

Abstract
We report here the clinical, genetics and molecular characterization of a five-generation Han Chinese family with Leber’s hereditary optic neuropathy (LHON). Strikingly, this family exhibits very high penetrance and occurrence of optic neuropathy. In particular, twenty-five (10 males/15 females) of 30 matrilineal relatives exhibited the variable severity, ranging from profound to mild of visual impairment. This penetrance of optic neuropathy in this Chinese family is much higher than those in many families with LHON worldwide. The age-at-onset for visual impairment in matrilineal relatives in this Chinese family varied from 7 to 24 years old, with the average of 15 years old. Furthermore, the ratio between affected male and female matrilineal relatives is 1:1.5 in the Chinese family. This observation is in contrast with the typical features in LHON pedigrees that there was predominance of affected males in LHON in many families from different ethnic origins. Molecular analysis of mitochondrial genome identified the known ND4 G11778A mutation and 51 variants, belonging to Asian haplogroup C4a1. The absence of other known secondary LHON-associated and functionally significant mtDNA mutations in this Chinese family suggested that mitochondrial variants may not play an important role in the phenotypic manifestation of the G11778A mutation in this Chinese family. Therefore, nuclear modifier gene(s) may be responsible for very high penetrance and occurrence of optic neuropathy in this Chinese pedigree.
INTRODUCTION
Leber’s hereditary optic neuropathy (LHON) is a maternally inherited eye disease that generally affects young adults with the rapid, painless, bilateral loss of central vision [1–3]. Mutations in mitochondrial DNA (mtDNA) are the molecular bases for this disorder [2, 4–6]. Since the landmark discovery of the LHON-associated ND4 G11778A mutation [4], more than 30 LHON-associated mtDNA mutations have been identified among various ethnic populations [7]. Of these, the ND1 G3460A, ND4 G11778A and ND6 T14484C mutations, which involve genes encoding the subunits of respiratory chain complex I, account for more than 95% of LHON pedigrees in some countries [3, 7–10]. Those LHON associated mtDNA mutations, unlike other pathogenic mtDNA mutations such as MELAS-associated tRNALeu(UUR) A3243G mutation present in heteroplasmy (mixture of mutated and wild-type molecules) [11], often occur in the nearly homoplasmy or homoplasmy. Typical features in LHON pedigrees are incomplete penetrance and male bias among the affected subjects, reflecting the complex etiology of this disease [12–14]. Matrilineal relatives within and among families, despite carrying the same LHON-associated mtDNA mutation(s), exhibited a wide range of severity, age-of-onset and penetrance of optic neuropathy. Therefore, other modifier factors including nuclear modifier genes, mitochondrial haplotypes, epigenetic factors and environmental factors should modulate the phenotypic manifestation of visual impairment associated with those primary mtDNA mutations [3, 15].
To further elucidate molecular basis of LHON in the Chinese population, a systematic and extended mutational screening of mtDNA has been initiated in the large clinical population of Ophthalmology Clinic at the Wenzhou Medical College, China [15–20]. In the previous investigation, we showed that the ND4 G11696A, tRNAMet A4435G and tRNAThr A15951G mutations contribute to the high penetrance and expressivity of vision loss in Chinese families [15,18,20]. On other hand, other nine Chinese families carrying the only G11778A mutation displayed extremely low penetrances of LHON [16,17]. In the present study, we performed the clinical, genetic and molecular characterization of another five-generation Han Chinese family with maternally transmitted LHON. Very strikingly, this family exhibited very higher penetrance of visual loss. In particular, 25 (10 males/15 females) of 30 matrilineal relatives in this five-generation family exhibited the variable severity and age-at-onset in visual loss. Mutational analysis of mtDNA demonstrated the presence of the ND4 G11778A mutation in this Chinese family. To elucidate the role of mitochondrial haplotype in the phenotypic manifestation of the G11778A mutation, we performed a PCR-amplification of fragments spanning entire mtDNA and subsequent DNA sequence analysis in the matrilineal relatives of this family.
MATERIALS AND METHODS
Patients and Subjects
We ascertained one Chinese family (Figure 1) through the School of Ophthalmology and Optometry, Wenzhou Medical College, Wenzhou, China. Informed consent, blood samples, and clinical evaluations were obtained from all participating family members, under protocols approved by the Cincinnati Children’s Hospital Medical Center Institute Review Board and the Wenzhou Medical College Ethics Committee. Members of these pedigrees were interviewed at length to identify both personal or family medical histories of visual impairments, and other clinical abnormalities.
Figure 1. One Chinese pedigree with Leber’s hereditary optic neuropathy.

Vision impaired individuals are indicated by filled symbols. Arrow denotes the proband.
Ophthalmological examinations
The ophthalmologic examinations of proband and other members of this family were conducted, including visual acuity, visual field examination (Humphrey Visual Field Analyzer IIi, SITA Standard; Carl Zeiss Meditec, Oberkochen, Germany), visual evoked potentials (VEP; RETI port gamma, flash VEP; Roland Consult, Brandenberg, Germany), and fundus photography (CR6–45NM fundus camera; Canon, Lake Success, NY). The degree of visual impairment was defined according to the visual acuity as follows: normal >0.3, mild =0.3–0.1; moderate<0.1–0.05; severe<0.05–0.02; and profound <0.02.
