

Abstract
Background
The association between hyperuricemia and incident heart failure (HF) is relatively unknown.
Methods
Of the 5461 community-dwelling older adults, ≥65 years, in the Cardiovascular Health Study without HF at baseline, 1505 had hyperuricemia (baseline serum uric acid ≥6 mg/dL for women and ≥7 mg/dL for men). Using propensity scores for hyperuricemia, estimated for each participant using 64 baseline covariates, we were able to match 1181 pairs of participants with and without hyperuricemia.
Results
Incident HF occurred in 21% and 18% of participants respectively with and without hyperuricemia during 8.1 years of mean follow-up (hazard ratio {HR} for hyperuricemia versus no hyperuricemia, 1.30; 95% confidence interval {CI}, 1.05–1.60; P=0.015). The association between hyperuricemia and incident HF was significant only in subgroups with normal kidney function (HR, 1.23; 95% CI, 1.02–1.49; P=0.031), without hypertension (HR, 1.31; 95% CI, 1.03–1.66; P=0.030), not receiving thiazide diuretics (HR, 1.20; 95% CI, 1.01–1.42; P=0.044), and without hyperinsulinemia (HR, 1.35; 95% CI, 1.06–1.72; P=0.013). Used as a continuous variable, each 1 mg/dL increase in serum uric acid was associated with a 12% increase in incident HF (HR, 1.12; 95% CI, 1.03–1.22; P=0.006). Hyperuricemia had no association with acute myocardial infarction or all-cause mortality.
Conclusions
Hyperuricemia is associated with incident HF in community-dwelling older adults. Cumulative data from our subgroup analyses suggest that this association is only significant when hyperuricemia is a marker of increased xanthine oxidase activity but not when hyperuricemia is caused by impaired renal elimination of uric acid.
Keywords: Uric acid, incident heart failure, kidney function, propensity score
1. Introduction
High serum uric acid or hyperuricemia is considered a surrogate marker of inflammation and has been associated with poor outcomes in heart failure (HF) [1-5]. Hyperuricemia has also been variably associated with cardiovascular morbidity and mortality [6-10]. However, the association between baseline hyperuricemia and new-onset HF has not been prospectively studied in community-dwelling older adults. We used a public-use copy of the Cardiovascular Health Study (CHS) data obtained from the National Heart, Lung and Blood Institute (NHLBI) to test the hypothesis that baseline hyperuricemia was associated with increased risk of new-onset HF in a well-balanced propensity-matched population.
2. Methods
2.1. Study design and participants
CHS is an NHLBI-funded, ongoing, community-based, epidemiologic study of 5,888 adults ≥65 years of age [11]. The purpose of the study was to investigate risk factors for cardiovascular morbidity and mortality in older adults. CHS participants were recruited from Forsyth County, North Carolina, Sacramento County, California, Washington County, Maryland, and Pittsburgh, Pennsylvania. An original cohort of 5,201 participants was recruited between 1989 and 1990, and a second cohort of 687 African American participants was recruited between 1992 and 1993. Detailed descriptions of the rationale, design, implementation, and results of the CHS have been previously reported [11]. Of the 5,888 original CHS participants, 5,795 consented to be included in the de-identified public-use copy of the dataset and are included in our analysis. From these, we excluded 79 participants without data on baseline serum uric acid and 255 participants with prevalent HF at baseline. The final sample size for the current analysis was 5,461. We restricted our main analysis to a subset of 1,181 pairs of propensity-matched participants with high and normal serum uric acid (described under Section 2.4).
2.2. Baseline serum uric acid and other baseline measurements
Serum uric acid levels were measured in a central blood analysis laboratory using Kodak Ektachem 700 analyzer assay (Eastman Kodak, Rochester, NY) [12]. Based on commonly used gender-based cut-offs, we defined hyperuricemia as serum uric acid levels ≥6 mg/dL for women and ≥7 mg/dL for men [7, 13]. Of the 5,461 participants, 1,505 (27.6%) had hyperuricemia. Data on socio-demographic, clinical, sub-clinical, and laboratory variables were also collected at baseline and have been previously described in detail [11]. Missing values for continuous variables were imputed based on values predicted by age, sex and race.
2.3. Study outcomes
The primary outcome for this study was definite new-onset HF during 8.1 years of mean follow-up. The process of adjudication of HF in CHS has been well documented in the literature [14-21]. Briefly, the CHS Events Committee adjudicated the diagnosis of HF based on participants’ self report of physician-diagnosed HF during semi-annual visits and their medical records for a constellation of symptoms, physical signs, and other supporting findings suggestive of HF, use of medications commonly used for HF, and follow-up surveillances [14, 15]. Secondary outcomes included all-cause mortality and acute myocardial infarction (AMI).
2.4. Assembly of study cohort using propensity score matching
We estimated propensity scores for hyperuricemia for each of the 5,461 participants using a non-parsimonious multivariable logistic regression model [22-24]. The propensity score for hyperuricemia for a participant is the conditional probability of having hyperuricemia given a set of measured baseline covariates of that participant [25, 26]. In our propensity score model, hyperuricemia was the dependent variable, and the 64 baseline characteristics displayed in Figure 1 were used as covariates. Several clinically important two-way interactions were tested and the only significant interaction term between age and baseline serum creatinine was included in the model. Using a greedy matching protocol described elsewhere we were able to match 1,181 pairs of participants with and without hyperuricemia who had similar propensity scores [22-24]. Absolute standardized differences for all 64 covariates were estimated to assess pre-match imbalances and the post-match balances achieved, and results were presented as a Love plot [22]. The Love plot was developed by Thomas E. Love to provide a concise and meaningful balance summary across a large set of covariates, both before and after matching [22]. An absolute standardized difference of 0% indicates no bias, and values <10% suggest inconsequential bias [22-24, 27].
Figure 1.
