

Abstract
Cardiovascular complications are the leading cause of death in patients with chronic kidney disease (CKD). Traditional causes such as diabetes, smoking, aging and hypertension do not fully explain the high rate of morbidity from cardiovascular disease seen in these patients. The renin-angiotensin-aldosterone system (RAAS) regulates extracellular volume homeostasis, which contributes to blood pressure stability. Overactivity of this system is involved in the pathophysiology of cardio-renal disease. New evidence suggests that vitamin D receptor activators (VDRAs) have a suppressive effect on the RAAS; However, VDRAs also have anti-inflammatory and anti-fibrotic effects. We have demonstrated that paricalcitol, a VDRA, ameliorates left ventricular hypertrophy (LVH) in uremic rats by up-regulating the VDR, decreasing myocardial PCNA and also decreasing myocardial oxidative stress.
Thus, paricalcitol can suppress the progression of LVH, myocardial and perivascular fibrosis and myocardial arterial vessel thickness presumably by up regulating the VDR. Paricalcitol may prove to have a substantial beneficial effect on cardiac disease and its outcome in patients with CKD. Prospective randomized studies in CKD patients are necessary to confirm these results.
1. Introduction
Uremic Cardiomyopathy is a common complication in patients with chronic kidney disease (CKD) and is associated with cardiovascular disease and the progression of renal disease, which is directly responsible for the high morbidity and mortality in these patients. Cardiomyopathy is characterized by cardiac fibrosis, cardiac hypertrophy and diastolic dysfunction. The renal involvement is characterized by glomerulosclerosis, interstitial infiltration of mononuclear cells, proteinuria, a progressive decrease in renal function and elevated blood pressure. Oxidative stress also plays an important role in the overall picture.
Recent studies in patients with ESRD have shown a survival benefit for patients receiving vitamin D analogs (1-4). This survival advantage is independent of the effects of these analogs on mineral metabolism. There is growing evidence that vitamin D has a beneficial effect on cardiovascular and renal disease in patients with CKD, which may, at least in part, explain the increased survival rate. The effects of vitamin D analogs go far beyond their traditional actions on mineral metabolism. Vitamin D analogs have significant pleiotropic effects on such functions as cell differentiation, immunomodulation, inflammation and fibrosis as well as on the endocrine, renal and cardiovascular systems(5). We therefore studied the effect of the vitamin D analog, paricalcitol, a VDR activator (VDRA), on the progression of cardiomyopathy in the 5/6 nephrectomized uremic model.
2. Materials and methods
Female Sprague-Dawley rats weighing from 225 to 250 grams underwent 5/6 nephrectomy. Rats were maintained on tap water ad lib and fed a high phosphorus (P) diet (1.2% P and 0.8% calcium (Ca)) for the duration of the study. At the onset of uremia, the rats were divided into the following treatment groups:
Group 1: Normal Control (NC) treated intraperitoneally (I.P.) with vehicle (100 μl of propylene glycol)
Group 2: Uremic Control (UC) treated I.P. with vehicle
Group 3: Uremic+ paricalcitol (UP) treated I.P. with paricalcitol (40 ng/injection)
Rats were treated 3 x week for 4 weeks. Twenty four hours after the last injection the rats were sacrificed under anesthesia by exsanguination. Blood was taken for determination of PTH and serum chemistries. Myocardium was taken for analysis of VDR, PCNA, interstitial fibrosis and vessel thickness. VDR was assessed by immunohistochemistry and western blot analysis and PCNA by immunohistochemistry. Fibrosis was assessed immunohistochemically using a Masson trichrome stain.
Western Blot Analysis
The protein expression of VDR and GAPDH was determined by Western blot analysis. Briefly, left ventricle tissue was homogenized in 2 ml of Cell Lysis Buffer (Cell Signaling Technology, Beverly, MA). Samples were centrifuged at 3000×g for 15 min, and the supernatants were assayed. After being mixed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and boiled for 5 min, samples (10 μg per lane) were electrophoresed on 4 to 12% SDS polyacrylamide gels and transferred to nitrocellulose membranes for 2 h at 30 V. Membranes were blocked for 30 min with Tris-buffered saline that contained 2% non-fat dry milk and 0.05% Tween 20 and incubated with diluted primary antibody overnight at room temperature. The source and the concentration of each antibody were as follows: Rabbit anti–VDR (Santa Cruz Biotechnology, Santa Cruz, CA; 1:200) and rabbit anti–GAPDH antibody (Cell Signaling Technology, 1:1000). The membranes were washed, and diluted secondary horseradish peroxidase (HRP)-conjugated antibodies were added (anti-rabbit IgG-HRP 1:1000; Santa Cruz Biotechnology). The membranes were again washed and developed using the enhanced chemiluminescence system (LumiGLO Reagent; Cell Signaling Technology).
