

Summary
Tissue damage predisposes humans to life-threatening disseminating infection by the opportunistic pathogen Pseudomonas aeruginosa. Bacterial adherence to host tissue is a critical first step in this infection process. It is well established that P. aeruginosa attachment to host cells involves type IV pili (TFP), which are retractile surface fibers. The molecular details of attachment and the identity of the bacterial adhesin and host receptor remain controversial. Using a mucosal epithelium model system derived from primary human tissue, we show that the pilus-associated protein PilY1 is required for bacterial adherence. We establish that P. aeruginosa preferentially binds to exposed basolateral host cell surfaces, providing a mechanistic explanation for opportunistic infection of damaged tissue. Further, we demonstrate that invasion and fulminant infection of intact host tissue requires the coordinated and mutually dependent action of multiple bacterial factors, including pilus fiber retraction and the host cell intoxication system, termed type III secretion. Our findings offer new and important insights into the complex interactions between a pathogen and its human host and provide compelling evidence that PilY1 serves as the principal P. aeruginosa adhesin for human tissue and that it specifically recognizes a host receptor localized or enriched on basolateral epithelial cell surfaces.
Introduction
Pseudomonas aeruginosa, an environmental bacterium, is a major source of fatal nosocomial infections and the primary cause of morbidity and mortality in patients with cystic fibrosis (CF). Surgery, severe wounds, burns and corneal abrasion predispose otherwise healthy individuals to disseminating tissue infection (Garau et al., 2003, Driscoll et al., 2007). The ability of P. aeruginosa to cause infection requires multiple virulence factors including type IV pili (TFP) and a contact-dependent type III secretion system (T3SS) (Sadikot et al., 2005). TFP are filamentous surface appendages expressed by a wide variety of human, animal and plant pathogens (Pelicic, 2008). In P. aeruginosa, TFP are specifically involved in adherence to host tissue, virulence and the formation of biofilms (Chi et al., 1991, Tang et al., 1995, O'Toole et al., 1998). In addition, P. aeruginosa TFP mediate a form of surface-associated bacterial movement known as twitching motility through extension and retraction of pilus fibers (Skerker et al., 2001).
The assembly and function of TFP in P. aeruginosa involves more than 40 gene products (Mattick, 2002). TFP are polymers composed primarily of a single repeating subunit termed pilin, which is encoded by the pilA gene (Strom et al., 1986). Pilin contains an extended, hydrophobic N-terminal α-helical region followed by a globular C-terminal domain terminating in a disulfide-bonded loop (Hazes et al., 2000, Craig et al., 2003). Pilin subunits are predicted to assemble such that the hydrophobic N-terminal α-helical regions form the core of the pilus, and the globular C-terminal domains are exposed on the exterior surface of the fiber (Craig et al., 2003). The pilus is assembled on the periplasmic surface of the inner membrane and extruded across the outer membrane via a pore complex composed of the secretin protein PilQ (Wolfgang et al., 2000, Collins et al., 2001). Although pilin is the major structural component of the pilus, other proteins are associated with the fiber and may play structural or functional roles. In Neisseria gonorrhoeae, six “pilin-like” proteins, sharing the highly conserved N-terminal α-helical region of pilin (PilH, PilI, PilJ, PilK, PilL, and PilV), are associated with the TFP fraction (Winther-Larsen et al., 2005). PilX, a N. meningitidis homolog of N. gonorrhoeae PilL, is tightly associated with pilus fibers and is proposed to be a minor TFP subunit (Helaine et al., 2007). An orthologous set of pilin-like proteins (FimU, PilV, PilW, PilX, PilE) is required for TFP biogenesis in P. aeruginosa (Alm et al., 1997); however, their association with the pilus fiber and specific function remain to be determined.
Extension and retraction of TFP involves two cytoplasmic membrane-associated ATPases, PilB and PilT. PilB is required for the assembly of pilin monomers into mature fibers (Turner et al., 1993, Chiang et al., 2008), while PilT is involved in the disassembly of polymerized pilin and subsequent retraction of TFP (Whitchurch et al., 1991, Chiang et al., 2008). Loss-of-function mutations in pilT results in hyperpiliation and loss of twitching motility due to the inability of formed pilus fibers to retract (Whitchurch et al., 1991). Inactivation of pilT was reported to result in the loss of cytotoxicity in vitro, presumably due to the inability of the TFP to retract and facilitate intimate contact between the bacteria and host cells, a process required for activation of the P. aeruginosa T3SS and subsequent delivery of anti-host effector proteins (Comolli et al., 1999a, Vallis et al., 1999, Sundin et al., 2002). Mutants lacking pilT are capable of establishing pulmonary infection in mice, but are unable to spread to peripheral organs, presumably due to their inability to undergo twitching motility and disseminate (Comolli et al., 1999a). Similarly, it has been shown that pilT mutants are unable to invade damaged corneal tissue in vivo (Zolfaghar et al., 2003).
In addition to their function in twitching motility, TFP also mediate adherence of P. aeruginosa to eukaryotic cells. While the molecular basis for this interaction has been the subject of many studies, the actual mechanisms underlying TFP-mediated P. aeruginosa adhesion remain controversial. Previous studies suggest that P. aeruginosa pilin may be directly involved in bacterial adherence to host cells. Specifically, purified P. aeruginosa pili bound to the GalNAcβ1-4Gal moiety of the non-sialylated glycosphingolipids asialo-GM1 and asialo-GM2, which are abundant on the apical surface of mammalian epithelial cells (Lee et al., 1994, Sheth et al., 1994). Fab fragments generated from monoclonal antibodies specific for the exposed C-terminal disulfide-bonded loop (DSL) region of pilin and a synthetic peptide corresponding to the C-terminal DSL, were reported to inhibit adhesion of purified P. aeruginosa TFP to human buccal epithelial cells and the disaccharide GalNAcβ1-4Gal, respectively (Doig et al., 1990, Sheth et al., 1994, Schweizer et al., 1998). Structural modeling suggested that the adhesive moiety of TFP (responsible for asialylated glycosphingolipid binding) is likely only exposed at the distal tip of purified TFP fibers (Lee et al., 1994). Based on these studies, it is widely assumed that the exposed C-terminal DSL of pilin serves as the primary P. aeruginosa adhesin for human tissue. However, more recent results have raised questions as to the role of pilin in TFP-mediated adherence as well as the identity of the pilus receptor. Specifically, Emam et al. (2006) showed that even though purified pili bound to asialo-GM1 or asialo-GM2, piliated P. aeruginosa failed to recognize the same receptor molecules. Second, it has been established that P. aeruginosa preferentially binds basolateral surfaces of epithelial cell monolayers or damaged areas of the monolayer where basolateral surfaces are exposed (Fleiszig et al., 1997, Lee et al., 1999). The fact that asialo-GM1 is predominantly localized to the apical compartment of polarized airway epithelial cells (Soong et al., 2004) suggests that asialo-GM1 may not be the primary P. aeruginosa host cell receptor. Finally, we recently showed that adherence of N. gonorrhoeae, expressing TFP composed of P. aeruginosa pilin, to primary human epithelial cells was dependent on the gonococcal TFP-associated adhesin, indicating that P. aeruginosa pilin is not sufficient for host cell adherence (Winther-Larsen et al., 2007).
Although the molecular interactions involved in P. aeruginosa adherence remain to be more clearly defined, the mechanism of TFP-mediated adhesion has been characterized in other Gram-negative bacteria. In N. gonorrhoeae, a minor TFP-associated protein PilC has been identified as the adhesin due to its ability to bind eukaryotic cells and competitively block the adherence of piliated gonococci (Rudel et al., 1995b). Association of PilC with gonoccocal TFP requires the pilin-like proteins PilH, PilI, PilJ, PilK, PilL, and PilV (Winther-Larsen et al., 2005). PilC was initially reported to be essential for TFP biogenesis (Rudel et al., 1995a, Morand et al., 2004), but work by Wolfgang et al. (1998) demonstrated that deletion of the pilus retraction gene pilT could suppress the TFP biogenesis defect of a pilC mutant. Additionally, pilin-like proteins in both N. gonorrhoeae and N. meningitidis are required for TFP production, but this requirement can also be suppressed by inactivation of pilT (Winther-Larsen et al., 2005, Carbonnelle et al., 2006). Therefore, neither PilC, nor the pilin-like proteins, are essential for TFP biogenesis in Neisseria species, but are believed to play a role in pilus homeostasis by antagonizing PilT-dependent retraction (Wolfgang et al., 2000, Morand et al., 2004).
P. aeruginosa expresses a TFP-associated protein designated PilY1 that shares limited sequence homology (Alm et al., 1996), but a high degree of structural similarity (Orans et al., 2010) with N. gonorrhoeae PilC. Interestingly, the pilY1 gene is located in an operon (fimUpilVWXY1Y2E) encompassing five genes that encode pilin-like proteins. A P. aeruginosa mutant carrying a polar insertion in pilY1 failed to produce TFP fibers but accumulated intracellular processed pilin subunits, suggesting pilY1 is required for fiber assembly (Alm et al., 1996). Given the likely orthologous nature of PilY1 and PilC, as well as the apparent similarities between the TFP systems of P. aeruginosa and N. gonorrhoeae, we hypothesized that PilY1 mediates adherence of P. aeruginosa to host epithelial cells. Here, we demonstrate that PilY1 is conditionally required for TFP expression, such that TFP biogenesis is PilY1-dependent but the defect can be suppressed by inactivation of pilT. This discovery facilitated direct examination of the role of TFP and PilY1 in bacterial-host interactions using a well-characterized in vitro model of human airway epithelium. Using this model, we demonstrate that P. aeruginosa infection is a multifactorial process that requires TFP, pilus retraction and T3S. Further, we demonstrate the requirement for PilY1 in robust TFP-associated adherence of P. aeruginosa to host cell basolateral surfaces. The latter result supports a model in which TFP-associated PilY1, rather than pilin, serves as the primary adhesin for differentiated human airway epithelial cells. Overall, our findings provide a mechanistic framework for understanding the complex interactions between a pathogen and host and the mutually dependent nature of bacterial virulence factors during infection.