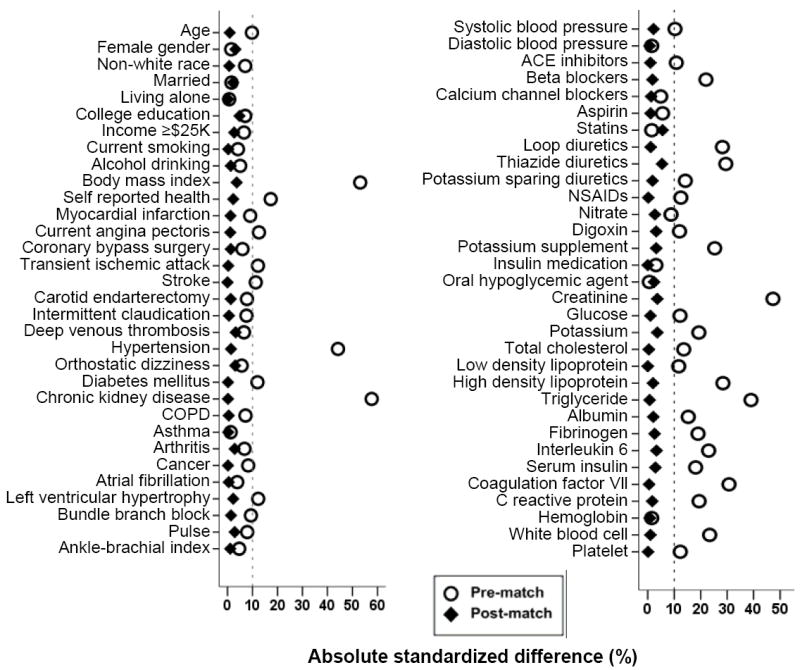
Love plots for absolute standardized differences for covariates between participants with and without hyperuricemia (defined as serum uric acid levels ≥6mg/dL for women and ≥7mg/dL for men), before and after propensity score matching (ACE=angiotensin-converting enzyme, COPD =chronic obstructive pulmonary disease, NSAID=non-steroidal anti inflammatory drug)
2.5. Statistical analysis
For descriptive analyses, we used Pearson Chi square and Wilcoxon rank-sum tests for the pre-match data, and McNemar’s test and paired sample t-test for post-match comparisons, as appropriate. Kaplan-Meier survival and matched Cox proportional hazard analyses were used to determine the association of hyperuricemia with outcomes during 8.1 years of mean follow-up. To assess the effect of loss of participants during matching, we repeated our analysis in all 5,461 pre-match participants using three different approaches: (1) unadjusted, (2) multivariable- adjusted, using all covariates used in the propensity score model, and (3) propensity score-adjusted. Select subgroup analyses were conducted to determine the heterogeneity of the association of hyperuricemia and incident HF using pre-match data, adjusting for propensity scores. All statistical tests were two-sided, and tests with p-value <0.05 were considered significant. Confidence intervals were computed based on a 95% confidence level. SPSS for Windows (Version 15) was used for all data analysis [28].
2.6. Sensitivity analyses
Even though our matched cohort was well balanced in a large number of socio-demographic, clinical, sub-clinical, and biochemical covariates between participants with and without hyperuricemia, we could not rule out the possibility of bias due to imbalances in unmeasured covariates. Therefore, we conducted a formal sensitivity analysis to quantify the degree of a hidden bias due to potential imbalance of an unmeasured covariate that would need to be present to invalidate our main conclusions [29].
3. Results
3.1. Baseline characteristics
Overall, matched participants had a mean age 73 (±6.0) years, 56% were women and 16% were African Americans. The mean (±SD) serum uric acid levels for patients with and without hyperuricemia were 7.5 (±1.0) and 5.0 (±1.0) mg/dL before matching (P<0.0001) and 7.3 (±1.0) and 5.2 (±0.9) mg/dL after matching (P<0.0001), respectively. Imbalances in baseline characteristics before matching and balances achieved after matching between patients with normal and high serum uric acid levels are displayed in Table 1 and Figure 1. After matching, standardized differences for all measured covariates were <10% (most were <5%), suggesting substantial covariate balance across the groups (Figure 1).
Table 1.
Baseline characteristics, by uric acid levels, before and after propensity score matching
| n (%) or mean (±SD) | Before matching | After matching | ||||
|---|---|---|---|---|---|---|
| Normal uric acid (n = 3,956) | High uric acid* (n = 1,505) | P value | Normal uric acid (n = 1,181) | High uric acid* (n = 1,181) | P value | |
| Age, years | 73 (±5) | 74 (±6) | 0.001 | 73 (±6) | 73 (±6) | 0.817 |
| Female | 2,268 (57%) | 874 (58%) | 0.620 | 652 (55%) | 671 (57%) | 0.450 |
| African American | 580 (15%) | 260 (17%) | 0.017 | 197 (17%) | 194 (16%) | 0.912 |
| Married | 2,647 (67%) | 996 (66%) | 0.608 | 802 (68%) | 790 (67%) | 0.629 |
| Living alone | 493 (13%) | 191 (13%) | 0.819 | 143 (12%) | 144 (12%) | 1.000 |
| College or higher education | 1,744 (44%) | 611 (41%) | 0.020 | 471 (40%) | 499 (42%) | 0.253 |
| Income ≥ $25 thousand | 1,482 (38%) | 516 (34%) | 0.029 | 412 (35%) | 427 (36%) | 0.546 |
| Current smoker | 495 (13%) | 168 (11%) | 0.172 | 133 (11%) | 134 (11%) | 1.000 |
| Alcohol, drinks per week | 2.4 (±6.1) | 2.8 (±7.3) | 0.081 | 3.1 (±7.7) | 3.0 (±7.4) | 0.750 |
| Body mass index, kg/m2 | 26 (±4) | 28 (±4) | <0.0001 | 28 (±4) | 28 (±4) | 0.323 |
| Self-reported fair to poor general health | 842 (21%) | 433 (29%) | <0.0001 | 312 (26%) | 300 (25%) | 0.602 |
| Prior myocardial infarction | 286 (7%) | 147 (10%) | 0.002 | 110 (9%) | 106 (9%) | 0.829 |