Immunohistochemistry
Immunohistochemical staining for VDR was performed by using a rat anti-VDR antibody (Chemicon, Temecula, CA) and a commercial staining kit (Histomouse-Max kit; Invitrogen, Carlsbad, CA). The sections were deparaffinized, rehydrated, and microwaved in 0.01 mol/L citrate buffer (pH 6.0) for 10 min to retrieve the antigens. The sections were then treated with 0.6% hydrogen peroxide in methanol for 10 min at room temperature to block endogenous peroxidase and subsequently blocked with 10% preimmune serum (goat) for 10 min at room temperature. The primary VDR antibody (1:100) was added followed by an overnight incubation at room temperature. Biotinylated secondary antibody was applied, followed by a streptavidin-HRP conjugate. The immune complexes were visualized with 3-amino-9-ethylcarbazole substrate-chromagen. Finally, all sections were counterstained with hematoxylin. Immunohistochemical staining for PCNA was performed by using a PCNA Staining Kit (Zymed®) (Invitrogen, CA) according to the manufacturer's instructions. The number of VDR-positive nuclei was counted manually in the 6 to 8 randomly selected areas of the samples at a magnification of×400. The average number of VDR-positive cell per section was calculated by dividing the sum of the VDR-positive cells by the sum of the total cell number, yielding the number of VDR-positive cell per 1000 cardiac cells. The semi quantification of PCNA-positive cell was performed in the same manner.
3. Results
Table 1 shows the blood chemistries. Serum creatinine increased from 0.75±0.02 in the (NC) group to 1.52±0.24 in the (UC) group and 1.35±0.08 mg/dl in the (UP) group. PTH was markedly increased in the uremic control groups compared to normal animals (NC: 58.1±22.1 vs. UC: 1580±415 pg/ml, p<0.01). Paricalcitol treatment greatly reduced PTH levels (250±63 pg/ml, p<0.01).
Table 1.
The Effect of Paricalcitol on Serum Chemistries in Uremic Rats
| Group | Cr (mg/dl) | ICa (mg/dl) | Total Ca (mg/dl) | P (mg/dl) | Ca × P (mg2/ml2) | PTH (pg/ml) |
|---|---|---|---|---|---|---|
| NC n=9 | 0.75 ± 0.02 | 4.67 ± 0.09 | 10.00 ± 0.21 | 5.04 ± 0.28 | 50.7 ± 3.7 | 58 ± 22.1 |
| UC n=9 | 1.52 ± 0.24 | 4.26 ± 0.14 | 9.44 ± 0.37 | 9.42 ± 2.13 | 90.9 ± 22.3 | 1580 ± 415 |
| Paricalcitol n=11 | 1.35 ± 0.08 | 4.93 ± 0.14 | 10.89 ± 0.32 | 8.06 ± .061 | 88.2 ± 7.64 | 250 ± 63 |
Figure 1 shows that after 4 weeks of uremia, left ventricular weight was significantly increased compared to the normal rats (4.01±0.25 vs. 3.01±0.06 mg/gBW, p<0.01), but treatment with paricalcitol, 3 times per week for 4 weeks, partially inhibited the development of LVH (3.53±0.86 mg/gBW).
Fig.1.
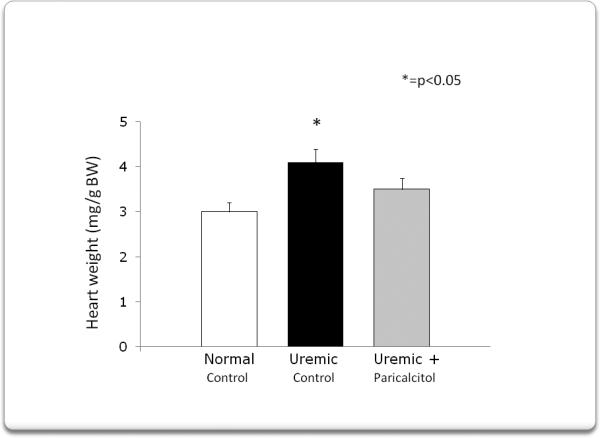
After four weeks of uremia, left ventricular weight significantly increased compared to normal rats, but treatment with paricalcitol 3 times per week for four weeks, minimized left ventricular hypertrophy.