Results
PilY1 is required for stable TFP production
It was previously reported that polar transposon insertions in P. aeruginosa pilY1 resulted in a defect in TFP production (Alm et al., 1996). To reassess the role of PilY1 in TFP biogenesis, we constructed a non-polar pilY1 deletion in wild type piliated P. aeruginosa strain PAK and examined the ability of the wild type parent and pilY1 mutant to produce TFP by transmission electron microscopy (TEM). Polar TFP were readily detected on the surface of the wild type strain (Fig. 1A). In contrast, the pilY1 mutant was devoid of TFP fibers and was indistinguishable from a non-piliated pilA mutant (Fig. 1A). To confirm the TEM results, the wild type and mutant strains were grown on glass coverslips and pilus fibers were labeled with pilin-specific antibody and examined by immunofluoresence microscopy (IF) (Fig. 1B). While the IF technique does not provide sufficient resolution to evaluate fiber morphology, it provides an unbiased assessment of TFP production in a large population of bacterial cells. The wild type strain produced abundant TFP, while no TFP could be detected in association with either the pilY1 or pilA mutants (Fig. 1B).
Fig. 1. PilY1 is conditionally required for TFP production.
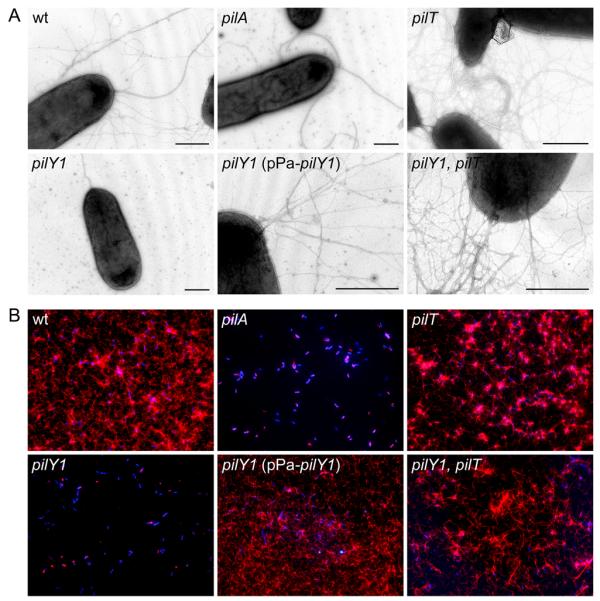
Surface TFP were visualized by (A) transmission electron microscopy and (B) immunofluorescence microscopy. Pilus fibers (thin) are visible on the surface of the wild type (wt) strain and a non-retractile pilT mutant. In contrast, a pilY1 mutant is devoid of TFP and indistinguishable from a non-piliated control strain (pilA). Wild type levels of TFP are restored in a pilY1 mutant complemented with plasmid-expressed pilY1 (pPa-pilY1). Pilus fibers are also restored in a pilY1 mutant when pilT is inactivated (pilY1, pilT), indicating PilY1 is not an essential TFP biogenesis factor. Strain genotype is indicated in each panel. For electron microscopy, scale bars equal 500 nm. Thick fibers visible on the wt strain and pilA, pilT and pilY1 mutants are flagella. For immunofluorescence, bacterial cells were labeled with DAPI (blue) and TFP were labeled with pilin-specific antibody, Alexa Red 594-conjugated goat anti-rabbit secondary antibody (red) and bacteria were imaged at 100X magnification.
To further validate microscopy-based assessment of TFP production, we performed semi-quantitative analysis of TFP production by comparing the relative amount of pilin in pilus fractions recovered from the surface of the wild type and mutant strains. Since pilin is the major structural subunit of TFP, the amount of pilin recovered in pilus fractions correlates directly with TFP abundance. Pilin was recovered from the wild type strain, but was not detected in the TFP fraction recovered from the pilY1 mutant (Fig. 2A). Comparison of the pilus preparation from the pilY1 mutant to a serial dilution of the pilus preparation from the wild type strain indicated a greater than 100-fold reduction in TFP produced by the pilY1 mutant (data not shown). Despite the lack of surface TFP, wild type levels of the pilin subunit were detected by immunoblot in whole bacterial cell lysates from the pilY1 mutant (Fig. 2C) demonstrating that the TFP defect was not due to the lack of pilin.
Fig. 2. PilY1 is conditionally required for TFP production and co-localizes with sheared surface fibers.

(A) Coomassie Blue-stained SDS-PAGE gel showing the relative abundance of recovered pilin in sheared pilus fractions. (B) Immunoblot of pilus fractions probed with PilY1-specific antiserum. (C-E) Immunoblots of whole bacterial cell lysates separated by SDS-PAGE and probed with pilin-specific (C), PilY1-specific (D) or PilT-specific (E) antiserum.
To rule out the possibility that the TFP biogenesis defect in the pilY1 mutant was due to a polar effect of the deletion on distal gene expression, we assessed the ability of cloned pilY1 to restore TFP production when expressed in trans. A pilY1 expression plasmid (pPa-pilY1) was created and transferred to the pilY1 mutant. Induction conditions were optimized such that wild type levels of PilY1 were detected in whole bacterial cell lysates by immunoblot using PilY1-specific antiserum (Fig. 2D). Under these conditions, expression of plasmid-borne pilY1 was sufficient to restore TFP production to a wild type level (Fig. 1 and 2A). Overall, these results provide definitive evidence that PilY1 is required for the production of P. aeruginosa TFP in a wild type background.
Inactivation of pilT suppresses the TFP production defect in a pilY1 mutant
Inactivation of P. aeruginosa pilT was previously reported to results in a non-twitching, hyperpiliated phenotype in P. aeruginosa (Whitchurch et al., 1991). In N. gonorrhoeae, inactivation of pilT suppresses the TFP biogenesis defect of a mutant lacking PilC (Wolfgang et al., 1998). Given the similarities between P. aeruginosa PilY1 and gonococcal PilC (Alm et al., 1996, Orans et al., 2010), we examined the effect of inactivation of pilT on TFP biogenesis in the absence of PilY1. As expected, deletion of pilT alone resulted in a hyperpiliated, non-twitching phenotype (Fig. 1, 2A and S1). Comparison of serial dilutions of TFP containing fractions indicates that the pilT mutant produced approximately 4-fold more surface TFP than the wild type strain (data not shown). Deletion of pilT in the non-polar pilY1 mutant background resulted in the restoration of TFP production to a level indistinguishable from the wild type (Fig. 2A). This result demonstrates that the TFP biogenesis defect of a pilY1 mutant is conditional and can be suppressed by inactivation of pilT. To confirm that the restoration of TFP production in the absence of PilY1 was due to pilT inactivation, we complemented the pilY1, pilT mutant with plasmid-expressed pilT (pPa-pilT). When wild type expression of PilT was restored (Fig. 2E), TFP production was abolished, resulting in a phenotype indistinguishable from that of a pilY1 mutant (Fig. 2A and S1). Conversely, complementation of the pilY1, pilT mutant with plasmid-borne pilY1 (Fig. 2D) resulted in restoration of the hyperpiliated pilT phenotype (Fig. 2A). Given that pilT is presumed to be involved in the retraction of assembled fibers, our data indicate the TFP defect seen in a pilY1 mutant is not due to an absolute defect in TFP fiber assembly. Instead, PilY1 appears to stabilize assembled pilus fibers, a process that likely antagonizes retraction mediated by PilT. The fact that the pilY1, pilT double mutant produces less TFP than the pilT mutant alone (Fig. 2A), suggests that PilY1 also plays a role in stabilizing TFP assembly in the absence of fiber retraction.
Co-localization of PilY1 with TFP requires additional genes in the pilY1-associated operon
Given the apparent similarities between PilY1 and the gonococcal TFP-associated PilC protein, we assessed whether PilY1 is localized to the sheared pilus fraction and whether other TFP-associated proteins contribute to PilY1 localization. To assess PilY1 localization, we used PilY1-specific antiserum to probe TFP fractions recovered from the surface of the wild type and mutant strains. PilY1 was present in the wild type TFP fraction, but not detected in fractions collected from the non-piliated pilY1 mutant or the piliated pilY1, pilT double mutant (Fig. 2B). Complementation of the pilY1 and pilY1, pilT mutants with plasmid-encoded pilY1 (pPa-pilY1) resulted in restoration of PilY1 to the TFP fractions (Fig. 2B). The TFP fraction from the non-piliated pilA mutant was devoid of PilY1 (Fig. 2B), despite the fact that PilY1 could be detected in whole bacterial cell lysate from the same strain (Fig. 2D). The latter result demonstrates that PilY1 localization to the TFP fraction requires the presence of surface pili. While not definitive, this result suggests that PilY1 may specifically associate with assembled pilus fibers, as is the case for gonococcal PilC (Rudel et al., 1995b).