Mutational analysis of the mitochondrial genome
Genomic DNA was isolated from whole blood of participants using the Puregene DNA Isolation Kits (Gentra Systems). For the examination of the ND4 G11778A mutation, the first PCR segments (803bp) were amplified using genomic DNA as template and oligodeoxynucleotides corresponding to mtDNA at positions 11295–12098 [21]. Then, the second PCR product (212bp) were amplified using the first PCR fragment as template and oligodeoxynucleotides corresponding to mtDNA at positions 11654–11865, and subsequently digested with a restriction enzymes Tsp45I as the G11778A mutation creates the site for this restriction enzyme [16]. Equal amounts of various digested samples were then analyzed by electrophoresis through 7% polyacrylamide gel. The proportions of digested and undigested PCR product were determined by the Image-Quant program after ethidium bromide staining to determine if the G11778A mutation is in the homoplasmy in these subjects. The entire mitochondrial genome of the proband IV-3 was PCR amplified in 24 overlapping fragments using sets of the light (L) strand and the heavy (H) strand oligonucleotide primers as described previously [22]. Each fragment was purified and subsequently analyzed by direct sequencing in an ABI 3700 automated DNA sequencer using the Big Dye Terminator Cycle sequencing reaction kit. These sequence results were compared with the updated consensus Cambridge sequence (GenBank accession number: NC_012920) [21]. DNA and polypeptide sequence alignments were carried out using seqweb program GAP (GCG).
Haplogroup analyses
The entire mtDNA sequence of the Chinese proband carrying the G11778A mutation was assigned to an Asian mitochondrial haplogroup by using the nomenclature of mitochondrial haplogroups [23–24].
RESULTS
Clinical presentation
The proband (IV-3) came to the Ophthalmology clinic of Wenzhou Medical College at the age of 28 year old. He began suffering from painless, progressive deterioration of bilateral visual impairment at the age of 20. His visual impairment occurred within 15 days, first in the right eye and then later in the left eye. He saw a dark cloud in the center of vision and had problems appreciating colors that all seemed a dark gray. The ophthalmological evaluation showed that his visual acuity was 0.02 and 0.04 at his right and left eyes, respectively. A fundus examination, as shown in Figure 2, showed that both his optic disks were abnormal: vascular tortuosity of the central retinal vessels, a circumpapillary telangiectatic microangiopathy, and swelling of the retinal nerve fiber layer. Visual field testing demonstrated large centrocaecal scotomata in both of his eyes. The flash VEP showed bilaterally decreased amplitudes with delayed latencies. Therefore, he exhibited a typical clinical feature of LHON. No other abnormality was found on radiological and neurological examination. Furthermore, he had no other significant medical history.
Figure 2. Fundus photograph of an affected subject (IV-3) and a married-in-control (V-1).
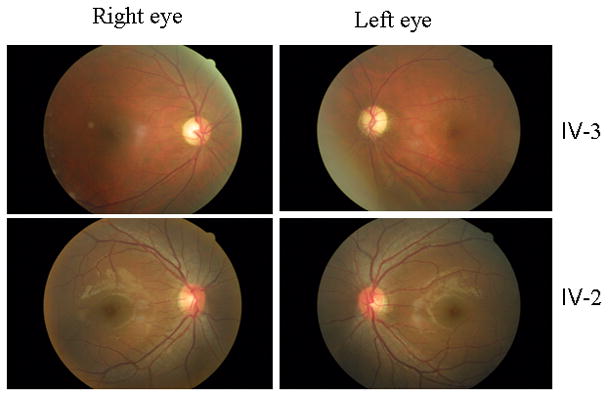
The figures were taken by the fundus photography (Cannon CR6-45NM fundus camera).
The family is originated from Shandong Province in Eastern China, and the majority of family members live in the same area. As shown in Figure 1, this familial history is consistent with a maternal inheritance. None of the offspring of a vision-impaired father has a visual impairment. Of other 29 matrilineal relatives, 9 of 12 male and 15 of 17 female matrilineal relatives exhibited the bilateral and symmetric visual impairment as the sole clinical symptom, whereas three male and two female matrilineal relatives had normal version. These affected matrilineal relatives of this family exhibited early-onset/progressive, but not congenital, visual impairment. As shown in table 1, visual acuity examination showed a variable severity of visual impairment in the maternal kindred, ranging from profound visual impairment (II-1, II-3, III-3, III-5, IV-4), to severe visual impairment (I-2, III-9, IV-3, IV-7, IV-12, IV-15), to moderate visual impairment (II-5, III-1, III-11, III-12, III-15, IV-13), to mild visual impairment (III-7, III-13, IV-1, IV-6, IV-8, IV-9, IV-10, IV-14), to completely normal vision (five matrilineal relatives). In addition, the age at onset of visual impairment in this family varies from 11 years to 24 years old, with the average of 15 years old. There is no evidence that any member of this family had any other known cause to account for visual impairment. Comprehensive family medical histories of these individuals showed no other clinical abnormalities, including diabetes, muscular diseases, hearing impairment and neurological disorders.