Figure 2 depicts a representative Western blot analysis for VDR in myocardial tissue showing that 4 weeks of uremia markedly decreased VDR expression compared to normal rats.
Fig.2.
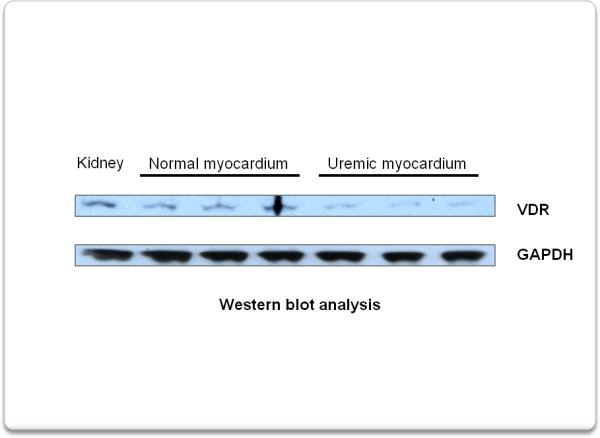
Western blot analysis shows that, compared to normal rats, 4 weeks of uremia causes a decrease in VDR expression in the myocardium.
Immunohistochemical analysis confirmed not only the decrease in VDR expression in myocardium after 4 weeks of uremia, but its prevention by the administration of paricalcitol (Fig 3). The quantification of VDR + cells is shown in figure 4. VDR expression decreased from 774±58 +cells/1000 cells in normal animals to 362±45 +cells /1000 cells (p<0.01) after 4 weeks of uremia (UC). Treatment with paricalcitol from the onset of uremia, prevented the decrease in VDR expression (755± 52 +cells/1000 cells, p<0.01).
Fig.3.
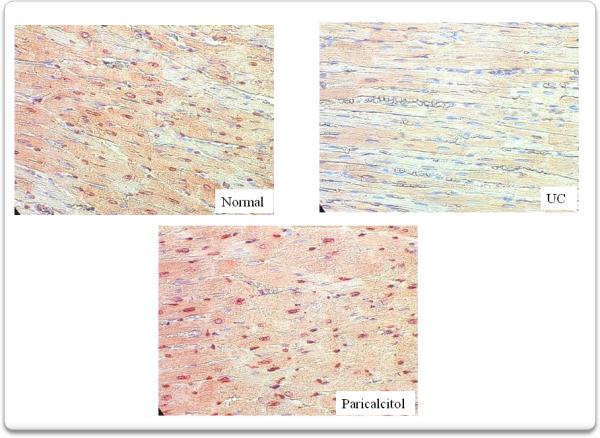
Immunohistochemistry confirmed the decrease in VDR expression in myocardium after 4 weeks of uremia (UC). Treatment with paricalcitol from the onset of uremia, for 4 weeks, prevented this decrease in VDR expression
Fig. 4.
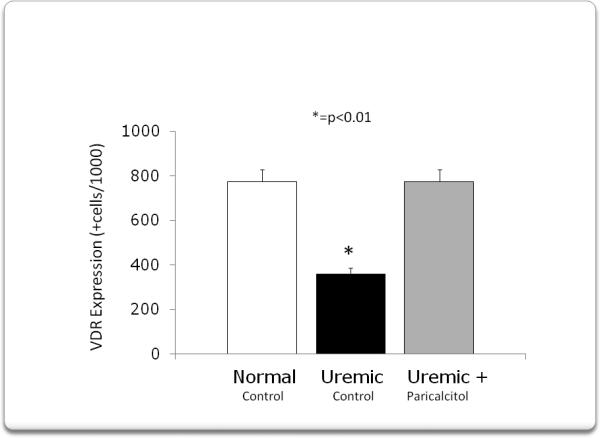
The quantification of the VDR in the myocardium is represented in this figure. VDR expression decreased from 774 ± 58 positive cells/1000 cells in normal animals to 362 ± 45 positive cells/1000 cells(p<0.01) after 4 weeks of uremia (UC). Treatment with paricalcitol from the onset of uremia, prevented the decrease in VDR expression (775 ± 52 positive cells/1000 cells, (p<0.01).