It is currently unclear if localization of PilY1 to the sheared TFP fraction requires additional proteins. However, pilY1 is located in an operon with six additional genes (fimU-pilVWXY1Y2E), five of which encode pilin-like proteins with homology to N. gonorrhoeae proteins required for localization of PilC (Fig. 3A) (Alm et al., 1997, Winther-Larsen et al., 2005, Belete et al., 2008). To determine if the other genes within the pilY1 operon are required for localization of PilY1 to the sheared pilus fraction, we created an unmarked deletion encompassing the entire pilY1 operon. The resulting mutant strain (fimU-pilE) was defective for TFP production (Fig. 3B). As was seen with the pilY1 mutant, wild type levels of the pilin subunit were detected by immuoblot in whole cell lysates from the fimU-pilE mutant (Fig. 3D), indicating that the defect in TFP biogenesis was not due to reduced pilin availability. Complementation of the fimU-pilE mutant with a plasmid-borne copy of the operon (pPa-fimU-pilE) was sufficient to restore TFP biogenesis (Fig. 3B) and localization of PilY1 to the TFP fraction (Fig. 3C). In contrast, complementation of the fimU-pilE mutant with plasmid-expressed pilY1 alone (Fig. 3E) was not sufficient to restore TFP production (Fig. 3B) or localization of PilY1 to the sheared surface fraction (Fig. 3C). While this result suggests that localization of PilY1 to surface fractions may require additional TFP biogenesis components, we cannot rule out the possibility that our fractionation technique is not sufficient for isolation of surface localized PilY1 in the absence of TFP fibers.
Fig. 3. Localization of PilY1 to the pilus fraction requires proteins encoded by the pilY1-associated operon.

(A) Organization of the pilY1-associated operon. Pilin-like genes are shaded grey. (B) Coomassie Blue-stained SDS-PAGE gel showing the relative abundance of recovered pilin in pilus fractions. (C) Immunoblot of surface pilus fractions probed with PilY1-specific antiserum. (D-E) Whole bacterial cell lysates separated by SDS-PAGE and probed with either pilin-specific (D) or PilY1-specific (E) antiserum by immunoblotting.
To determine whether other genes in the pilY1 operon are conditionally required for TFP biogenesis, we introduced the fimU-pilE deletion into the pilT mutant background. The resulting strain (fimU-pilE, pilT) produced TFP (Fig. 3B), indicating that none of the products of the pilY1 operon are essential for TFP biogenesis. Restoration of TFP production in the absence of the pilin-like genes (fimU-pilE, pilT) provided us with the opportunity to determine the role of these proteins in PilY1 localization in an otherwise piliated background. Complementation of the fimU-pilE, pilT mutant with plasmid-expressed pilY1 resulted in wild type levels of PilY1 in whole cell lysates (Fig. 3E); however, PilY1 was not detectable in the sheared TFP fraction (Fig. 3C) indicating that localization of PilY1 to surface TFP fractions requires both the production of TFP fibers and at least one of the pilin-like proteins encoded by the pilY1-associated operon.
TFP and pilus retraction are required for P. aeruginosa interaction with A549 cells
Previous studies have shown that TFP are required for P. aeruginosa adherence to mammalian cells (Doig et al., 1988, Chi et al., 1991). In addition, TFP-dependent host cell adherence is required for in vivo activation of the P. aeruginosa contact-dependent T3SS, which causes host cell cytotoxicity (Comolli et al., 1999b, Sundin et al., 2002, Wolfgang et al., 2003). Although our pilY1 mutant fails to express TFP in vitro, the defect appears to be at the level of fiber stabilization and not TFP biogenesis. We hypothesized that transient fiber production in the presence of the appropriate host cell receptor may be sufficient to promote host cell interaction. To assess the role of PilY1 in host cell interactions, we measured the ability of the pilY1 mutant to adhere to and cause cytotoxicity in A549 cells (human type II pneumocyte-like carcinoma cells).
Consistent with previous reports (Chi et al., 1991, Comolli et al., 1999a), wild type P. aeruginosa adhered to A549 cells to a greater extent than the non-piliated pilA mutant (Fig. 4). In addition, the wild type strain was more cytotoxic than the pilA mutant or a pscC mutant, which lacks a functional T3SS (Wolfgang et al., 2003) (Fig. 4). The pilY1 mutant showed a significant reduction in both adherence and cytotoxicity compared to the wild type strain, and was indistinguishable from the pilA mutant (Fig. 4). Complementation of the pilY1 mutant with plasmid-encoded pilY1 restored both adherence and cytotoxicity.
Fig. 4. Retractile TFP and pilus-associated PilY1 are necessary for productive interactions with A549 epithelial cells.

Bacterial adherence (black bars; mean +/− SEM; n=5) and cytotoxicity (grey bars; mean +/− SEM; n=3) to A549 cells is shown. Bacterial adherence was determined based on the average number of bacteria bound per A549 cell. Adherence to A549 cells was significantly reduced (*; p<0.001) in the non-pilated pilA and pilY1 mutants and in the piliated non-retractile pilT and pilY1, pilT mutants compared to wild type. Cytotoxicity was determined based on the percentage (%) of lactate dehydrogenase (LDH) released from A549 cells following bacterial infection relative to the amount of LDH released following cell lysis with 0.25% Triton X-100. The ability to elicit a cytotoxic effect was significantly reduced (#; p<0.001) in the non-adherent strains and in a T3S mutant (pscC) compared to wild type.
These findings indicate that pilY1 is required for P. aeruginosa to establish pathogenic interactions with host cells, but it is not clear whether the requirement is solely at the level of stable TFP production or whether PilY1 directly mediates TFP-dependent adherence. To distinguish between these possibilities, we assessed adherence and cytotoxicity of piliated pilT mutants in the presence or absence of pilY1 (pilT versus pilY1, pilT) (Fig. 4). Both mutants had significantly reduced adherence and cytotoxicity compared to the wild type strain. While the pilT mutant showed greater adherence than the pilY1, pilT double mutant the difference was not significant. The reduced capacity of the of pilT mutants to adhere despite being piliated suggests that PilT, and presumably TFP retraction, is a prerequisite for interaction with A549 cells, and prevented us from drawing any conclusions about the role of PilY1 in adherence in this system.
Retractile TFP and T3S are required for infection and invasion of human ciliated airway epithelium in vitro
Mutants lacking the pilus retraction protein PilT were previously reported to display reduced adherence to non-polarized or transformed epithelial cell lines (Comolli et al., 1999a, Sundin et al., 2002). The lack of binding to A549 cells exhibited by the piliated pilT and pilY1, pilT mutants (Fig. 4) may be due to a defect in pilus retraction (Fig. S1), a process that is likely to facilitate tight association of P. aeruginosa with host cells. Experiments by Fleiszig et al. (1997) and Lee et al. (1999) demonstrated preferential binding of P. aeruginosa to the basolateral surfaces of well-differentiated mammalian epithelial cells. Based on these studies, we hypothesized that polarized or well-differentiated airway epithelial cell cultures may provide a more biologically relevant model for determining the role of PilY1 in TFP-mediated adherence. To more accurately model the host environment, we evaluated P. aeruginosa infection in an in vitro model of human ciliated airway epithelium (HAE) (Fulcher et al., 2005). HAE cultures are derived from freshly isolated epithelial cells from human conducting airways, which are allowed to propagate and differentiate into a pseudostratified epithelium that mimics the organization and structure of the ciliated human airway mucosal epithelium in vivo (Fig. 5).
Fig. 5. Invasive infection of Human Airway Epithelium (HAE) cultures requires retractile TFP and Type III Secretion.

Histological cross-sections of HAE cultures inoculated with wild type (wt), pilA, pilT and pscC strains. Representative images taken at 3, 6, and 12 hours post-infection are shown. Wild type P. aeruginosa rarely adhered to the ciliated mucosal surfaces but local infection foci could be detected after 3 hours. After breaching the mucosal barrier, the wild type strain interacted efficiently with the basolateral surfaces of ciliated cells and the underlying basal epithelial cells. Bacterial adherence was associated with host cell rounding and detachment. By 12 hours, the wild type infection appeared to spread between cells to encompass the entire HAE culture. In contrast, the non-piliated pilA mutant, the piliated non-retractile pilT mutant and the T3S mutant (pscC) did not interact with the epithelium or cause invasive infection. However, unlike the pilA mutant, the pilT and pscC mutants retained the ability to interact with shed or extruding epithelial cells (bottom right panels).