Table 1.
Summary of Clinical Data for Some Members in one Chinese Pedigree
| Patient | Gender | Age of test (yrs) | Age of onset (yrs) | Visual acuity right | Visual acuity left | Level of visual impairment |
|---|---|---|---|---|---|---|
| I-2 | F | 35 | 18 | 0.03 | 0.05- | severe- |
| II-1 | F | 75 | 8 | 0.01 | 0.01 | profound |
| II-3 | F | 72 | 7 | 0.01 | 0.01 | profound- |
| II-5 | M | 24 | 23 | 0.07 | 0.1 | moderate- |
| III-1 | F | 53 | 9 | 0.08 | 0.05 | moderate |
| III-3 | F | 51 | 18 | 0.02 | 0.01 | profound |
| III-5 | F | 42 | 24 | 0.01 | 0.01 | profound |
| III-7 | F | 39 | 12 | 0.3 | 0.25 | mild |
| III-9 | M | 14 | 12 | 0.03 | 0.02 | severe- |
| III-10 | M | 45 | - | 1.2 | 0.4 | normal |
| III-11 | M | 25 | 17 | 0.05 | 0.07 | moderate |
| III-12 | M | 20 | 20 | 0.05- | 0.10- | moderate |
| III-13 | F | 19 | 19 | 0.1 | 0.2 | mild |
| III-15 | F | 24 | 22 | 0.08 | 0.09 | moderate |
| IV-1 | F | 31 | 7 | 0.12 | 0.6 | mild |
| IV-3 | M | 28 | 20 | 0.02 | 0.04 | severe |
| IV-4 | M | 30 | 18 | 0.01 | 0.02 | profound |
| IV-6 | M | 27 | 18 | 0.4 | 0.15 | mild |
| IV-7 | F | 20 | 9 | 0.1 | 0.6 | severe |
| IV-8 | F | 19 | 9 | 0.4 | 0.12 | mild |
| IV-9 | F | 18 | 10 | 0.25 | 0.5 | mild |
| IV-10 | M | 14 | 9 | 0.6 | 0.3 | mild |
| IV-11 | M | 17 | - | 0.6 | 0.5 | normal |
| IV-12 | M | 20 | 18 | 0.03 | 0.05 | severe |
| IV-13 | F | 16 | 16 | 0.08 | 0.09 | moderate |
| IV-14 | M | 17 | 17 | 0.1 | 0.2 | mild |
| IV-15 | F | 19 | 19 | 0.05 | 0.03 | severe |
| V-1 | F | 7 | - | 0.8 | 1 | normal |
| V--2 | F | 7 | - | 1.2 | 1.2 | normal |
| V-3 | M | 3 | - | 1.2 | 1.2 | normal |
F= Female; M= Male;
Mitochondrial DNA analysis
The maternal transmission of visual dysfunction in this family suggested the mitochondrial involvement and led us to analyze the mitochondrial genome of matrilineal relatives. First, we examined three commonly known LHON-associated mtDNA mutations (G3460A, G11778A and T14484C) by PCR amplification and subsequent restriction enzyme digestion analysis of PCR fragments derived from four matrilineal relatives (proband IV-3, his mother III-1, unaffected male III-10, affected female III-13) and two unrelated Chinese controls. The results revealed the presence of G11778A mutation, but the absence of the G3460A and T14484C mutations in those subjects.
To further determine the presence and amount of the G11778A mutation in these matrilineal relatives, nested PCR amplification, as detailed in Materials and Methods section, was performed to rule out the possible co-amplification of nuclear pseudogenes [25]. The resultant 212bp PCR segments corresponding to mtDNA at positions 11654–11865 were then digested by another restriction enzyme Tsp45I, and separated them by electrophoresis using a 7% polyacrylamide gel. There was not detectable wild type DNA, indicating that the G11778A mutation appears to be homoplasmy in these matrilineal relatives of this Chinese family (data not shown). As expected, the G11778A mutation was absent in PCR products derived from married-in controls in this family. This strongly indicated that the levels of G11778A mutation in those matrilineal relatives did not correlate with the variability of visual dysfunction including the severity and age-of-onset of visual impairment.