| Current angina pectoris | 618 (16%) | 308 (21%) | <0.0001 | 220 (19%) | 225 (19%) | 0.832 |
| Hypertension | 1,492 (38%) | 892 (59%) | <0.0001 | 618 (52%) | 626 (53%) | 0.755 |
| Diabetes mellitus | 568 (14%) | 283 (19%) | <0.0001 | 213 (18%) | 212 (18%) | 1.000 |
| Atrial fibrillation by EKG | 77 (2%) | 38 (3%) | 0.183 | 30 (3%) | 31 (3%) | 1.000 |
| LVH by EKG | 140 (4%) | 93 (6%) | <0.0001 | 60 (5%) | 66 (6%) | 0.648 |
| Transient ischemic attack | 182 (5%) | 113 (8%) | <0.0001 | 74 (6%) | 73 (6%) | 1.000 |
| Stroke | 125 (3%) | 82 (5%) | <0.0001 | 52 (4%) | 52 (4%) | 1.000 |
| Intermittent claudication | 72 (2%) | 45 (3%) | 0.008 | 28 (2%) | 27 (2%) | 1.000 |
| Chronic obstructive pulmonary disease | 462 (12%) | 212 (14%) | 0.016 | 165 (14%) | 167 (14%) | 0.953 |
| Chronic kidney disease | 560 (14%) | 581 (39%) | <0.0001 | 349 (30%) | 350 (30%) | 1.000 |
| Cancer | 532 (13%) | 247 (16%) | 0.005 | 183 (16%) | 182 (15%) | 1.000 |
| Medications | ||||||
| ACE inhibitors | 240 (6%) | 134 (9%) | <0.0001 | 93 (8%) | 90 (8%) | 0.878 |
| Beta blockers | 421 (11%) | 276 (18%) | <0.0001 | 198 (17%) | 190 (16%) | 0.695 |
| Calcium channel blockers | 477 (12%) | 206 (14%) | 0.104 | 153 (13%) | 158 (13%) | 0.806 |
| Aspirin | 150 (4%) | 42 (3%) | 0.073 | 30 (3%) | 32 (3%) | 0.894 |
| Statin | 86 (2%) | 36 (2%) | 0.626 | 23 (2%) | 33 (3%) | 0.229 |
| Loop diuretics | 115 (3%) | 146 (10%) | <0.0001 | 70 (6%) | 73 (6%) | 0.858 |
| Thiazide diuretics | 338 (9%) | 279 (19%) | <0.0001 | 197 (17%) | 174 (15%) | 0.206 |
| Potassium sparing diuretics | 16 (0.4%) | 29 (1.9%) | <0.0001 | 10 (1%) | 12 (1%) | 0.832 |
| NSAID | 454 (12%) | 237 (16%) | <0.0001 | 167 (14%) | 168 (14%) | 1.000 |
| Pulse, beats per minute | 68 (±11) | 68 (±11) | 0.117 | 68 (±11) | 68 (±11) | 0.792 |
| Average blood pressure (mm Hg) | ||||||
| Systolic | 136 (±22) | 138 (±22) | 0.001 | 138 (±22) | 138 (±21) | 0.580 |
| Diastolic | 71 (±11) | 71 (±11) | 0.617 | 72 (±11) | 72 (±11) | 0.869 |
| Serum creatinine, mg/dL | 0.90 (±0.35) | 1.09 (±0.43) | <0.0001 | 1.01 (±0.47) | 1.03 (±0.38) | 0.226 |
| Serum glucose, mg/dL | 110 (±37) | 114 (±36) | <0.0001 | 113 (±38) | 114 (±38) | 0.809 |
| Serum insulin, mcU/ml | 15 (±23) | 20 (±27) | <0.0001 | 19 (±30) | 18 (±18) | 0.479 |
| Serum potassium, mEq/L | 4.19 (±0.35) | 4.11 (±0.43) | <0.0001 | 4.13 (±0.38) | 4.14 (±0.42) | 0.364 |
| Total cholesterol, mg/dL | 210 (±38) | 216 (±42) | <0.0001 | 213 (±40) | 214 (±41) | 0.917 |
| Triglyceride, mg/dL | 131 (±73) | 162 (±83) | <0.0001 | 153 (±93) | 154 (±76) | 0.868 |
| Albumin, g/dL | 3.98 (±0.29) | 4.03 (±0.30) | <0.0001 | 4.02 (±0.29) | 4.02 (±0.29) | 0.605 |
| Fibrinogen, mg/dL | 319 (±65) | 332 (±68) | <0.0001 | 324 (±70) | 326 (±69) | 0.517 |
| C reactive protein, mg/dL | 4.2 (±8) | 5.8 (±8) | <0.0001 | 5.2 (±9) | 5.4 (±8) | 0.669 |
| Hemoglobin, gram/dL | 14.0 (±1.40) | 14.1 (±1.42) | 0.618 | 14.1 (±1.41) | 14.1 (±1.38) | 0.837 |
| White blood cells, 103/μL | 6.14 (±1.74) | 6.68 (±2.80) | <0.0001 | 6.48 (±1.90) | 6.46 (±2.02) | 0.811 |
| Platelets, 103/μL | 242 (±64) | 250 (±74) | <0.0001 | 249 (±68) | 249 (±73) | 0.990 |
High uric acid was defined as serum uric acid levels ≥6mg/dL for women and ≥7mg/dL for men
ACE=angiotensin converting enzyme, EKG=electrocardiography, LVH=left ventricular hypertrophy, MI=myocardial infarction, NSAID=non-steroidal anti-inflammatory drug
3.2. Association between serum uric acid and new-onset HF
During 8.1 years of mean follow-up, new-onset HF developed in 19% of matched participants. Incident HF occurred in 21% (rate, 2,623/100,000 person-years of follow-up) and 18% (rate, 2,130/100,000 person-years of follow-up) of matched participants with and without hyperuricemia (hazard ratio {HR} when hyperuricemia was compared with normal serum uric acid, 1.30; 95% confidence interval {CI}, 1.05–1.60; P=0.015 (Figure 2 and Table 2). When serum uric acid was used as a continuous variable, every 1 mg/dL increase in serum uric acid level was associated with significant increase in the risk of incident HF (HR, 1.12; 95% CI, 1.03–1.22; P =0.006).
Figure 2.