To determine the feasibility of using HAE cultures for testing P. aeruginosa interactions with host cells, we performed initial infection experiments with wild type, piliated P. aeruginosa. Following apical inoculation of the HAE culture, the wild type strain rarely adhered to the ciliated mucosal surface, but by 3 hours it had invaded the epithelium and caused local infection foci (Fig. 5). By 6 hours, wild type P. aeruginosa spread to the basolateral compartment of the HAE cultures and appeared to interact efficiently with newly exposed basolateral surfaces of ciliated cells as well as the underlying basal epithelial cells (Fig. 5). Subsequent to invasion of the epithelium, bacterial adherence was associated with host cell rounding and detachment or extrusion. By 12 hours, the wild type infection spread to encompass the entire HAE culture. Given the efficiency with which the wild type strain was able to invade and interact with the epithelium, we expanded our study to examine the role of TFP in the infection process. In stark contrast to the wild type strain, mutants lacking TFP (pilA), or expressing TFP but defective for pilus retraction/twitching motility (pilT) did not cause invasive infection after 12 hours and could be completely removed from the intact ciliated mucosal surface of the epithelium by gentle washing (Fig. 5). Similar to the pilA mutant, the non-piliated pilY1 mutant was unable to interact with the surface of the intact epithelium (data not shown). Previously published results indicate that T3S is necessary for disruption of epithelial barrier function and paracellular invasion of polarized epithelial cell monolayers (Soong et al., 2008). Because pilA and pilT mutants fail to elicit T3S-dependent cytotoxicity due to their respective TFP defects (Fig. 4), we also examined the potential contribution of T3S to the invasion phenotype in the HAE model. A T3S mutant (pscC) was unable to invade HAE cultures and was indistinguishable from the pilA and pilT mutants (Fig. 5). These results demonstrate that invasion of the HAE is a multifactorial process that requires retractile TFP and T3S and suggests that these virulence factors are mutually dependent during infection of well-differentiated and intact tissue. The apparent lack of high affinity binding of the wild type strain or mutants to the mucosal surface of the epithelium prevented us from distinguishing between adherence and invasion in this model. However, we did observe that the pilT and the pscC mutants were capable of binding efficiently to cells that were being extruded onto the surface of the epithelium (Fig. 5). In an intact epithelium, the natural turnover of epithelial cells proceeds by extrusion of the dying cells onto the mucosal surface (Mayhew et al., 1999, Pentecost et al., 2006). While a relatively rare event, this observation strongly suggested that the pilT and pscC mutants were capable of binding HAE cells, despite their inability to penetrate the mucosal barrier. Our findings demonstrate that the mucosal surface of polarized and differentiated airway cells represents a substantial barrier to infection, and that once breached, P. aeruginosa preferentially binds to exposed basolateral surfaces.
PilY1 is required for adherence of P. aeruginosa to injured HAE cell cultures
The results presented above are consistent with previous studies showing that P. aeruginosa preferentially adheres to injured or remodeling epithelial tissue (Ramphal et al., 1980, de Bentzmann et al., 1996, Lee et al., 1999). To determine the role of TFP and pilus-associated PilY1 in adherence to host basolateral cell surfaces, we modified the HAE cultures to artificially expose the underlying basal epithelial cells. Specifically, the columnar epithelial cells were disrupted by mechanical abrasion prior to P. aeruginosa infection. To assess adherence, injured HAE cultures were exposed to bacterial strains expressing green fluorescent protein (GFP) for 45 minutes and adherent bacteria were visualized by fluorescence microscopy. Wild type P. aeruginosa adhered to the injured regions of the HAE, but not to the adjacent intact ciliated epithelium (Fig. 6). Similarly, the T3S mutant (pscC), which was unable to invade and interact with the mucosal surface of the intact epithelium (Fig. 5), showed robust adherence to the injured regions (Fig. 6), a finding that is consistent with the ability of this mutant to interact with cells extruded from the intact HAE culture. In contrast, the non-piliated pilY1 and pilA mutants did not bind either injured or non-injured regions of the HAE (Fig. 6), indicting a requirement for TFP. Adherence of the pilY1 mutant could be restored by complementation with plasmid-encoded pilY1 (Fig. 6).
Fig. 6. TFP and pilus-associated PilY1 are required for adherence of P. aeruginosa to injured HAE cultures.
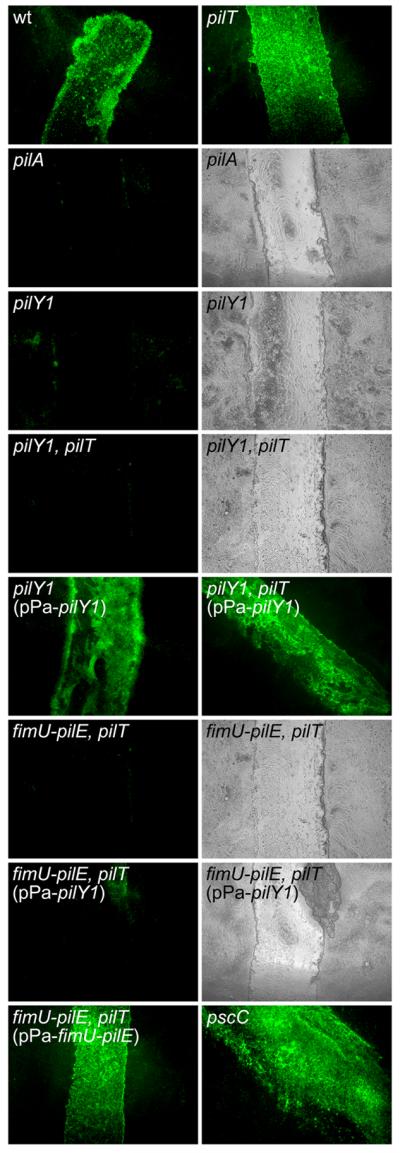
Injured HAE cultures were imaged (en face) by fluorescence microscopy 45 minutes after inoculation with bacterial strains expressing GFP. The wild type (wt) strain, the piliated pilT mutant and a non-cytotoxic T3S mutant (pscC) attached efficiently to the injured tissue but showed limited interaction with the adjacent intact epithelium. The non-piliated (pilY1) and piliated (pilY1, pilT) pilY1 mutants did not adhere to the injured tissue and were indistinguishable from the non-piliated control mutant (pilA). Adherence was restored for both the non-piliated and piliated pilY1 mutants following complementation with plasmid-expressed PilY1 (pPa-pilY1). The piliated fimU-pilE, pilT mutant was unable to adhere to the damaged tissue; complementation with the entire fimU operon (pPa-fimU-pilE) but not pilY1 alone (pPa-pilY1) restored adherence. For non-adherent strains, tissue damage was confirmed by examining the HAE cultures by both fluorescence and light microscopy.
To directly examine the role of PilY1 in TFP-mediated adherence, we compared binding of the piliated non-retractile pilT mutant to the piliated pilY1, pilT double mutant. The pilT mutant, which displayed poor binding to A549 cells (Fig. 4), adhered efficiently to the injured region of the HAE (Fig. 6). Conversely, the piliated pilY1 mutant (pilY1, pilT) mutant showed little or no binding to the injured or non-injured regions of the HAE (Fig. 6). Complementation of the pilY1, pilT mutant with plasmid-expressed pilY1 restored adherence (Fig. 6). To determine whether localization of PilY1 to the pilus fraction is necessary for adherence, we assessed binding of the piliated fimU-pilE, pilT mutant and found it was unable to bind the injured epithelium (Fig. 6). Complementation with the entire pilY1-associated operon (pPa-fimU-pilE), but not pilY1 alone (pPa-pilY1) restored adherence (Fig. 6). These results demonstrate that the adherence of P. aeruginosa to injured HAE cultures requires PilY1 localization to the surface TFP fraction and strongly suggest that PilY1 functions as a TFP-associated adhesin. Further, the inability of the piliated, PilY1-lacking strain (pilY1, pilT) to adhere to host cells indicates that pilin, in its polymerized form, is not sufficient for adherence to well-differentiated human airway epithelial cells.
To confirm the results of the injured HAE binding experiments, we directly visualized bacterial-host cell interactions in fixed histological sections of the injured HAE regions by light microscopy. In all cases, the bacterial strains that adhered to the injured regions of the HAE were specifically associated with exposed basal epithelial cells (Fig. 7). In contrast, the non-adherent mutants (pilA, pilY1 and pilY1, pilT) were rarely detected in association with exposed underlying host cell surfaces (Fig. 7). Interestingly, adherent strains that retained cytotoxicity (T3S) and twitching motility (Fig. 4 and S1; wild type and the complemented pilY1 mutant) appeared to further disrupt the integrity of the basal cell layer and penetrated to the level of the culture membrane support during the 45 minute infection (Fig. 7). In contrast, the adherent twitching-defective strains (Fig. S1; pilT and pilY1, pilT complemented with plasmid-expressed pilY1) did not further disrupt the integrity of or penetrate the exposed basal cell layer (Fig. 7). In addition, the non-cytotoxic T3S mutant (pscC) interacted with the superficial exposed or damaged basal cells but did not penetrate further into the exposed basal cell layers (Fig. 7). These result indicates that although binding of P. aeruginosa to exposed basal cells does not require pilus retraction or T3S, subsequent intercellular spread within the damaged tissue depends on twitching motility and the ability to deliver toxic effector proteins.
Fig. 7. PilY1 is required for adherence to exposed basal epithelial cells of injured HAE cultures.

Histological sections of HAE cultures show the presence of bacteria on newly exposed basal epithelial cells after injury. The wild type strain (wt), the piliated pilT mutant and a non-cytotoxic T3S mutant (pscC) showed robust adherence, while the non-piliated (pilY1) and piliated (pilY1, pilT) PilY1-lacking strains were defective for binding to basal epithelial cells at the injury site. Adherence was restored for both the non-piliated and piliated pilY1 mutants following complementation with plasmid-expressed pilY1 (pPa-pilY1). Only strains which retained twitching motility and cytotoxicity (wt and the complemented pilY1 mutant) were able to further penetrate the exposed basal cell layers.