To determine the role of mitochondrial haplotypes in the phenotypic manifestation of the G11778A mutation, the DNA fragments spanning the entire mtDNA of an affected patient IV-3 were PCR amplified. Each fragment was purified and subsequently analyzed by direct sequence. The comparison of the resultant sequences with the Cambridge consensus sequence [21] identified a number of nucleotide changes as shown in Table 2. All of those nucleotide changes were verified in 3 additional matrilineal relatives of this family his mother III-1, unaffected male III-10, affected female III-13) by sequence analysis and appeared to be homoplasmy. Sequence analysis confirmed the presence of the G11778A mutation in matrilineal relatives of this family. In addition to the identical G11778A mutation, as shown in Table 2, these subjects exhibited a set of mtDNA polymorphisms. Of other nucleotide changes, there are 12 known variants in the D-loop, 2 known variants in 12S rRNA gene, 2 known variants in the 16S rRNA gene, the known T15968C variant in the tRNAPro gene [26], 24 known silent variants in the protein encoding genes as well as 10 known missense mutations in the protein encoding genes [7]. These missense mutations are the G8584A (A20T), A8701G (T59A) and A8860G (T112A) in the A6 gene, the A10398G (T114A) in the ND3 gene, the G11969A (A406T) in the ND4 gene, and the T14318C (N119S) in the ND6 gene and the C14766T (T7I), T15204C (I153T), G15323A (A193T) and A15326G (T194A) in the Cytb gene. These variants in RNAs and polypeptides were further evaluated by phylogenetic analysis of these variants and sequences from other organisms including mouse [27], bovine [28], and Xenopus laevis[29]. However, none of those variants showed evolutionary conservation. Based on the nomenclature of mitochondrial haplogroups [23,24], we used the mtDNA sequence variations in this Chinese proband to establish the haplogroup affiliation of each mtDNA. Here, mtDNAs of this pedigree belong to the Eastern Asian haplogroup C4a1.
TABLE 2.
mtDNA Variants in the Chinese Family with Leber Hereditary Optic Neuropathy.
| Gene | Position | Replacement | Conservation (H/B/M/X)* | Previously reported† |
|---|---|---|---|---|
| D-loop | 73 | A to G | Yes | |
| 195 | T to C | Yes | ||
| 249 | A to Del | Yes | ||
| 263 | A to G | Yes | ||
| 310 | T to TC | Yes | ||
| 489 | T to C | Yes | ||
| 522 | C to Del | Yes | ||
| 523 | A to Del | Yes | ||
| 16093 | T to C | Yes | ||
| 16129 | G to A | Yes | ||
| 16189 | T to C | Yes | ||
| 16519 | T to C | Yes | ||
| 12S rRNA | 750 | A to G | A/A/A/- | Yes |
| 1438 | A to G | A/A/A/G | Yes | |
| 16S rRNA | 1715 | C to T | C/A/A/T | Yes |
| 2706 | A to G | A/G/A/A | Yes | |
| ND1 | 3552 | T to A | Yes | |
| ND2 | 4715 | A to G | Yes | |
| 4769 | A to G | Yes | ||
| CO1 | 6026 | G to A | Yes | |
| 7001 | A to G | Yes | ||
| 7028 | C to T | Yes | ||
| 7196 | C to A | Yes | ||
| CO2 | 7999 | T to C | Yes | |
| ATP6 | 8584 | G to A (Ala to Thr) | A/V/V/I | Yes |
| 8701 | A to G (Thr to Ala) | T/S/L/Q | Yes | |
| 8860 | A to G (Thr to2 Ala) | T/A/A/T | Yes | |
| CO3 | 9540 | T to C | Yes | |
| 9545 | A to G | Yes | ||
| ND3 | 10398 | A to G (Thr to Ala) | T/T/T/A | Yes |
| 10400 | C to T | Yes | ||
| ND4L | 10688 | G to A | Yes | |
| ND4 | 10873 | T to C | Yes | |
| 11719 | G to A | Yes | ||
| 11778 | G to A (Arg to His) | R/R/R/R | Yes | |
| 11914 | G to A | Yes | ||
| 11969 | G to A (Ala to Thr) | A/A/G/A | Yes | |
| ND5 | 12672 | A to G | Yes | |
| 12705 | C to T | Yes | ||
| 13263 | A to G | Yes | ||
| ND6 | 14318 | T to C (Asn to Ser) | N/N/D/S | Yes |
| Cytb | 14766 | C to T (Thr to Ile) | T/S/T/S | Yes |
| 14783 | T to C | Yes | ||
| 15043 | G to A | Yes | ||
| 15109 | T to C | Yes | ||
| 15139 | T to C | Yes | ||
| 15204 | T to C (Ile to Thr) | I/I/I/K | Yes | |
| 15301 | G to A | Yes | ||
| 15323 | G to A (Ala to Thr) | A/A/A/S | Yes | |
| 15326 | A to G (Thr to Ala) | T/M/I/I | Yes | |
| 15487 | A to T | Yes | ||
| tRNAPro | 15968 | T to C | T/T/A/G | Yes |
Conservation of amino acid for polypeptides or nucleotide for RNAs in human (H), bovine (B), mouse (M) and Xenopus laevis (X).