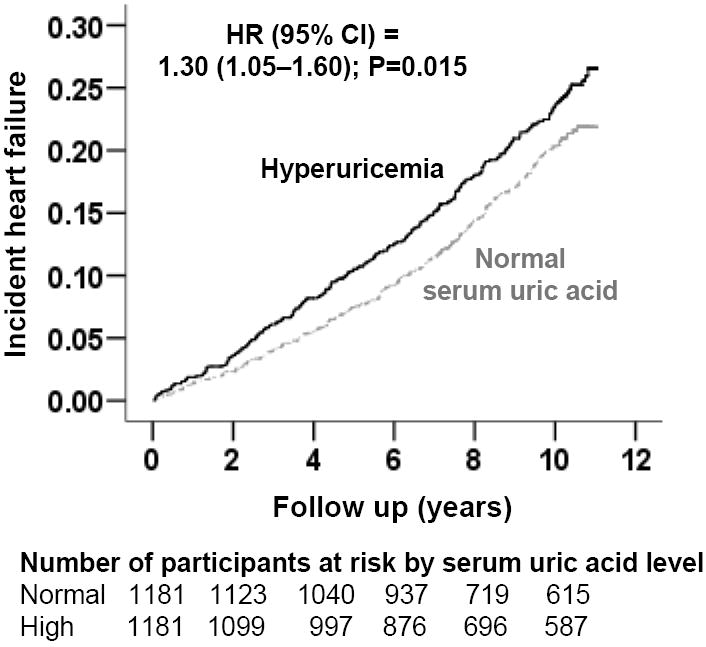
Kaplan-Meier plots for incident heart failure in participants with and without hyperuricemia (defined as serum uric acid levels ≥6mg/dL for women and ≥7mg/dL for men) (HR=hazard ratio; CI=confidence interval)
Table 2.
High serum uric acid* and outcomes
| Outcomes | Rate, per 100,000 person-years (Events / total follow up years) | Absolute rate difference** (per 100,000 person-years) | Matched hazard ratio† (95% confidence interval) | P value | |
|---|---|---|---|---|---|
| Normal uric acid (n = 1,181) | High uric acid (n = 1,181) | ||||
| New-onset heart failure | 2,130 (208 / 9,767) | 2,623 (248 / 9,452) | + 494 | 1.30 (1.05–1.60) | 0.015 |
| Acute myocardial infarction | 1,043 (104 / 9,974) | 1,233 (121 / 9,817) | + 190 | 1.23 (0.91–1.65) | 0.178 |
| All-cause mortality | 3,917 (404 / 10,313) | 4,215 (430 / 10,202) | + 292 | 1.14 (0.98–1.34) | 0.090 |
High uric acid was defined as serum uric acid levels ≥6mg/dL for women and ≥7mg/dL for men.
Absolute rate differences were calculated by subtracting the rates of new-onset heart failure in the high serum uric acid group from the rate of new-onset heart failure in the normal serum uric acid group (before values were rounded)
In the pre-match cohort of 5,461 participants, incident HF occurred in 22% and 15% of participants respectively with and without hyperuricemia (unadjusted HR, 1.63; 95% CI, 1.42–1.86; P<0.0001). Multivariable-adjusted and propensity-adjusted HRs for hyperuricemia-associated incident HF were respectively 1.18 (95% CI, 1.01–1.36; P=0.033) and 1.15 (95% CI, 0.99–1.35; P=0.077). The propensity-adjusted association between hyperuricemia and incident HF became significant when we restricted our analysis to those without baseline CKD (HR, 1.23; 95% CI, 1.02–1.49; P=0.031) and when uric acid was used as a continuous variable (HR, 1.08; 95% CI, 1.03–1.13; P=0.002).
3.3. Results of sensitivity analysis
In the absence of hidden bias, a sign-score test for matched data with censoring provides strong evidence (P =0.0145) that participants with normal serum uric acid clearly had fewer incident HF than those with hyperuricemia. A hidden covariate, that is a near-perfect predictor of incident HF and is not strongly associated with any of the 64 baseline covariates used in our study, would need to increase the odds of hyperuricemia by 5.2% before it could potentially explain away this association
3.4. Findings from subgroup analyses
The association of hyperuricemia with incident HF was not significant in subgroups of participants in whom hyperuricemia may have been caused by impaired renal elimination of uric acid, viz. hypertension (or the use of thiazide diuretics), chronic kidney disease (CKD), as defined by an estimated glomerular filtration rate <60 ml/min/1.73 m2, and hyperinsulinemia [30-33]. The association between hyperuricemia and incident HF was significant only in subgroups with normal kidney function (HR, 1.23; 95% CI, 1.02–1.49; P=0.031), without hypertension (HR, 1.31; 95% CI, 1.03–1.66; P=0.030), not receiving thiazide diuretics (HR, 1.20; 95% CI, 1.01–1.42; P=0.044), and without hyperinsulinemia (HR, 1.35; 95% CI, 1.06–1.72; P=0.013; Figure 3).
Figure 3.
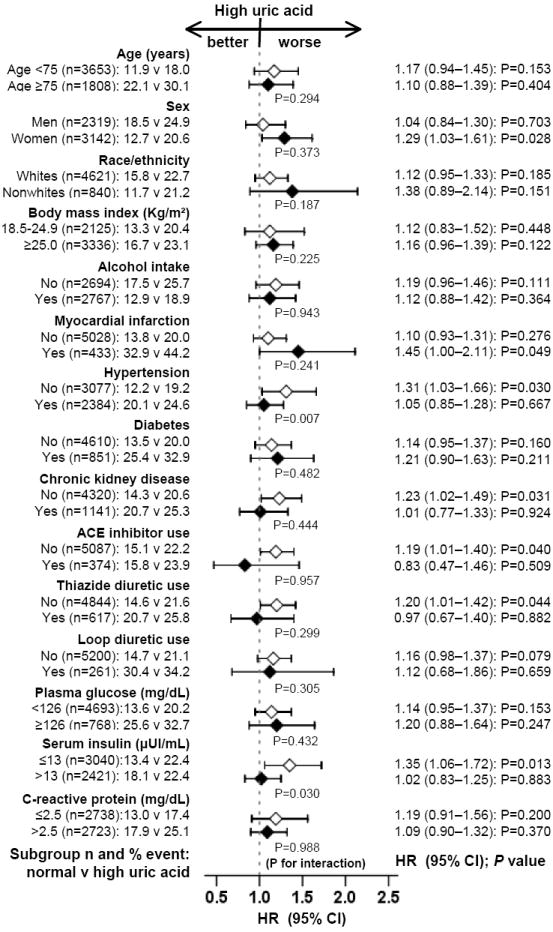
Association between hyperuricemia (defined as serum uric acid levels ≥6mg/dL for women and ≥7mg/dL for men) and incident heart failure in subgroups of participants (HR=hazard ratio; CI=confidence interval)
3.5. Association of serum uric acid with other outcomes
There were 225 cases of incident AMI, all of which occurred in 2,146 matched participants without a prior AMI. AMI occurred in 10% (rate, 1,233 /100,000 person-years of follow-up) and 9% (rate, 1,043 /100,000 person-years of follow-up) of participants respectively with and without hyperuricemia (HR for hyperuricemia, 1.23; 95% CI, 0.91–1.65; P=0.178; Table 2). Overall, 834 matched participants died from all causes, which occurred in 36% (rate, 4,215 /100,000 person-years of follow-up) and 34% (rate, 3,917 /100,000 person-years of follow-up) of participants respectively with and without hyperuricemia (HR, 1.14; 95% CI, 0.98–1.34; P=0.090; Table 2).