Discussion
Adherence to and infection of mucosal epithelium is a crucial event in the pathogenesis of human disease caused by the opportunistic pathogen P. aeruginosa. However, the factors contributing to this event have yet to be clearly defined. Here, we examined the role of the PilY1 protein and pilus retraction in TFP-mediated adherence using a genetic approach. Initially, we examined the involvement P. aeruginosa PilY1 in TFP biogenesis and function. We found that PilY1 is not a canonical pilus assembly factor but rather acts as a conditional effector of pilus homeostasis by promoting extension/polymerization events in the presence of the pilus retraction ATPase PilT, a function previously demonstrated for the related Neisseria PilC proteins (Wolfgang et al., 1998, Carbonnelle et al., 2006). In addition, we confirmed that PilY1 co-purifies with TFP in P. aeruginosa and further showed that this localization requires at least one of a set of genes linked to and co-transcribed with pilY1 (fimU-pilVWXY1Y2E). As previously noted, the fimU operon displays synteny with the N. gonorrhoeae pilH-L locus whose products are necessary for localization of PilC to the TFP fraction (Winther-Larsen et al., 2005). In contrast to our results and previous results (Alm et al., 1996), Bohn and colleagues (2009) detected a truncated form of PilY1 in concentrated P. aeruginosa culture supernatants but were unable to detect full-length PilY1 in extracellular fractions by immunnoblot or in association with TFP fibers by immunogold labeling and electron microscopy. Technical differences between the studies, including the fractionation methods employed and the reagents and bacterial strains used likely account for the discrepancy.
Transcription of the fimU-pilVWXY1Y2E operon, but not the pilA gene (encoding pilin), is controlled by intracellular cyclic AMP in combination with the cyclic AMP receptor protein Vfr and the AlgZ/AlgR two component regulatory system (Lizewski et al., 2002, Wolfgang et al., 2003, Belete et al., 2008), suggesting that the level of PilY1 and associated proteins can be modulated independent of the rest of the TFP biogenesis machinery. This circuitry may allow P. aeruginosa to alter PilY1-dependent TFP production and associated phenotypes (twitching motility and host cell adherence) in response to environmental conditions. Regulation of pilY1 expression may be analogous to pilC regulation in N. meningitidis where host cell contact results in reduced pilC transcription (Taha et al., 1998, Morand et al., 2004). Interestingly, it has been reported that deletion of pilY1 results in the altered expression of numerous P. aeruginosa genes including activation of lipC, encoding a secreted lipase and the fimU-pilVWXY1Y2E operon (Martinez et al., 1999, Bohn et al., 2009). Furthermore, it was shown that pilY1 mutant strains have increased autolysis and reduced secretion of secondary metabolites including the redox-activated pigment pyocyanin and 4-hydroxy-2-alkylquinolines (Bohn et al., 2009). These results suggest that the expression and/or localization of PilY1 may serves as a feedback signal for altering gene expression. While it remains to be determined whether pilus retraction or bacterial attachment elicit the same response as pilY1 deletion, it is conceivable that these mechanical processes result in the altered localization of PilY1, which in turn acts as a signal for changes in gene expression and/or production and secretion of virulence factors and toxic secondary metabolites.
Next, we evaluated the role of PilY1 in P. aeruginosa adherence to human cells by using a piliated non-retractile pilT mutant background in which TFP expression was maintained in the absence of PilY1. In our studies with the A549 carcinoma cell line, the piliated pilT and pilY1, pilT mutants showed significantly reduced adherence compared to the wild type strain, which precluded any conclusions regarding the role of PilY1 in adherence. Similarly, the piliated pilT and pilY1, pilT mutants were unable to interact with the mucosal surface of intact HAE cultures or cause invasive infection. However, injury of the HAE cultures supported robust adherence of the pilT mutant to regions of exposed basal cells. The fact that the piliated pilY1, pilT mutant, the piliated fimU-pilE, pilT mutant and the piliated fimU-pilE, pilT mutant complemented with plasmid-expressed pilY1 (in which PilY1 does not localize with TFP) all failed to bind the injured HAE cultures provides genetic evidence that PilY1 serves as a TFP-associated adherence factor in this model. Although these results do not formally rule out the contribution of an integral adhesive component in pilin, they do question the relevance of such an activity as it is unclear why it would only be manifest in conjunction with PilY1 expression.
Our results suggest that bacterial adherence to non-polarized transformed cell line and injured HAE cultures involve different adhesin-receptor interactions. We hypothesize that the interaction between P. aeruginosa and primary well-differentiated human airway epithelial cells more accurately reflects the in vivo situation. The ability to P. aeruginosa to recognize a high affinity/abundant receptor on the basolateral surfaces of HAE cells is consistent with previous work showing that P. aeruginosa preferentially infects polarized epithelium via a basolateral route (Fleiszig et al., 1997, Lee et al., 1999). The involvement of distinct adhesin-receptor interactions is supported by our observation that the piliated non-retractile pilT mutant bound to the basolateral surface of primary epithelial cells but not to A549 cells. This result suggests that pilus retraction may increase the affinity of an adhesin-receptor interaction during attachment to non-polarized transformed cells. An analogous scenario has been described for Escherichia coli lectin-like adhesin FimH, where shear forces dramatically increase adhesin-receptor affinity by a catch-bond mechanism (Thomas et al., 2002). Alternatively, pilus retraction may facilitate additional bacterial-host cell interactions necessary to support adherence to transformed cells, similar to the cascade of adherence events that occur during colonization of intestinal epithelial cells by enteropathogenic E. coli (EPEC). Initial or localized adherence of EPEC involves bacterial attachment to an N-acetyllactosamine contain receptor via α-bundlin, the major subunit of bundle-forming pili (Hyland et al., 2008). Following localized adherence, EPEC established intimate attachment, a process mediated by intimin, an EPEC outer membrane protein that specifically binds the Tir receptor, a bacterial protein delivered to host cells by the EPEC T3SS (Nougayrede et al., 2003, Cleary et al., 2004).
Our HAE culture studies indicate that P. aeruginosa gains access to the basolateral compartment following inoculation of the apical surface of the intact epithelium (Fig. 5). Pathogenic bacteria have evolved a number of discrete mechanisms to gain access to these privileged basolateral sites including invading M cells in order to traverse the epithelium or by disrupting the integrity of epithelial cell tight junctions (Jensen et al., 1998, Soong et al., 2008, van Alphen et al., 2008). In the case of Listeria monocytogenes, the bacteria appear to invade epithelial junctions at sites of cell extrusion and expulsion occurring during epithelial renewal (Pentecost et al., 2006). Such an event results in the transient exposure of basolateral cell surfaces and components (Pentecost et al., 2006) and may represent a similar route used by P. aeruginosa to breach the epithelial barrier. Alternatively, it has been proposed that P. aeruginosa-induced tissue damage alters the N-glycoprotein and heparan sulfate proteoglycan receptor composition of the apical and basolateral compartments, thereby allowing bacterial access to basolateral cell surfaces via receptor-mediated bacterial binding (Bucior et al., 2010). Once the epithelial barrier has been bypassed, we hypothesize that interaction between P. aeruginosa and components present on host basolateral membranes facilitates intercellular spread of the bacteria, similar to the basolateral-specific invasion mechanisms used by L. monocytogenes, Shigella flexneri, and Campylobacter jejuni (Mounier et al., 1992, Gaillard et al., 1996, Monteville et al., 2002). Further, our observation that P. aeruginosa preferentially adheres to mechanically injured epithelial tissue in vitro (Fig. 6) is consistent with studies showing “opportunistic adherence” to damaged areas of mammalian tissue (Ramphal et al., 1980, de Bentzmann et al., 1996, Lee et al., 1999) and provides a mechanistic explanation for the increased rate of P. aeruginosa infection following tissue damage (wounds, corneal abrasion, burns, surgery and endotracheal intubation).
Based on our studies with intact and damaged HAE cultures, we propose a sequence of events leading to fulminant tissue infection by P. aeruginosa (Fig. 8). We hypothesize that P. aeruginosa infection of intact mucosal epithelium can be initiated by a relatively rare adherence event (Fig. 8A) that evolves into basolateral infection (Fig. 8B and C). While we were unable to detect the earliest adherence events preceding tissue invasion by wild type P. aeruginosa, we did capture these events with the pilT and pscC mutants. Both the piliated non-retractile pilT mutant and the piliated but non-cytotoxic pscC mutant bound to epithelial cells that had been extruded onto the surface of the HAE cultures or were undergoing extrusion from the otherwise intact epithelium (Fig. 5). Although the pilT and pscC mutants adhered to extruded cells, these strains were unable to penetrate the mucosal barrier to cause invasive infection, indicating an early block in the infection process. Given that non-piliated mutant strains (pilA and pilY1) were unable to bind mechanically exposed basal cells (Fig. 6 and 7) and that adherence of the pilT mutant to exposed basal cells required TFP-associated PilY1 (Fig. 6 and 7), we propose that TFP and TFP-associated PilY1 are required for initial binding to the mucosal surface at the site of cell extrusion. Subsequent interactions between P. aeruginosa and the intact apical surface lead to a breach of the epithelial barrier. This process likely involves T3S-dependent alteration of tight junctions (Soong et al., 2008) or the relocalization of apical and basolateral receptors in response to bacterial-induced host cell damage (Bucior et al., 2010). Regardless of the initiating mechanism for apical penetration, we hypothesize that subsequent access of P. aeruginosa to normally masked basolateral receptors allows attachment and bacteria-induced cytotoxicity requiring PilT-dependent pilus retraction and T3S (Fig. 8B). Assuming the requirement for PilT reflects its role in TFP retraction (Merz et al., 2000), two independent functions can be envisioned: 1) pilus retraction brings the bacteria into intimate contact with host cells and thus facilitates activation of the contact-dependent T3SS and efficient effector delivery, and/or 2) pilus retraction promotes intercellular spread of the bacteria via twitching motility. Further dissemination of the bacteria within the basal compartment of the stratified tissue (Fig. 8C) requires PilY1, PilT and T3S such that bacterial adherence, tissue damage and dissemination mutually fuel a positive feedback loop that culminates in fulminant infection.