See the online mitochondrial genome database http://www.mitomap.org and http://www.genpat.uu.se/mtDB
DISCUSSION
In the present study, we have performed the clinical, genetic, and molecular characterization of a five-generation Han Chinese family with Leber’s hereditary optic neuropathy. Visual impairment as a sole clinical phenotype was only present in the maternal lineage of this pedigree, suggesting that the mtDNA mutation(s) is the molecular basis for this disorder. In fact, the molecular analysis identified the homoplasmic G11778A mutation in the ND4 gene in matrilineal relatives of this Chinese family. Clinical and genetic evaluations revealed the variable severity and age-of-onset in visual impairment in these matrilineal relatives, although these subjects share some common features: being the rapid, painless, bilateral loss of central vision. Strikingly, this Chinese family exhibited very high penetrance and occurrence of visual impairment. In particular, 25 (10 males/15 females) of 30 matrilineal relatives in this family exhibited the variable severity of visual impairment. However, as shown in Table 3, the penetrances of visual impairment in other 11 Chinese pedigrees carrying only the G11778A mutation ranged from 5.3% to 60%, with the average of 19.2% [16,17,19], and those in another five Chinese families carrying one of functionally significant mtDNA variants varied from 33% to 65.6%, respectively, with the average of 45% [15,18,19,20,30]. However, ~50% males and ~10% females in several cohorts of Caucasians population carrying one of LHON associated G3460A, G11778A and T14484C mutations indeed developed the optic neuropathy [2,31,32], whereas the penetrance of optical atrophy was 27% in males and 8% in females among 36 LHON families in Finland [33]. Therefore, the penetrance of visual loss in this Chinese family is much higher than those in other families carrying the G11778A mutation. Interestingly, the average-age-at-onset for visual impairment in matrilineal relatives in this Chinese family was 15 years old. On the other hand, as shown in Table 3, the average-age-at-onset of visual impairment in other 11 Chinese pedigrees carrying only the G11778A mutation ranged from 14 to 27 years old, with the average of 18 year olds [16,17,19], and another five Chinese families carrying one of functionally significant mtDNA variants varied from 14 to 18, with the average of 17 years old [15,18,19,20,30], while the average of age-of-onset for visual loss were 24 and 28 years old from matrilineal relatives of 66 and 49 Caucasian pedigrees carrying the G11778A mutation, respectively [31,32,34]. Thus, the average age-of-onset of visual impairment among matrilineal relatives in this Chinese family was younger than other families carrying the G11778A mutation. Furthermore, the ratio between affected male and female matrilineal relatives is 1:1.5 in the Chinese family in the present study, while the ratios varied from 1:1 to 6:0 in other 16 Chinese families carrying the G11778A mutation [15–20]. However, this ratio was 4.5:1 and 3.7:1 from two large cohorts of Caucasian pedigrees carrying the G11778A mutation, respectively [31,32,33]. Hence, the ratio between affected male and female matrilineal relatives in this family was in contrast with typical features in LHON that there was predominance of affected males in LHON in many families carrying the G11778A mutation [3,33,34].
Table 3.
Summary of Clinical and Molecular Data for 17 Chinese Families Carrying the G11778A Mutation.
| Pedigree | Ratio (Affected Male/Female) | Average Age of Onset (y) | Matrilineal Relatives (n) | Penetrance (%)* | Secondary mtDNA mutations | mtDNA Haplogroup |
|---|---|---|---|---|---|---|
| WZ419 | 1:1.5 | 15 | 30 | 83.3 | C4a1 | |
| WZ116 | 6:0 | 22 | 38 | 15.8 | B5 | |
| WZ215 | 3:1 | 14 | 14 | 57.1 | A4435G | D5 |
| WZ320 | 3:0 | 17 | 9 | 33.3 | A15951G | F1 |
| WZ419 | 2:1 | 19 | 10 | 60.0 | D4 | |
| WZ5 | 1:1 | 20 | 14 | 35.7 | D4b2b | |
| WZ6 | 1.2:1 | 18 | 31 | 35.5 | T14502C | M10a |
| WZ1218 | 3.5:1 | 18 | 30 | 33.0 | G11696A | D4 |
| WZ3130 | 1.4:1 | 18 | 29 | 65.6 | 6480/12811/15935 | M7b |
| WZ4117 | 1:0 | 18 | 7 | 14.3 | M8a2 | |
| WZ42 | 2:0 | 14.5 | 25 | 8.0 | D4g2 | |
| WZ43 | 1:0 | 15 | 9 | 11.1 | B4a1c | |
| WZ44 | 1:0 | 15 | 17 | 5.9 | B5b | |
| WZ45 | 2:0 | 16 | 12 | 16.7 | N9a1 | |
| WZ46 | 2:1 | 14 | 19 | 15.8 | D4b2b | |
| WZ47 | 2:1 | 27 | 11 | 27.3 | C | |
| WZ48 | 1:0 | 16 | 19 | 5.3 | M7b1 |
Affected matrilineal relatives/total matrilineal relatives.