4. Discussion
4.1. Key findings
The findings from the current analysis demonstrate that in a propensity-matched population of community-dwelling older adults, who were well balanced in 64 measured baseline demographic, clinical, subclinical, and biochemical covariates, baseline hyperuricemia was associated with new-onset HF but had no association with myocardial infarction or mortality. The association between hyperuricemia and incident HF seemed to be significant only in subgroups of participants in whom hyperuricemia was likely due to increased production of uric acid rather than impaired renal clearance of uric acid. The cumulative evidence from our subgroups analyses suggests that uric acid may lack an intrinsic association with HF and that hyperuricemia may be a predictor of HF when it is marker of increased xanthine oxidase activity.
4.2. Possible mechanistic explanations
Because of an evolutionary loss of hepatic uricase, uric acid elimination in humans is nearly completely dependent on the kidneys, yet due to a highly efficient renal tubular reabsorption of filtered uric acid, the renal elimination of uric acid is imperfect [33, 34]. Therefore, hyperuricemia in humans is more often due to an impaired excretion of uric acid than its increased production. However, hyperuricemia can also occur due to excess production of uric acid by increased xanthine oxidase activity during conditions of ischemia and/or hypoxia [35, 36]. Thus, the association of hyperuricemia and incident HF observed in our study may be explained by an increased xanthine oxidase activity (increased production), by a high serum uric acid level (a direct effect), by confounding, or combinations thereof. However, cumulative findings from our subgroup analyses suggest that increased production of uric acid via increased xanthine oxidase activity may provide a mechanistic insight into the association between hyperuricemia and incident HF found in our study.
Xanthine oxidase, a flavoprotein enzyme that catalyzes the oxidation of xanthine to uric acid, also generates free oxygen radicals and causes inflammation [4, 5]. In laboratory animals, xanthine oxidase depresses myocardial excitation-contraction coupling and causes oxidative damage during myocardial ischemia-reperfusion [37, 38]. Xanthine oxidase inhibition, on the other hand, improves myofilament responsiveness to calcium in the ischemic, as well as, in the normal cardiomyocyte [38, 39]. Animal models of HF are associated with increased xanthine oxidase activity, inhibition of which improves left ventricular performance and outcomes [40]. Endothelial function is improved by allopurinol, a xanthine oxidase inhibitor, but not by probenecid, a uricosuric drug [41-43]. Therefore, it is plausible that increased xanthine oxidase activity may be responsible for diastolic and/or systolic dysfunction resulting in clinical HF. Cumulative evidence from our subgroup analyses supports this xanthine oxidase hypothesis. Because renal excretion of uric acid is expected to be normal in participants without hypertension (and in those not receiving thiazide diuretics), without hyperinsulinemia, and in those with normal kidney function, hyperuricemia in these participants is likely due to increased production of uric acid rather than impaired renal excretion. However, as renal excretion of serum uric acid is impaired in patients with hypertension, hyperinsulinemia, CKD, and in those receiving thiazide diuretics, these patients may develop hyperuricemia despite normal uric acid production and normal xanthine oxidase activity [30-32, 44]. In fact, despite a higher mean serum uric acid levels in those with CKD (6.5 versus 6.2 mg/dL in those without CKD; P<0.0001), high serum uric acid had no association with incident HF in these patients, pointing to a lack of an intrinsic effect of uric acid.
In-vivo studies of the direct effect of high serum uric acid are complicated by difficulties in isolating the effect of uric acid from those of xanthine oxidase, and its apparent opposing effects in laboratory animals and humans. In cultures of rat vascular smooth muscles and cardiomyocytes, the addition of uric acid resulted in the release of monocyte chemoattractant protein-1, an important chemokine in the pathogenesis of atherosclerosis and myocardial fibrosis [45, 46]. In laboratory animals, artificial hyperuricemia induced by oral uricase inhibitor, led to hypertension and renal damage [47, 48]. In those studies, the adverse effects of hyperuricemia could be prevented by both allopurinol and benziodarone, an uricosuric drug, suggesting a direct effect of uric acid. In humans, on the other hand, an acute systemic administration of uric acid has been shown to improve endothelial function [49]. It has been suggested that serum uric acid may be elevated as a compensatory mechanism to attenuate oxidative damage related to atherosclerosis [50, 51]. Endothelial nitric oxide plays an important role in inhibiting xanthine oxidase activity. However, under conditions of oxidative stress, reduced vascular nitric oxide activity stimulates increased xanthine oxidase activity and elevates serum uric acid levels, which in turn reduces the oxidative stress [52, 53]. Therefore, acute elevation of serum uric acid is unlikely to have an adverse cardiovascular effect. However, the effect of chronic uric acid elevation in humans remains an open question.
Finally, the association of hyperuricemia with incident cardiovascular morbidity and mortality has been attributed to confounding risk factors such as CKD and hypertension [6, 7, 54, 55]. However, our matched participants with high and normal serum uric acid were well balanced in 64 baseline covariates including major traditional risk factors (Figure 1) suggesting that our findings may not be explained by baseline imbalances in any of those measured covariates. However, bias due to imbalances in unmeasured covariates is possible. While sensitivity analysis cannot determine whether an unmeasured confounder exists, it suggests that for an unmeasured covariate to explain away the association between hyperuricemia and incident HF observed in our study, it would need to increase the odds of hyperuricemia by 5.2% [29]. Further, to be a confounder, that unmeasured covariate would also need to be a near-perfect predictor of incident HF and could not be strongly correlated with any of the measured covariates in our study.