Fig. 8. Model of events leading to invasive infection of the human airway epithelium by P. aeruginosa.

The three different phases of epithelial infection (apical interaction, penetration and dissemination) and the P. aeruginosa factors required for each phase are indicated. (A) Apical interaction. Productive interactions between P. aeruginosa and the intact mucosal epithelium is a relatively rare event exploiting transiently exposed basolateral surfaces during cell extrusion. This event is dependent on bacterial adherence mediated by TFP and TFP-associated PilY1. (B) Penetration. Subsequent penetration of the mucosal barrier requires retractile TFP and T3S. PilT-dependent pilus retraction may facilitate contact-dependent T3S and TFP-mediated bacterial motility. Cytotoxicity mediated by T3S causes additional tissue damage and disruption of host cell junctions and exposure of additional basolateral host receptors. (C) Dissemination. Following the formation of a focal infection, PilY1-mediated adherence, retractile TFP and T3S act synergistically to cause fulminant infection and dissemination into deeper tissue.
In summary, we have identified and characterized a novel, multifactorial strategy by which P. aeruginosa gains access to and damages human mucosal tissue. In addition, this study provides compelling evidence that TFP-associated PilY1 is an adhesin that specifically recognizes a basolateral host cell receptor. Further biochemical and structural studies of PilY1 should reveal how it exerts its adherence-promoting effect and undoubtedly facilitate the identification of the host ligand(s) involved.
Experimental Procedures
Bacterial Strains and Growth Conditions
All P. aeruginosa strains and plasmids used in this study are described in Table S1. Unmarked, non-polar deletion alleles were constructed as previously described (Wolfgang et al., 2003) using the oligonucleotides listed in Table S2. The pilY1 deletion allele carries an in-frame stop codon (TGA) in place of the nucleotides encoding amino acid residues 51 through 801 of the strain PAK pilY1 coding sequence (GenBank #EU234515). The fimU-pilE deletion was engineered to remove the entire pilY1 operon (fimUpilVWXY1Y2E) sequence between the fimU start codon (ATG) to the pilE stop codon (TGA). Deletion alleles were introduced onto the chromosome of P. aeruginosa strain PAK as described (Wolfgang et al., 2003) and confirmed by PCR and DNA sequencing. P. aeruginosa strains were routinely grown at 37°C in Luria Bertani (LB) broth or on LB agar. The pMMB-based expression plasmids were maintained in P. aeruginosa with 150 μg ml−1 carbenicillin (Cb), except where noted. Bacterial growth in broth culture was assessed by optical density at 600 nm (OD600). Constitutive GFP expression was achieved by transferring plasmid pSMC21 into wild type and mutant P. aeruginosa strains as previously described (Bloemberg et al., 1997).
Expression Plasmid Construction
The open reading frames (ORFs) of pilY1 and pilT and the entire pilY1-associated operon (fimUpilVWXY1Y2E) were amplified from strain PAK chromosomal DNA and cloned into the expression plasmid pMMBV2GW as described previously (Wolfgang et al., 2003), to yield pPa-pilY1, pPa-pilT and pPa-fimU-pilE, respectively. Oligonucleotide primers used to generate expression plasmids are indicated (Table S2). Plasmid pMMBV2GW was generated from the broad host-range expression plasmid pMMBGW (Wolfgang et al., 2003) by changing the sequence of the −35 and −10 regions of the tac promoter from TTGACA to TTTACA and from TATAAT to CATTAT, respectively (S. Lory, Harvard Medical School, unpublished). The resulting tac promoter modifications result in tighter transcriptional repression of cloned down-stream genes in the absence of isopropyl-β-D-thiogalactopyranoside (IPTG) induction (data not shown). All expression plasmids were transferred to the appropriate strain by conjugation (Furste et al., 1986) followed by selection on LB agar plates containing 150 μg ml−1 Cb and 25 μg ml−1 irgasan. Following selection, expression plasmid carrying strains were grown in the presence of 30 μg ml−1 Cb. To achieve wild type expression of the cloned genes, strains containing expression plasmids were grown in the presence of 20 (pPa-pilT) or 75 μM IPTG (pPa-pilY1 and pPa-fimU-pilE).
Electron and Immunoflourescence Microscopy
Transmission electron microscopy (TEM) was performed as described previously (Winther-Larsen et al., 2007), with the exception that grids were placed on a drop of bacterial suspension at 22°C for 10 minutes and the samples were stained with an aqueous 0.5% ammonium molybdate solution for 10 minutes. Immunofluorescence (IF) microscopy was performed as described previously (Winther-Larsen et al., 2005), except that the P. aeruginosa strains were grown until OD600 = 0.2 prior to incubating the bacteria on poly-L-lysine-coated glass cover slips. P. aeruginosa pilin-specific antiserum (gift of E. C. Gotschlich, Rockefeller University) was used as a primary antibody for TFP labeling, followed by an Alexa Red 594-conjugated goat anti-rabbit IgG (Molecular Probes). P. aeruginosa cells were strained with 4′-6-diamidino-2-phenylindole (DAPI) at 1 μg ml−1 in Mowiol Mounting Medium (Sigma) containing 2% 1,4-diazabicyclo(2,2,2)octane (DABCO) prior to viewing with a Nikon Eclipse C400 fluorescence microscope.
Pilus Purification
P. aeruginosa strains were growth on LB agar plates; plasmid-harboring strains were grown on plates agar containing 30 μg ml−1 Cb and the indicated amount of IPTG. After incubation at 37°C for 18 hours, bacteria were collected and suspended in 10 ml of 0.15 M NaCl and 0.2% formaldehyde. The suspensions were vortexed for 1 minute to release surface TFP and bacterial cells were removed by centrifugation at 12,000 × g for 5 minutes. Supernatants were transferred to 15 ml glass Corex tubes, adjusted to 0.1 M MgCl2, and incubated at 4°C for 3 hours. Following centrifugation 12,000 × g for 5 minutes, the resulting TFP pellets were washed and suspended in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, resolved on an 18% polyacrylamide gel and visualized by GelCode Blue Stain (Pierce).
Twitching Motility Assay
P. aeruginosa strains were stab-inoculated to the bottom of 100 mm tissue culture-treated dishes (Corning) containing 5 ml LB plus 1% agar. For assays with plasmid-harboring strains, bacteria were grown in the presence of 30 μg ml−1 Cb and the indicated amount of IPTG. Plates were incubated for 48 hours at 37°C in a humidified chamber and the zone of subsurface bacterial growth radiating from the point of inoculation was measured.
Immunoblotting
Whole cell lysates were prepared from bacteria grown in LB broth to mid-exponential growth phase (OD600 = 1). Bacteria were collected by centrifugation, suspended in 50 μl of SDS-PAGE sample buffer and total cellular proteins were separated by SDS-polyacrylamide gels and transferred to nitrocellulose. Membranes were probed with the following primary antibodies: PKL1 anti-pilin mouse monoclonal antibody (Yu et al., 1994) (1:30,000 dilution, gift of Randall Irvin, University of Alberta), anti-PilT rabbit serum (1:30,000 dilution, gift of Katrina Forest, University of Wisconsin), or anti-PilY1 rabbit serum (1:4000 dilution) generated against a purified C-terminal portion of PilY1. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (for PilT and PilY1) and goat anti-mouse IgG (for pilin) were used as secondary antibodies. Blots were developed with enhanced chemiluminescence reagents (Millipore) and visualized by autoradiography.
A549 Bacterial Adherence and Cytotoxicity Assays
Routine passage of A549 cells was performed as previously described (Ichikawa et al., 2005). For bacterial challenge assays, A549 cells were seeded and grown on sterile glass coverslips placed in 6-well tissue culture plates (adherence assays) or in 24-well tissue culture plates (cytotoxicity assays). For adherence assays, bacteria were grown to mid-log growth phase in LB broth and washed and diluted in Hank's Balanced Salt Solution (HBSS) plus supplements (1 mM CaCl2, 2 mM MgCl2 and 20 mM HEPES). Bacteria (2 ml) were introduced at a multiplicity of infection (MOI) of 50 bacteria per A549 cell and the infection was allowed to proceed for 1 hour at 37°C in 5% CO2. The A549 cells were washed 3 times with 1 ml of HBSS plus supplements to remove unattached bacteria. The co-cultures were fixed in 2.5% glutaraldehyde in PBS (pH 7.4) for 1 hour and then in 5% formaldehyde, 5% glacial acetic acid and 70% methanol for 1 hour, followed by staining with Giemsa stain for 10 minutes. Coverslips were mounted on glass slides, viewed by light microscopy at 100 X magnification and the eukaryotic cells and associated bacteria from 6 fields, representing different regions of the coverslip, were enumerated and reported as number of bacteria per A549 cell. Cytotoxicity assays were performed using bacteria grown to mid-log phase in LB broth and washed in Dulbecco's Modified Eagle's Medium. A549 cells were infected with bacteria (500 μl) at an MOI of 10 for 10 hours at 37°C in 5% CO2. Cytotoxicity was assessed by measuring host cell lactate dehydrogenase (LDH) release using a commercially available LDH assay kit (Takara Bio). LDH release from infected cultures was reported relative to the percent of LDH released from A549 cells treated with 0.25% Triton X-100.