Our previous investigations revealed that nine pedigrees carrying the G11778A mutation exhibited extremely low penetrance of visual loss, indicating that the G11778A mutation, similar to ND1 G3460A and ND6 T14484C mutations [35, 36], was necessary but itself insufficient to produced a clinical phenotype [16, 17]. The extremely high penetrance and occurrence of optic neuropathy in this Chinese family indicated the involvement of other environmental and genetic factors including nuclear modifier genes and mitochondrial haplotypes in the phenotypic manifestation of LHON–associated G11778A mutation. The mitochondrial variants/haplotypes were implicated to influence the penetrance and expressivity of visual impairment associated with the primary LHON-associated mtDNA mutations. In particular, secondary LHON mutations at ND1 T4216C and ND5 G13708A may increase the penetrance and expressivity of LHON associated with the primary LHON mutations including G11778A and T14484C [9,37–40]. Furthermore, the G7444A mutation in the CO1 and tRNASer(UCN) genes has also been implicated to influence the penetrance and phenotypic expression of visual loss associated with the primary LHON mutations [9]. In addition, the ND4 G11696A, tRNAMet A4435G and tRNAThr A15951G mutations may increase the penetrance and expressivity of vision loss in other Chinese families [15,18,20]. In this Chinese family carrying the G11778A mutation, none of other mtDNA variants belonging to the halpogroup C4a1 showed evolutionary conservation and was implicated to have functional significance. These suggest that these mtDNA haplogroup-specific variants, unlike other mtDNA variants [15,18,20,41], may not play an important role in the phenotypic expression of the G11778A mutation in this Chinese family. Furthermore, it is possible that environmental factors may also contribute to the phenotypic variability of visual impairment in matrilineal relatives of these families.
The phenotypic variability of matrilineal relatives within this family including the variable severity of vision impairment suggests an important role of nuclear modifier genes in the phenotypic manifestation of the G11778A mutation. In fact, the predominance of affected males in LHON in other families carrying the G11778A mutation suggests the existence of a recessive X-linked susceptibility gene acting in synergy with the mtDNA mutation to precipitate the optic neuropathy [42,43]. In particular, the development of blindness in males is consistent with the simultaneous inheritance of an X-linked visual loss allele and the primary LHON-associated mtDNA mutation [42]. An X-chromosomal gene closely linked to DXS7 region was proposed to be a nuclear modifier for the phenotypic expression of LHON-associated G11778A mutation [44]. Most recently, two overlapping disease loci with highly significant LOD scores at Xp21–Xq21 [45] and Xq25–27.2 [46] were identified using a larger number of more extensively defined LHON pedigrees. However, the genetic etiology of LHON might be more complex, with epistatic interaction of these multiple nuclear susceptibility loci and genetic heterogeneity. Here, the fact that extremely high penetrance of visual loss and ratio between affected male and female matrilineal relatives is 1:1.5 in this Chinese family strongly suggested the involvement of autosomal recessive or X-linked modifier genes in the phenotypic manifestation in this Chinese family.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants RO1DC05230 and RO1DC07696 from the National Institute on Deafness and Other Communication Disorders to M.X.G., a Chinese Young Scholar Award (30628013) from National Science Foundation of China and a key research grant Z204492 from Zhejiang Provincial Natural Science Foundation of China to M.X.G., a project grant ZB0202 from Zhejiang Provincial Natural Science Foundation of China and a Key Research and Development Program project grant 2004C14005 from Zhejiang Province, China to J.Q.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Newman NJ. Leber’s hereditary optic neuropathy. Ophthalmol Clin NA. 1993;4:431–447. [Google Scholar]
- 2.Brown MD, Wallace DC. Spectrum of mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Clin Neurosci. 1994;2:134–145. [Google Scholar]
- 3.Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J Med Genet. 2009;46:145–158. doi: 10.1136/jmg.2007.054270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 5.Howell N. LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol. 2003;37:94–108. doi: 10.1159/000072041. [DOI] [PubMed] [Google Scholar]
- 6.Servidei S. Mitochondrial encephalomyopathies: gene mutation. Neuromuscul Disord. 2004;14:107–116. doi: 10.1016/s0960-8966(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 7.Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC. MITOMAP:a human mitochondrial genome database--2004 update. Nucleic Acids Res. 2005;33:D611–613. doi: 10.1093/nar/gki079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackey DA, Oostra RJ, Rosenberg T, Nikoskelainen E, Bronte-Stewart J, Poulton J, Harding AE, Govan G, Bolhuis PA, Norby S, Bleeker-Wagemakers EM, Savontaus ML, Cahn C, Howell N. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet. 1996;59:481–485. [PMC free article] [PubMed] [Google Scholar]