4.3. Comparison with other published studies
The associations of high serum uric acid with cardiovascular morbidity and mortality have been studied in a number of population-based studies [6, 7, 9, 56]. Even though HF was part of the composite cardiovascular outcome in many of those studies, none examined incident HF as a separate outcome. Further, the use of propensity matching and adjustment for 64 baseline characteristics distinguishes our study from all prior studies.
4.4. Clinical and public health implications
Findings of the current analysis are important as they identify subsets of community-dwelling older adults in whom hyperuricemia may be a potential marker of incident HF. They also provide insights as to why interventions using xanthine oxidase inhibitors may not have been successful in improving outcomes in patients with HF [55]. The prevalence of CKD is high in HF in whom hyperuricemia is more likely due to reduced renal excretion of uric acid. If hyperuricemia is due to impaired renal excretion and not due to increased xanthine oxidase activity, the use of xanthine oxidase inhibitors is unlikely to improve outcomes. It remains to be seen, however, if xanthine oxidase inhibitors would improve outcomes in HF patients without CKD in whom hyperuricemia is likely due to increased xanthine oxidase activity and not to impaired renal excretion of uric acid.
4.5. Study limitations
We were able to match 78% of participants with hyperuricemia. Therefore, any effect due to loss of participants during matching would be minimal. Further, we were able to reproduce our key findings in the pre-match dataset. We had no data on gout or the use of xanthine oxidase inhibitors. Participants with normal and high serum uric acid at baseline may have developed high or normal serum uric acid respectively during follow-up. This regression dilution may have underestimated the true associations observed in our study [57].
4.6. Conclusions
In a well-balanced propensity-matched population of community-dwelling older adults, baseline hyperuricemia was associated with incident HF. However, cumulative data from our subgroup analyses indicate that hyperuricemia may predict incident HF when it is due to increased production of uric acid reflecting an increased xanthine oxidase activity and not when it is due to impaired renal excretion of uric acid suggesting lack of a direct intrinsic effect. These conclusions are hypothesis generating and need to be replicated in other patient populations using propensity score methods.
Acknowledgments
“The Cardiovascular Health Study (CHS) was conducted and supported by the NHLBI in collaboration with the CHS Investigators. This manuscript was prepared using a limited access dataset obtained by the NHLBI and does not necessarily reflect the opinions or views of the CHS Study or the NHLBI.”
The authors wish to thank Casey Daniel for her careful review of the manuscript.
Funding/Support: Dr. Ahmed is supported by the National Institutes of Health through a grant from the National Heart, Lung, and Blood Institute (5-R01-HL085561-03) and a generous gift from Ms. Jean B. Morris of Birmingham, Alabama
Footnotes
Author Contributions Dr. Ahmed conceived the study hypothesis and design. Drs. Ahmed, Dell’Italia and Ekundayo wrote the first draft. Dr. Ahmed performed statistical analyses in collaboration with Drs. Ekundayo, Aban and Love. All authors interpreted the data, participated in critical revision of the paper for important intellectual content, and approved the final version of the article. Drs. Ahmed and Ekundayo had full access to all data.
An abstract based on the current analysis was presented at the American College of Cardiology annual meeting in Chicago in March 2008.
‘The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology’ [58].
Conflict of Interest Disclosures: None
References
- 1.Anker SD, Leyva F, Poole-Wilson PA, Kox WJ, Stevenson JC, Coats AJ. Relation between serum uric acid and lower limb blood flow in patients with chronic heart failure. Heart. 1997;78:39–43. doi: 10.1136/hrt.78.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leyva F, Anker SD, Godsland IF, et al. Uric acid in chronic heart failure: a marker of chronic inflammation. Eur Heart J. 1998;19:1814–22. doi: 10.1053/euhj.1998.1188. [DOI] [PubMed] [Google Scholar]
- 3.Anker SD, Doehner W, Rauchhaus M, et al. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation. 2003;107:1991–7. doi: 10.1161/01.CIR.0000065637.10517.A0. [DOI] [PubMed] [Google Scholar]
- 4.Doehner W, Anker SD. Uric acid in chronic heart failure. Semin Nephrol. 2005;25:61–6. doi: 10.1016/j.semnephrol.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Hare JM, Johnson RJ. Uric acid predicts clinical outcomes in heart failure: insights regarding the role of xanthine oxidase and uric acid in disease pathophysiology. Circulation. 2003;107:1951–3. doi: 10.1161/01.CIR.0000066420.36123.35. [DOI] [PubMed] [Google Scholar]
- 6.Culleton BF, Larson MG, Kannel WB, Levy D. Serum uric acid and risk for cardiovascular disease and death: the Framingham Heart Study. Ann Intern Med. 1999;131:7–13. doi: 10.7326/0003-4819-131-1-199907060-00003. [DOI] [PubMed] [Google Scholar]
- 7.Fang J, Alderman MH. Serum uric acid and cardiovascular mortality the NHANES I epidemiologic follow-up study, 1971-1992. National Health and Nutrition Examination Survey. JAMA. 2000;283:2404–10. doi: 10.1001/jama.283.18.2404. [DOI] [PubMed] [Google Scholar]