We then assessed PCNA expression in myocardium. Immunohistochemical analysis, figure 5, showed that compared to normal rats, 4 weeks of uremia increased PCNA expression, while treatment with paricalcitol abolished this increase. Figure 6 shows the quantification of PCNA+ cells. PCNA+ cells increased from: 81±21 in normal rats to 299±69 +cells/1000 cells (p<0.01) in uremic control rats. Paricalcitol treatment abolished this increase (82± 6 +cells/1000 cells, p<0.01).
Fig. 5.
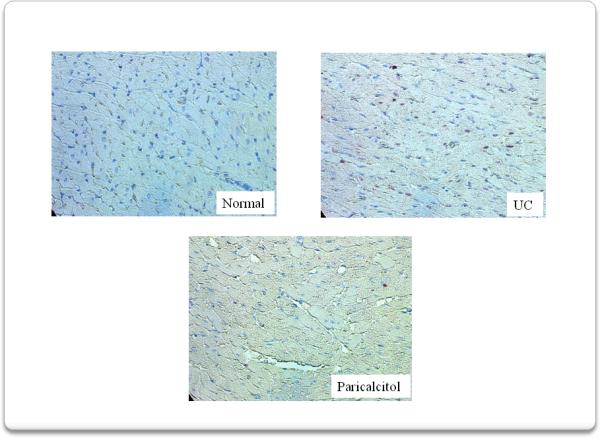
Immunohistochemistry analysis shows that compared to normal rats, 4 weeks of uremia increased PCNA expression, while treatment with paricalcitol abolished this increase.
Fig. 6.
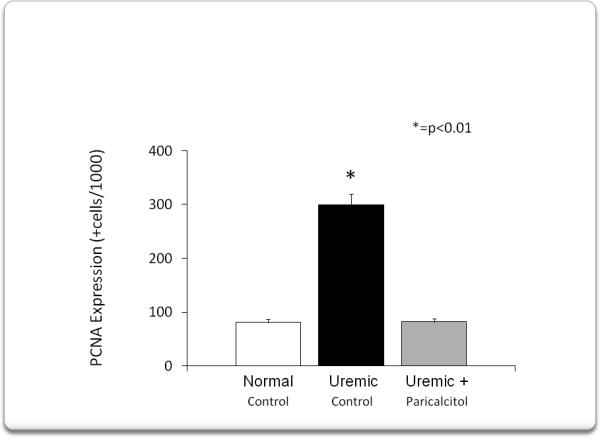
Shows the quantification of PCNA positive cells. PCNA positive cells increased from 81 ± 21 cells/1000 cells in normal rats to 299 ± 69 cells /1000 cells (p<0.01). Paricalcitol treatment abolished this increase (82 ± 6 cells/ 1000 cells, p<0.01).
We then analyzed left ventricular fibrosis (figure 7) and vessel thickness (figure 8) since they represent important pathogenetic mechanisms in the development of uremic cardiomyopathy. Mason trichrome stained sections of myocardium show that compared to normal rats the UC group developed marked interstitial and perivascular fibrosis as well as increased vessel thickness. These abnormalities were greatly ameliorated by paricalcitol.
Fig. 7.
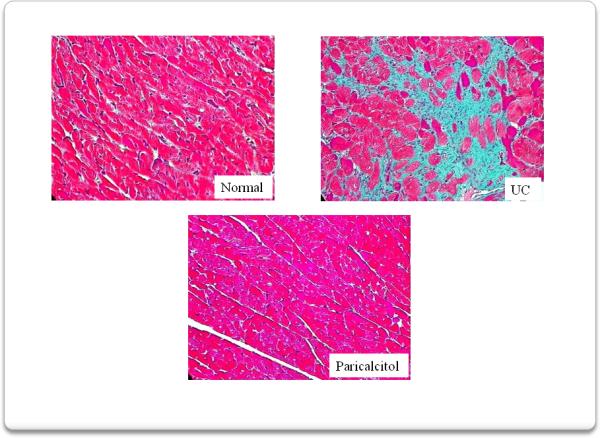
Interstitial fibrosis was assessed using a Masson trichrome stain. Compared to normal rats, 4 weeks of uremia produced marked interstitial fibrosis in the myocardium. Myocardium from rats treated with paricalcitol, from the onset of uremia, displayed virtually no interstitial fibrosis.