HAE Cultures and P. aeruginosa Infection and Binding Assays
Human airway tracheobronchial epithelial cells were isolated from excess tissue following lung transplantation under University of North Carolina at Chapel Hill Institutional Review Board-approved protocols by the UNC Cystic Fibrosis Center Tissue Procurement and Cell Culture Core. Primary cells were expanded on plastic and plated on permeable Transwell-Clear membrane supports (Corning, Inc.). HAE cultures were grown in custom media (ALI) with provision of an air-liquid interface for 4 to 6 weeks to form differentiated, polarized cultures that resemble in vivo pseudostratified mucociliary epithelium, as previously described (Fulcher et al., 2005). HAE cultures were transferred to antibiotic-free media 1 day prior to bacterial infection. For infection of intact HAE cultures, wild type and mutant P. aeruginosa strains were grown to mid-exponential growth phase (OD600 = 1.0), washed twice with sterile PBS and suspended in antibiotic-free ALI at a concentration of 4 × 107 CFU ml−1. For each strain, 25 μl of bacterial suspension was added to the mucosal surface of the HAE cultures at an approximate MOI of 10. Infected cultures were incubated at 37°C and 5% CO2 for 3, 6 or 12 hours, washed twice with pre-warmed antibiotic-free ALI and then fixed in a solution of 2% formaldehyde, 2% paraformaldehyde overnight. For infection of injured HAE cultures, the cultures were damaged by lightly scraping a sterile pipette tip across the mucosal surface to remove ciliated columnar epithelial cells. GFP-expressing P. aeruginosa strains were grown in the presence of 50 μg ml−1 kanamycin (to ensure maintenance of plasmid pSCM21), washed twice with sterile PBS and suspended in antibiotic-free ALI to a concentration of 4 × 108 CFU ml−1. Bacterial suspensions (250 μl) were added to the apical surface of the injured HAE cultures at an approximate MOI of 1000. After incubation at 37°C and 5% CO2 for 45 minutes, HAE cultures were washed six times with pre-warmed antibiotic-free ALI. After the final wash, all media was removed from the surface of the HAE cultures. Binding of GFP P. aeruginosa to the injury site was recorded using a Leica DMIRB Inverted Fluorescence/DIC microscope with a black/white digital camera. Phase contrast images of the infected cultures were taken in parallel to locate the site of injury. After imaging, infected HAE cultures were fixed in a solution of 2% formaldehyde, 2% paraformaldehyde overnight. Fixed HAE cultures, (intact and injured) were embedded in epon resin and sectioned for histological examination. Semi-thin sections were stained with Richardson's stain to allow identification of single bacteria and HAE cells. Representative sections were visualized by light microscopy and imaged with a digital camera.
Supplementary Material
Subsurface twitching motility measured as the zone of bacterial expansion at the agar-plastic interface. Mutants lacking TFP (pilA, pilY1 and fimU-pilE) or expressing non-retractile TFP due to deletion of the pilus retraction gene pilT, show an absolute defect in twitching motility. Data are represented as mean +/− SEM (n=3).
Acknowledgements
We would like to thank to the Directors and staff of the UNC Cystic Fibrosis Center Core Facilities (Tissue Procurement and Cell Culture Core, Morphology and Morphometry Core and the Michael Hooker Microscopy Facility) for supplying reagents and technical expertise, Norbert Roos and Susan Burkett for technical assistance and Nanette Fulcher for critical reading of the manuscript. This work was supported by National Institutes of Health Grants AI069116 (M.C.W.), HL084934 (R.J.P.) and HL080098 (R.J.P.). H.C.W.-L. and M.K. were supported in part by the Research Council of Norway Functional Genomics initiative (FUGE) directed through The Consortium of Advances Microbial Sciences and Technologies (CAMST), Research Council of Norway grants 166931, 152020, 183613 and funds from the Department of Molecular Biosciences and Center for Molecular Biology and Neurosciences at the University of Oslo.
References
- Alm RA, Hallinan JP, Watson AA, Mattick JS. Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol Microbiol. 1996;22:161–173. doi: 10.1111/j.1365-2958.1996.tb02665.x. [DOI] [PubMed] [Google Scholar]
- Alm RA, Mattick JS. Genes involved in the biogenesis and function of type-4 fimbriae in Pseudomonas aeruginosa. Gene. 1997;192:89–98. doi: 10.1016/s0378-1119(96)00805-0. [DOI] [PubMed] [Google Scholar]
- Belete B, Lu H, Wozniak DJ. Pseudomonas aeruginosa AlgR regulates type IV pilus biosynthesis by activating transcription of the fimU-pilVWXY1Y2E operon. J Bacteriol. 2008;190:2023–2030. doi: 10.1128/JB.01623-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg GV, O'Toole GA, Lugtenberg BJ, Kolter R. Green fluorescent protein as a marker for Pseudomonas spp. Appl Environ Microbiol. 1997;63:4543–4551. doi: 10.1128/aem.63.11.4543-4551.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn YS, Brandes G, Rakhimova E, Horatzek S, Salunkhe P, Munder A, et al. Multiple roles of Pseudomonas aeruginosa TBCF10839 PilY1 in motility, transport and infection. Mol Microbiol. 2009;71:730–747. doi: 10.1111/j.1365-2958.2008.06559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucior I, Mostov K, Engel JN. Pseudomonas aeruginosa-mediated damage requires distinct receptors at the apical and basolateral surfaces of the polarized epithelium. Infect Immun. 2010;78:939–953. doi: 10.1128/IAI.01215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonnelle E, Helaine S, Nassif X, Pelicic V. A systematic genetic analysis in Neisseria meningitidis defines the Pil proteins required for assembly, functionality, stabilization and export of type IV pili. Mol Microbiol. 2006;61:1510–1522. doi: 10.1111/j.1365-2958.2006.05341.x. [DOI] [PubMed] [Google Scholar]
- Chi E, Mehl T, Nunn D, Lory S. Interaction of Pseudomonas aeruginosa with A549 pneumocyte cells. Infect Immun. 1991;59:822–828. doi: 10.1128/iai.59.3.822-828.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang P, Sampaleanu LM, Ayers M, Pahuta M, Howell PL, Burrows LL. Functional role of conserved residues in the characteristic secretion NTPase motifs of the Pseudomonas aeruginosa type IV pilus motor proteins PilB, PilT and PilU. Microbiology. 2008;154:114–126. doi: 10.1099/mic.0.2007/011320-0. [DOI] [PubMed] [Google Scholar]
- Cleary J, Lai LC, Shaw RK, Straatman-Iwanowska A, Donnenberg MS, Frankel G, Knutton S. Enteropathogenic Escherichia coli (EPEC) adhesion to intestinal epithelial cells: role of bundle-forming pili (BFP), EspA filaments and intimin. Microbiology. 2004;150:527–538. doi: 10.1099/mic.0.26740-0. [DOI] [PubMed] [Google Scholar]
- Collins RF, Davidsen L, Derrick JP, Ford RC, Tonjum T. Analysis of the PilQ secretin from Neisseria meningitidis by transmission electron microscopy reveals a dodecameric quaternary structure. J Bacteriol. 2001;183:3825–3832. doi: 10.1128/JB.183.13.3825-3832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect Immun. 1999a;67:3625–3630. doi: 10.1128/iai.67.7.3625-3630.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comolli JC, Waite LL, Mostov KE, Engel JN. Pili binding to asialo-GM1 on epithelial cells can mediate cytotoxicity or bacterial internalization by Pseudomonas aeruginosa. Infect Immun. 1999b;67:3207–3214. doi: 10.1128/iai.67.7.3207-3214.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig L, Taylor RK, Pique ME, Adair BD, Arvai AS, Singh M, et al. Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin-coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol Cell. 2003;11:1139–1150. doi: 10.1016/s1097-2765(03)00170-9. [DOI] [PubMed] [Google Scholar]
- de Bentzmann S, Roger P, Puchelle E. Pseudomonas aeruginosa adherence to remodelling respiratory epithelium. Eur Respir J. 1996;9:2145–2150. doi: 10.1183/09031936.96.09102145. [DOI] [PubMed] [Google Scholar]
- Doig P, Sastry PA, Hodges RS, Lee KK, Paranchych W, Irvin RT. Inhibition of pilus-mediated adhesion of Pseudomonas aeruginosa to human buccal epithelial cells by monoclonal antibodies directed against pili. Infect Immun. 1990;58:124–130. doi: 10.1128/iai.58.1.124-130.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig P, Todd T, Sastry PA, Lee KK, Hodges RS, Paranchych W, Irvin RT. Role of pili in adhesion of Pseudomonas aeruginosa to human respiratory epithelial cells. Infect Immun. 1988;56:1641–1646. doi: 10.1128/iai.56.6.1641-1646.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- Emam A, Yu AR, Park HJ, Mahfoud R, Kus J, Burrows LL, Lingwood CA. Laboratory and clinical Pseudomonas aeruginosa strains do not bind glycosphingolipids in vitro or during type IV pili-mediated initial host cell attachment. Microbiology. 2006;152:2789–2799. doi: 10.1099/mic.0.28863-0. [DOI] [PubMed] [Google Scholar]