- 8.Strasak AM, Kelleher CC, Brant LJ, et al. Serum uric acid is an independent predictor for all major forms of cardiovascular death in 28,613 elderly women: a prospective 21-year follow-up study. Int J Cardiol. 2008;125:232–9. doi: 10.1016/j.ijcard.2007.11.094. [DOI] [PubMed] [Google Scholar]
- 9.Meisinger C, Koenig W, Baumert J, Doring A. Uric Acid Levels Are Associated With All-Cause and Cardiovascular Disease Mortality Independent of Systemic Inflammation in Men From the General Population. The MONICA/KORA Cohort Study. Arterioscler Thromb Vasc Biol. 2008 doi: 10.1161/ATVBAHA.107.160184. [DOI] [PubMed] [Google Scholar]
- 10.Lazzeri C, Valente S, Chiostri M, Sori A, Bernardo P, Gensini GF. Uric acid in the acute phase of ST elevation myocardial infarction submitted to primary PCI: Its prognostic role and relation with inflammatory markers A single center experience. Int J Cardiol. 2008 Aug 4; doi: 10.1016/j.ijcard.2008.06.024. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 11.Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–76. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 12.Cushman M, Cornell ES, Howard PR, Bovill EG, Tracy RP. Laboratory methods and quality assurance in the Cardiovascular Health Study. Clin Chem. 1995;41:264–70. [PubMed] [Google Scholar]
- 13.Krishnan E, Kwoh CK, Schumacher HR, Kuller L. Hyperuricemia and incidence of hypertension among men without metabolic syndrome. Hypertension. 2007;49:298–303. doi: 10.1161/01.HYP.0000254480.64564.b6. [DOI] [PubMed] [Google Scholar]
- 14.Psaty BM, Kuller LH, Bild D, et al. Methods of assessing prevalent cardiovascular disease in the Cardiovascular Health Study. Ann Epidemiol. 1995;5:270–7. doi: 10.1016/1047-2797(94)00092-8. [DOI] [PubMed] [Google Scholar]
- 15.Ives DG, Fitzpatrick AL, Bild DE, et al. Surveillance and ascertainment of cardiovascular events. The Cardiovascular Health Study. Ann Epidemiol. 1995;5:278–85. doi: 10.1016/1047-2797(94)00093-9. [DOI] [PubMed] [Google Scholar]
- 16.Gottdiener JS, Arnold AM, Aurigemma GP, et al. Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol. 2000;35:1628–37. doi: 10.1016/s0735-1097(00)00582-9. [DOI] [PubMed] [Google Scholar]
- 17.Kitzman DW, Gardin JM, Gottdiener JS, et al. Importance of heart failure with preserved systolic function in patients > or = 65 years of age. CHS Research Group. Cardiovascular Health Study. Am J Cardiol. 2001;87:413–9. doi: 10.1016/s0002-9149(00)01393-x. [DOI] [PubMed] [Google Scholar]
- 18.Gottdiener JS, McClelland RL, Marshall R, et al. Outcome of congestive heart failure in elderly persons: influence of left ventricular systolic function. The Cardiovascular Health Study. Ann Intern Med. 2002;137:631–9. doi: 10.7326/0003-4819-137-8-200210150-00006. [DOI] [PubMed] [Google Scholar]
- 19.Schellenbaum GD, Rea TD, Heckbert SR, et al. Survival associated with two sets of diagnostic criteria for congestive heart failure. Am J Epidemiol. 2004;160:628–35. doi: 10.1093/aje/kwh268. [DOI] [PubMed] [Google Scholar]
- 20.Schellenbaum GD, Heckbert SR, Smith NL, et al. Congestive heart failure incidence and prognosis: case identification using central adjudication versus hospital discharge diagnoses. Ann Epidemiol. 2006;16:115–22. doi: 10.1016/j.annepidem.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 21.de Simone G, Gottdiener JS, Chinali M, Maurer MS. Left ventricular mass predicts heart failure not related to previous myocardial infarction: the Cardiovascular Health Study. Eur Heart J. 2008;29:741–7. doi: 10.1093/eurheartj/ehm605. [DOI] [PubMed] [Google Scholar]
- 22.Ahmed A, Husain A, Love TE, et al. Heart failure, chronic diuretic use, and increase in mortality and hospitalization: an observational study using propensity score methods. Eur Heart J. 2006;27:1431–9. doi: 10.1093/eurheartj/ehi890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed A, Zannad F, Love TE, et al. A propensity-matched study of the association of low serum potassium levels and mortality in chronic heart failure. Eur Heart J. 2007;28:1334–43. doi: 10.1093/eurheartj/ehm091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed A, Aban IB, Vaccarino V, et al. A propensity-matched study of the effect of diabetes on the natural history of heart failure: variations by sex and age. Heart. 2007;93:1584–90. doi: 10.1136/hrt.2006.113522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenbaum PR, Rubin DB. The central role of propensity score in observational studies for causal effects. Biometrika. 1983;70:41–55. [Google Scholar]
- 26.Rubin DB. Using propensity score to help design observational studies: Application to the tobacco litigation. Health Services and Outcomes Research Methodology. 2001;2:169–188. [Google Scholar]
- 27.Normand ST, Landrum MB, Guadagnoli E, et al. Validating recommendations for coronary angiography following acute myocardial infarction in the elderly: a matched analysis using propensity scores. J Clin Epidemiol. 2001;54:387–98. doi: 10.1016/s0895-4356(00)00321-8. [DOI] [PubMed] [Google Scholar]
- 28.SPSS for Windows, Rel. 15 program] SPSS Inc.; Chicago, IL: 2007. [Google Scholar]
- 29.Rosenbaum PR. Sensitivity to Hidden Bias. In: Rosenbaum PR, editor. Observational Studies. 2. New York: Springer-Verlag; 2002. pp. 110–124. [Google Scholar]
- 30.Facchini F, Chen YD, Hollenbeck CB, Reaven GM. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA. 1991;266:3008–11. [PubMed] [Google Scholar]
- 31.Ferrannini E, Buzzigoli G, Bonadonna R, et al. Insulin resistance in essential hypertension. N Engl J Med. 1987;317:350–7. doi: 10.1056/NEJM198708063170605. [DOI] [PubMed] [Google Scholar]
- 32.Quinones-Galvan A, Natali A, Baldi S, et al. Effect of insulin on uric acid excretion in humans. Am J Physiol. 1995;268:E1–5. doi: 10.1152/ajpendo.1995.268.1.E1. [DOI] [PubMed] [Google Scholar]