Fig.8.
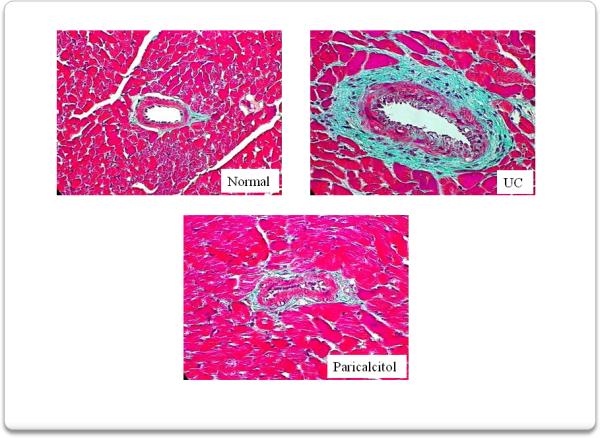
Perivascular fibrosis was also assessed using a Masson trichrome stain. Compared to normal rats, 4 weeks of uremia produced significant areas of perivascular fibrosis well as increased vessel thickness in the myocardium from these rats. Treatment with paricalcitol, from the onset of uremia, markedly reduced the development of perivascular fibrosis and the increase in the vessel thickness.
4. Discussion
The key findings of the present report are: (1) the administration of paricalcitol to uremic rats attenuated the increase in LV weight and prevented the increase in PCNA expression seen in untreated uremic rats. (2) Paricalcitol prevented the decrease in myocardial VDR expression seen in the vehicle-treated uremic rats, and (3) Paricalcitol significantly decreased interstitial and perivascular fibrosis and prevented the increase in vessel thickness seen in myocardium of vehicle-treated uremic rats.
The VDR is expressed in cardiac myocytes (6-7). Thus, the heart is believed to be a vitamin D target organ. Chen et al (8) demonstrated the presence of the 1-hydroxylase and 24-hydroxylase in the myocardium of Wistar rats and the VDR is shown to interact directly with the human B-natriuretic gene promoter, a surrogate marker of the transcriptional response to hypertrophy. Recent studies (9, 10) suggest that vitamin D plays an important role in the regulation of blood pressure. VDR-/- mice have an increase in the RAAS with consequent development of cardiac hypertrophy. Xiang et al (11) demonstrated that VDR-/- mice have an increase in the ratio of heart weight to BW, which is well correlated with an increase in the size of the left ventricular cardiomyocytes and the up regulation of ANP, which can be attenuated by captopril treatment. Thus, VDR-null mice develop ANGII-dependent hypertrophy. The same authors demonstrated that VDR-/- mice have an increase in renin production. Similarly, vitamin D deficient rats also show a marked increase in plasma renin activity. There is evidence that 1,25-(OH)2D3 can suppress renin gene transcription. Work by Yuan et al (12) demonstrated that the cAMP-PKA pathway plays a key role in the stimulation of renin gene expression. This pathway activates cAMP response element (CREB) binding by phosphorylation, leading to the recruitment of the co activator CBP/p300 and its binding to the cAMP-RE. In the presence of 1,25-(OH)2D3, ligand-VDR interacts with CREB and blocks the cAMP-RE, leading to a reduction in renin gene transcription. Several studies have shown that calcitriol and its analogs are also renoprotective in experimental models of primary glomerular sclerosis. In the rat remnant kidney model (5/6th nephrectomy) vitamin D compounds reduce albuminuria, prevent podocyte injury and attenuate glomerulosclerosis. We have recently demonstrated (13) that the combination of enalapril and paricalcitol, a vitamin D receptor activator, greatly reduces glomerulosclerosis, proteinuria and inflammation as assessed by ED-1 and monocyte chemo attractant protein-1 (MCP-1) in uremic rats. The mechanism responsible for the correction of these abnormalities is mediated by suppression of the TGFβ-1 message and protein and of Smad2. Moreover, we also demonstrated a significant reduction in myocardial oxidative stress in these rats (14).