- 9.Mashima Y, Yamada K, Wakakura M, Kigasawa K, Kudoh J, Shimizu N, Oguchi Y. Spectrum of pathogenic mitochondrial DNA mutations and clinical features in Japanese families with Leber’s hereditary optic neuropathy. Curr Eye Res. 1998;17:403–408. doi: 10.1080/02713689808951221. [DOI] [PubMed] [Google Scholar]
- 9.Brown MD, Torroni A, Reckord CL, Wallace DC. Phylogenetic analysis of Leber’s hereditary optic neuropathy mitochondrial DNA’s indicates multiple independent occurrences of the common mutations. Hum Mut. 1995;6:311–325. doi: 10.1002/humu.1380060405. [DOI] [PubMed] [Google Scholar]
- 10.Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787:518–528. doi: 10.1016/j.bbabio.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 11.Goto Y, Noaka L, Horai S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;148:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 12.Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol. 1991;111:750–762. doi: 10.1016/s0002-9394(14)76784-4. [DOI] [PubMed] [Google Scholar]
- 13.Nikoskelainen EK. Clinical pictures of LHON. Clin Neurosci. 1994;2:115–120. [Google Scholar]
- 14.Riordan-Eva P, Sanders MD, Govan GG, Sweeney MG, Da Costa J, Harding AE. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118:319–337. doi: 10.1093/brain/118.2.319. [DOI] [PubMed] [Google Scholar]
- 15.Qu J, Li R, Zhou X, Tong Y, Lu F, Qian Y, Hu Y, Mo JQ, West CE, Guan MX. The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Invest Ophth Vis Sci. 2006;47:475–483. doi: 10.1167/iovs.05-0665. [DOI] [PubMed] [Google Scholar]
- 16.Qu J, Li R, Tong Y, Hu Y, Zhou X, Qian Y, Lu F, Guan MX. Only male matrilineal relatives with Leber’s hereditary optic neuropathy in a large Chinese family carrying the mitochondrial DNA G11778A mutation. Biochem Biophys Res Commun. 2005;328:1139–1145. doi: 10.1016/j.bbrc.2005.01.062. [DOI] [PubMed] [Google Scholar]
- 17.Qu J, Zhou X, Zhang J, Zhao F, Sun YH, Tong Y, Wei QP, Cai W, Yang L, West CE, Guan MX. Extremely low penetrance of Leber’s hereditary optic neuropathy in eight Han Chinese families carrying the ND4 G11778A mutation. Ophthalmology. 2009;116:558–564. doi: 10.1016/j.ophtha.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qu J, Li R, Zhou X, Tong Y, Yang L, Chen J, Zhao F, Qian Y, Lu F, West CE, Guan MX. Cosegregation of the ND4 G11696A mutation with the LHON-associated ND4 G11778A mutation in a four generation Chinese family. Mitochondrion. 2007;7:140–146. doi: 10.1016/j.mito.2006.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian Y, Zhou X, Hu Y, Tong Y, Li R, Lu F, Yang H, Mo JQ, Qu J, Guan MX. Clinical evaluation and mitochondrial DNA sequence analysis in three Chinese families with Leber’s hereditary optic neuropathy. Biochem Biophys Res Commun. 2005;332:614–621. doi: 10.1016/j.bbrc.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Li R, Qu J, Zhou X, Tong Y, Lu F, Qian Y, Hu Y, Mo JQ, West CE, Guan MX. The mitochondrial tRNAThr A15951G mutation may influence the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Gene. 2006;376:79–86. doi: 10.1016/j.gene.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Andrews RM, Kubacka I, Chinerry PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 22.Rieder MJ, Taylor SL, Tobe VO, Nickerson DA. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1981;26:967–973. doi: 10.1093/nar/26.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka M, Cabrera VM, Gonzalez AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M, Shinoda K, Umetsu K, Yamada Y, Oshida Y, Sato Y, Hattori N, Mizuno Y, Arai Y, Hirose N, Ohta S, Ogawa O, Tanaka Y, Kawamori R, Shamoto-Nagai M, Maruyama W, Shimokata H, Suzuki R, Shimodaira H. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 2004:1832–1850. doi: 10.1101/gr.2286304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong QP, Bandelt HJ, Sun C, Yao YG, Salas A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, Torroni A, Zhang YP. Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Hum Mol Genet. 2006;15:2076–2086. doi: 10.1093/hmg/ddl130. [DOI] [PubMed] [Google Scholar]
- 25.Woischnik M, Moraes CT. Pattern of organization of human mitochondrial pseudogenes in the nuclear genome. Genome Res. 2002;12:885–893. doi: 10.1101/gr.227202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan H, Chen J, Liu X, Cheng XJ, Wang X, Yang L, Yang S, Cao J, Kang D, Dai P, Zhai S, Han D, Young WY, Guan MX. Coexistence of mitochondrial 12S rRNA C1494T and CO1/tRNASer(UCN) G7444A mutations in two Han Chinese pedigrees with aminoglycoside-induced and non-syndromic hearing loss. Biochem Biophys Res Commun. 2007;362:94–100. doi: 10.1016/j.bbrc.2007.07.161. [DOI] [PubMed] [Google Scholar]