- 33.Sanders PW. Uric acid: an old dog with new tricks? J Am Soc Nephrol. 2006;17:1767–8. doi: 10.1681/ASN.2006050455. [DOI] [PubMed] [Google Scholar]
- 34.Wu XW, Lee CC, Muzny DM, Caskey CT. Urate oxidase: primary structure and evolutionary implications. Proc Natl Acad Sci U S A. 1989;86:9412–6. doi: 10.1073/pnas.86.23.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terada LS, Guidot DM, Leff JA, et al. Hypoxia injures endothelial cells by increasing endogenous xanthine oxidase activity. Proc Natl Acad Sci U S A. 1992;89:3362–6. doi: 10.1073/pnas.89.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambers DE, Parks DA, Patterson G, et al. Xanthine oxidase as a source of free radical damage in myocardial ischemia. J Mol Cell Cardiol. 1985;17:145–52. doi: 10.1016/s0022-2828(85)80017-1. [DOI] [PubMed] [Google Scholar]
- 37.Khan SA, Lee K, Minhas KM, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–8. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kogler H, Fraser H, McCune S, Altschuld R, Marban E. Disproportionate enhancement of myocardial contractility by the xanthine oxidase inhibitor oxypurinol in failing rat myocardium. Cardiovasc Res. 2003;59:582–92. doi: 10.1016/s0008-6363(03)00512-1. [DOI] [PubMed] [Google Scholar]
- 39.Perez NG, Gao WD, Marban E. Novel myofilament Ca2+-sensitizing property of xanthine oxidase inhibitors. Circ Res. 1998;83:423–30. doi: 10.1161/01.res.83.4.423. [DOI] [PubMed] [Google Scholar]
- 40.Minhas KM, Saraiva RM, Schuleri KH, et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res. 2006;98:271–9. doi: 10.1161/01.RES.0000200181.59551.71. [DOI] [PubMed] [Google Scholar]
- 41.George J, Carr E, Davies J, Belch JJ, Struthers A. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation. 2006;114:2508–16. doi: 10.1161/CIRCULATIONAHA.106.651117. [DOI] [PubMed] [Google Scholar]
- 42.Doehner W, Schoene N, Rauchhaus M, et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation. 2002;105:2619–24. doi: 10.1161/01.cir.0000017502.58595.ed. [DOI] [PubMed] [Google Scholar]
- 43.Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106:221–6. doi: 10.1161/01.cir.0000022140.61460.1d. [DOI] [PubMed] [Google Scholar]
- 44.Langford HG, Blaufox MD, Borhani NO, et al. Is thiazide-produced uric acid elevation harmful? Analysis of data from the Hypertension Detection and Follow-up Program. Arch Intern Med. 1987;147:645–9. doi: 10.1001/archinte.147.4.645. [DOI] [PubMed] [Google Scholar]
- 45.Kanellis J, Watanabe S, Li JH, et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension. 2003;41:1287–93. doi: 10.1161/01.HYP.0000072820.07472.3B. [DOI] [PubMed] [Google Scholar]
- 46.Cheng TH, Lin JW, Chao HH, et al. Uric acid activates extracellular signal-regulated kinases and thereafter endothelin-1 expression in rat cardiac fibroblasts. Int J Cardiol. 2008 doi: 10.1016/j.ijcard.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 47.Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38:1101–6. doi: 10.1161/hy1101.092839. [DOI] [PubMed] [Google Scholar]
- 48.Kang DH, Nakagawa T, Feng L, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002;13:2888–97. doi: 10.1097/01.asn.0000034910.58454.fd. [DOI] [PubMed] [Google Scholar]
- 49.Waring WS, McKnight JA, Webb DJ, Maxwell SR. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes. 2006;55:3127–32. doi: 10.2337/db06-0283. [DOI] [PubMed] [Google Scholar]
- 50.Nieto FJ, Iribarren C, Gross MD, Comstock GW, Cutler RG. Uric acid and serum antioxidant capacity: a reaction to atherosclerosis? Atherosclerosis. 2000;148:131–9. doi: 10.1016/s0021-9150(99)00214-2. [DOI] [PubMed] [Google Scholar]
- 51.Tan S, Radi R, Gaudier F, et al. Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatr Res. 1993;34:303–7. doi: 10.1203/00006450-199309000-00013. [DOI] [PubMed] [Google Scholar]
- 52.Maxwell AJ, Bruinsma KA. Uric acid is closely linked to vascular nitric oxide activity. Evidence for mechanism of association with cardiovascular disease. J Am Coll Cardiol. 2001;38:1850–8. doi: 10.1016/s0735-1097(01)01643-6. [DOI] [PubMed] [Google Scholar]
- 53.Sevanian A, Davies KJ, Hochstein P. Serum urate as an antioxidant for ascorbic acid. Am J Clin Nutr. 1991;54:1129S–1134S. doi: 10.1093/ajcn/54.6.1129s. [DOI] [PubMed] [Google Scholar]
- 54.Freedman DS, Williamson DF, Gunter EW, Byers T. Relation of serum uric acid to mortality and ischemic heart disease. The NHANES I Epidemiologic Follow-up Study. Am J Epidemiol. 1995;141:637–44. doi: 10.1093/oxfordjournals.aje.a117479. [DOI] [PubMed] [Google Scholar]
- 55.Moriarity JT, Folsom AR, Iribarren C, Nieto FJ, Rosamond WD. Serum uric acid and risk of coronary heart disease: Atherosclerosis Risk in Communities (ARIC) Study. Ann Epidemiol. 2000;10:136–43. doi: 10.1016/s1047-2797(99)00037-x. [DOI] [PubMed] [Google Scholar]
- 56.Sakata K, Hashimoto T, Ueshima H, Okayama A. Absence of an association between serum uric acid and mortality from cardiovascular disease: NIPPON DATA 80, 1980-1994. National Integrated Projects for Prospective Observation of Non-communicable Diseases and its Trend in the Aged. Eur J Epidemiol. 2001;17:461–8. doi: 10.1023/a:1013735717961. [DOI] [PubMed] [Google Scholar]
- 57.Clarke R, Shipley M, Lewington S, et al. Underestimation of risk associations due to regression dilution in long-term follow-up of prospective studies. Am J Epidemiol. 1999;150:341–53. doi: 10.1093/oxfordjournals.aje.a010013. [DOI] [PubMed] [Google Scholar]
- 58.Coats AJ. Ethical authorship and publishing. Int J Cardiol. 2009;131:149–50. doi: 10.1016/j.ijcard.2008.11.048. [DOI] [PubMed] [Google Scholar]