–315. [Google Scholar]
- 94.Craig AM, Smith JH, Denhardt DT. Osteopontin, a transformation-associated cell adhesion phosphoprotein, is induced by 12-O-tetradecanoylphorbol 13-acetate in mouse epidermis. J Biol Chem. 1989;264(16):9682–9689. [PubMed] [Google Scholar]