- 27.Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 28.Gadaleta G, Pepe G, De Candia G, Quagliariello C, Sbisa E, Saccone C. The complete nucleotide sequence of the Rattus norvegicus mitochondrial genome: cryptic signals revealed by comparative analysis between vertebrates. J Mol Evol. 1989;28:497–516. doi: 10.1007/BF02602930. [DOI] [PubMed] [Google Scholar]
- 29.Roe A, Ma DP, Wilson RK, Wong JF. The complete nucleotide sequence of the Xenopus laevis mitochondrial genome. J Biol Chem. 1985;260:9759–9774. [PubMed] [Google Scholar]
- 30.Cai W, Fu Q, Zhou X, Qu J, Tong Y, Guan MX. Mitochondrial variants may influence the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated ND4 G11778A mutation. J Genet Genomics. 2008;35:649–655. doi: 10.1016/S1673-8527(08)60086-7. [DOI] [PubMed] [Google Scholar]
- 31.Harding AE, Sweeney MG, Govan GG, Riordan-Eva P. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet. 1995;57:77–86. [PMC free article] [PubMed] [Google Scholar]
- 32.Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol. 1991;111:750–762. doi: 10.1016/s0002-9394(14)76784-4. [DOI] [PubMed] [Google Scholar]
- 33.Puomila A, Hamalainen P, Kivioja S, Savontaus ML, Koivumäki S, Huoponen K, Nikoskelainen E. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 2007;15:1079–1089. doi: 10.1038/sj.ejhg.5201828. [DOI] [PubMed] [Google Scholar]
- 34.Nikoskelainen NK. Clinical picture of LHON. Clin Neurosci. 1994;2:115–120. [Google Scholar]
- 35.Qu J, Zhou X, Zhao F, Liu X, Zhang M, Sun YH, Liang M, Yuan M, Liu Q, Tong Y, Wei QP, Yang L, Guan MX. Low penetrance of Leber’s hereditary optic neuropathy in ten Han Chinese families carrying the ND6 T11484C mutation. Biochim Biophys Acta. 2010;1800:304–311. doi: 10.1016/j.bbagen.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong Y, Sun YH, Zhou X, Zhao F, Mao Y, Wei QP, Yang L, Qu J, Guan MX. Very low penetrance of Leber’s hereditary optic neuropathy in five Han Chinese families carrying the ND1 G3460A mutation. Mol Genet Metab. 2010;99:417–424. doi: 10.1016/j.ymgme.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howell N, Kubacka I, Xu M, McCullough DA. Leber hereditary optic neuropathy: involvement of the mitochondrial ND1 gene and evidence for an intragenic suppressor mutation. Am J Hum Genet. 1991;48:935–942. [PMC free article] [PubMed] [Google Scholar]
- 38.Johns DR, Berman J. Alternative, simultaneous Complex I mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Biochem Biophys Res Commun. 1991;174:1324–1330. doi: 10.1016/0006-291x(91)91567-v. [DOI] [PubMed] [Google Scholar]
- 39.Torroni A, Petrozzi M, D’Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, De Negri A, Scozzari R. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet. 1997;60:1107–1121. [PMC free article] [PubMed] [Google Scholar]
- 40.Brown MD, Starikovskaya E, Derbeneva O, Hosseini S, Allen JC, Mikhailovskaya IE, Sukernik RI, Wallace DC. The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup. J Hum Genet. 2002;110:130–138. doi: 10.1007/s00439-001-0660-8. [DOI] [PubMed] [Google Scholar]
- 41.Hudson G, Carelli V, Spruijt L, Gerards M, Mowbray C, Achilli A, Pyle A, Elson J, Howell N, La Morgia C, Valentino ML, Huoponen K, Savontaus ML, Nikoskelainen E, Sadun AA, Salomao SR, Belfort R, Jr, Griffiths P, Man PY, de Coo RF, Horvath R, Zeviani M, Smeets HJ, Torroni A, Chinnery PF. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet. 2007:228–233. doi: 10.1086/519394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bu XD, Rotter JI. X chromosome-linked and mitochondrial gene control of Leber hereditary optic neuropathy: evidence from segregation analysis for dependence on X chromosome inactivation. Proc Natl Acad Sci USA. 1991;88:8198–8202. doi: 10.1073/pnas.88.18.8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura M, Fujiwara Y, Yamamoto M. The two locus control of Leber hereditary optic neuropathy and a high penetrance in Japanese pedigrees. Hum Genet. 1993;91:339–341. doi: 10.1007/BF00217353. [DOI] [PubMed] [Google Scholar]
- 44.Shankar SP, Fingert JH, Carelli V, Valentino ML, King TM, Daiger SP, Saloma SR, Berezovsky A, Belfort R, Jr, Braun TA, Sheffield VC, Sadun AA, Stone EM. Evidence for a novel x-linked modifier locus for leber hereditary optic neuropathy. Ophthalmic Genet. 2008;29:17–24. doi: 10.1080/13816810701867607. [DOI] [PubMed] [Google Scholar]
- 45.Hudson G, Carelli V, Horvath R, Zeviani RM, Smeets HJ, Chinnery PF. X-Inactivation patterns in females harboring mtDNA mutations that cause Leber hereditary optic neuropathy. Mol Vis. 2007;13:2339–2343. [PubMed] [Google Scholar]
- 46.Hudson G, Keers S, Yu Wai Man P, Griffiths P, Huoponen K, Savontaus ML, Nikoskelainen E, Zeviani M, Carrara F, Horvath R, Karcagi R, Spruijt L, de Coo IF, Smeets HJ, Chinnery PF. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am J Hum Genet. 2005;77:1086–1091. doi: 10.1086/498176. [DOI] [PMC free article] [PubMed] [Google Scholar]