Thus, the up regulation of the myocardial VDR and suppression of PCNA by paricalcitol in uremic rats makes this analog a potential tool for the treatment of cardiomyopathy secondary to chronic renal failure in patients. Further clinical trials are necessary to confirm our preliminary results.
Acknowledgments
This research was support in part by grants from Research in Renal Diseases, Washington University School of Medicine and from Abbott Pharmaceutical. We thank the WUCKDR O'Brien Center (P30DK079333) for technical assistance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Presented at the 14th Workshop on Vitamin D (Bruges, Belgium, 4-8 October 2009)
Disclosures
E.S. is a consultant/speaker for Genzyme Corporation and Abbott Pharmaceutical. E.S. and Washington University may receive income based on a license of related technology by the University of Wisconsin
References
- 1.Teng M, Wolf M, Lowrie E, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446–56. doi: 10.1056/NEJMoa022536. [DOI] [PubMed] [Google Scholar]
- 2.Teng M, Wolf M, Ofsthun MN, et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16:1115–25. doi: 10.1681/ASN.2004070573. [DOI] [PubMed] [Google Scholar]
- 3.Kalantar-Zadeh K, Kuwaen, Regidor D, et al. Survival predictability time-varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int. 2006;70:771–780. doi: 10.1038/sj.ki.5001514. [DOI] [PubMed] [Google Scholar]
- 4.Tentori F, Hunt WC, Stidley CA, et al. Mortality risk among hemodialysis patients receiving different vitamin D analogs. Kidney Int. 2006;70:1858–65. doi: 10.1038/sj.ki.5001868. [DOI] [PubMed] [Google Scholar]
- 5.Dusso A, Brown AJ. Slatopolsky E: Vitamin D. Am J Physiol- Renal Physiol. 2005;289:8028. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 6.Fraga C, Blanco M, Vigo E, et al. Autogenesis of the vitamin D receptor in the rat heart. Histochem Cell Biol. 2002;117:547–550. doi: 10.1007/s00418-002-0413-3. [DOI] [PubMed] [Google Scholar]
- 7.Simpson RU, Thomas GA, Arnold AJ. Identification of 1,25 dihydroxyvitamin D3 receptors and activities in muscle. J Biol Chem. 1985;260:8882–8891. [PubMed] [Google Scholar]
- 8.Cheng S, Glenn DJ, Grigbsby L, et al. Expression of the vitamin D receptor is increased in the hypertropic heart. Hypertension. 2008;52:1160–1112. doi: 10.1161/HYPERTENSIONAHA.108.119602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li YC, Kong J, Wei M, et al. 1,25 Dihydroxyvitamin D3 is a negative endocrine regulator of the rennin-angiotensin system. J Clin Invest. 2002;110:229–238. doi: 10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong J, Li YC. Effect of angiotensin-converting enzyme inhibitor in vitamin D receptor null mice. Am J Physiol, Regul Inter Comp Physiol. 2003;285:R255–R261. doi: 10.1152/ajpregu.00517.2002. [DOI] [PubMed] [Google Scholar]
- 11.Xiang W, Kong J, Chen S, et al. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac rennin-angiotensin systems. Am J Physiol Endocrinol Metab. 2005;288:E125–E132. doi: 10.1152/ajpendo.00224.2004. [DOI] [PubMed] [Google Scholar]
- 12.Yuan W, Pan W, ZHeng V, et al. 1,15 Dihydroxyvitamin D3 suppresses rennin gene transcription by blocking the activity of the cyclic AMP response element in the rennin gene promoter. J Biol Chem. 2007;282:29821–29830. doi: 10.1074/jbc.M705495200. [DOI] [PubMed] [Google Scholar]
- 13.Mizobuchi M, Morrissey J, Finch J, et al. Combination therapy with an angiotensin-converting enzyme inhibitor and vitamin D analog suppress the progression of renal insufficiency in the rat. J Am Soc Nephrol. 2007;18:1796–1806. doi: 10.1681/ASN.2006091028. [DOI] [PubMed] [Google Scholar]
- 14.Husain K, Ferder L, Mizobuchi M, et al. Combination therapy with paricalcitol and enalapril ameliorates cardiac oxidative injury in uremic rats. Am J Nephrol. 2009;29:1–16. doi: 10.1159/000178251. [DOI] [PubMed] [Google Scholar]