- Fleiszig SM, Evans DJ, Do N, Vallas V, Shin S, Mostov KE. Epithelial cell polarity affects susceptibility to Pseudomonas aeruginosa invasion and cytotoxicity. Infect Immun. 1997;65:2861–2867. doi: 10.1128/iai.65.7.2861-2867.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- Furste JP, Pansegrau W, Frank R, Blocker H, Scholz P, Bagdasarian M, Lanka E. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene. 1986;48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- Gaillard JL, Finlay BB. Effect of cell polarization and differentiation on entry of Listeria monocytogenes into the enterocyte-like Caco-2 cell line. Infect Immun. 1996;64:1299–1308. doi: 10.1128/iai.64.4.1299-1308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garau J, Gomez L. Pseudomonas aeruginosa pneumonia. Curr Opin Infect Dis. 2003;16:135–143. doi: 10.1097/00001432-200304000-00010. [DOI] [PubMed] [Google Scholar]
- Hazes B, Sastry PA, Hayakawa K, Read RJ, Irvin RT. Crystal structure of Pseudomonas aeruginosa PAK pilin suggests a main-chain-dominated mode of receptor binding. J Mol Biol. 2000;299:1005–1017. doi: 10.1006/jmbi.2000.3801. [DOI] [PubMed] [Google Scholar]
- Helaine S, Dyer DH, Nassif X, Pelicic V, Forest KT. 3D structure/function analysis of PilX reveals how minor pilins can modulate the virulence properties of type IV pili. Proc Natl Acad Sci U S A. 2007;104:15888–15893. doi: 10.1073/pnas.0707581104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland RM, Sun J, Griener TP, Mulvey GL, Klassen JS, Donnenberg MS, Armstrong GD. The bundlin pilin protein of enteropathogenic Escherichia coli is an N-acetyllactosamine-specific lectin. Cell Microbiol. 2008;10:177–187. doi: 10.1111/j.1462-5822.2007.01028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa JK, English SB, Wolfgang MC, Jackson R, Butte AJ, Lory S. Genome-wide analysis of host responses to the Pseudomonas aeruginosa type III secretion system yields synergistic effects. Cell Microbiol. 2005;7:1635–1646. doi: 10.1111/j.1462-5822.2005.00581.x. [DOI] [PubMed] [Google Scholar]
- Jensen VB, Harty JT, Jones BD. Interactions of the invasive pathogens Salmonella typhimurium, Listeria monocytogenes, and Shigella flexneri with M cells and murine Peyer's patches. Infect Immun. 1998;66:3758–3766. doi: 10.1128/iai.66.8.3758-3766.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami Y, Ratliff M, Surber M, Martinez A, Nunn DN. Type II protein secretion by Pseudomonas aeruginosa: genetic suppression of a conditional mutation in the pilin-like component XcpT by the cytoplasmic component XcpR. Mol Microbiol. 1998;27:221–233. doi: 10.1046/j.1365-2958.1998.00679.x. [DOI] [PubMed] [Google Scholar]
- Lee A, Chow D, Haus B, Tseng W, Evans D, Fleiszig S, et al. Airway epithelial tight junctions and binding and cytotoxicity of Pseudomonas aeruginosa. Am J Physiol. 1999;277:L204–217. doi: 10.1152/ajplung.1999.277.1.L204. [DOI] [PubMed] [Google Scholar]
- Lee KK, Sheth HB, Wong WY, Sherburne R, Paranchych W, Hodges RS, et al. The binding of Pseudomonas aeruginosa pili to glycosphingolipids is a tip-associated event involving the C-terminal region of the structural pilin subunit. Mol Microbiol. 1994;11:705–713. doi: 10.1111/j.1365-2958.1994.tb00348.x. [DOI] [PubMed] [Google Scholar]
- Lizewski SE, Lundberg DS, Schurr MJ. The transcriptional regulator AlgR is essential for Pseudomonas aeruginosa pathogenesis. Infect Immun. 2002;70:6083–6093. doi: 10.1128/IAI.70.11.6083-6093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Ostrovsky P, Nunn DN. LipC, a second lipase of Pseudomonas aeruginosa, is LipB and Xcp dependent and is transcriptionally regulated by pilus biogenesis components. Mol Microbiol. 1999;34:317–326. doi: 10.1046/j.1365-2958.1999.01601.x. [DOI] [PubMed] [Google Scholar]
- Mattick JS. Type IV pili and twitching motility. Annu Rev Microbiol. 2002;56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- Mayhew TM, Myklebust R, Whybrow A, Jenkins R. Epithelial integrity, cell death and cell loss in mammalian small intestine. Histol Histopathol. 1999;14:257–267. doi: 10.14670/HH-14.257. [DOI] [PubMed] [Google Scholar]
- Merz AJ, So M, Sheetz MP. Pilus retraction powers bacterial twitching motility. Nature. 2000;407:98–102. doi: 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
- Monteville MR, Konkel ME. Fibronectin-facilitated invasion of T84 eukaryotic cells by Campylobacter jejuni occurs preferentially at the basolateral cell surface. Infect Immun. 2002;70:6665–6671. doi: 10.1128/IAI.70.12.6665-6671.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morand PC, Bille E, Morelle S, Eugene E, Beretti JL, Wolfgang M, et al. Type IV pilus retraction in pathogenic Neisseria is regulated by the PilC proteins. Embo J. 2004;23:2009–2017. doi: 10.1038/sj.emboj.7600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier J, Vasselon T, Hellio R, Lesourd M, Sansonetti PJ. Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infect Immun. 1992;60:237–248. doi: 10.1128/iai.60.1.237-248.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nougayrede JP, Fernandes PJ, Donnenberg MS. Adhesion of enteropathogenic Escherichia coli to host cells. Cell Microbiol. 2003;5:359–372. doi: 10.1046/j.1462-5822.2003.00281.x. [DOI] [PubMed] [Google Scholar]
- O'Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- Orans J, Johnson MD, Coggan KA, Sperlazza JR, Heiniger RW, Wolfgang MC, Redinbo MR. Crystal structure analysis reveals Pseudomonas PilY1 as an essential calcium-dependent regulator of bacterial surface motility. Proc Natl Acad Sci U S A. 2010;107:1065–1070. doi: 10.1073/pnas.0911616107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicic V. Type IV pili: e pluribus unum? Mol Microbiol. 2008;68:827–837. doi: 10.1111/j.1365-2958.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- Pentecost M, Otto G, Theriot JA, Amieva MR. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog. 2006;2:e3. doi: 10.1371/journal.ppat.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramphal R, Small PM, Shands JW, Jr., Fischlschweiger W, Small PA., Jr. Adherence of Pseudomonas aeruginosa to tracheal cells injured by influenza infection or by endotracheal intubation. Infect Immun. 1980;27:614–619. doi: 10.1128/iai.27.2.614-619.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel T, Boxberger HJ, Meyer TF. Pilus biogenesis and epithelial cell adherence of Neisseria gonorrhoeae pilC double knock-out mutants. Mol Microbiol. 1995a;17:1057–1071. doi: 10.1111/j.1365-2958.1995.mmi_17061057.x. [DOI] [PubMed] [Google Scholar]
- Rudel T, Scheurerpflug I, Meyer TF. Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature. 1995b;373:357–359. doi: 10.1038/373357a0. [DOI] [PubMed] [Google Scholar]
- Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2005;171:1209–1223. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer F, Jiao H, Hindsgaul O, Wong WY, Irvin RT. Interaction between the pili of Pseudomonas aeruginosa PAK and its carbohydrate receptor beta-D-GalNAc(1-->4)beta-D-Gal analogs. Can J Microbiol. 1998;44:307–311. [PubMed] [Google Scholar]
- Sheth HB, Lee KK, Wong WY, Srivastava G, Hindsgaul O, Hodges RS, et al. The pili of Pseudomonas aeruginosa strains PAK and PAO bind specifically to the carbohydrate sequence beta GalNAc(1-4)beta Gal found in glycosphingolipids asialo-GM1 and asialo-GM2. Mol Microbiol. 1994;11:715–723. doi: 10.1111/j.1365-2958.1994.tb00349.x. [DOI] [PubMed] [Google Scholar]
- Skerker JM, Berg HC. Direct observation of extension and retraction of type IV pili. Proc Natl Acad Sci U S A. 2001;98:6901–6904. doi: 10.1073/pnas.121171698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong G, Parker D, Magargee M, Prince AS. The type III toxins of Pseudomonas aeruginosa disrupt epithelial barrier function. J Bacteriol. 2008;190:2814–2821. doi: 10.1128/JB.01567-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong G, Reddy B, Sokol S, Adamo R, Prince A. TLR2 is mobilized into an apical lipid raft receptor complex to signal infection in airway epithelial cells. J Clin Invest. 2004;113:1482–1489. doi: 10.1172/JCI20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom MS, Lory S. Cloning and expression of the pilin gene of Pseudomonas aeruginosa PAK in Escherichia coli. J Bacteriol. 1986;165:367–372. doi: 10.1128/jb.165.2.367-372.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundin C, Wolfgang MC, Lory S, Forsberg A, Frithz-Lindsten E. Type IV pili are not specifically required for contact dependent translocation of exoenzymes by Pseudomonas aeruginosa. Microb Pathog. 2002;33:265–277. doi: 10.1006/mpat.2002.0534. [DOI] [PubMed] [Google Scholar]
- Taha MK, Morand PC, Pereira Y, Eugene E, Giorgini D, Larribe M, Nassif X. Pilus-mediated adhesion of Neisseria meningitidis: the essential role of cell contact-dependent transcriptional upregulation of the PilC1 protein. Mol Microbiol. 1998;28:1153–1163. doi: 10.1046/j.1365-2958.1998.00876.x. [DOI] [PubMed] [Google Scholar]
- Takeya K, Amako K. A rod-shaped Pseudomonas phage. Virology. 1966;28:163–165. doi: 10.1016/0042-6822(66)90317-5. [DOI] [PubMed] [Google Scholar]
- Tang H, Kays M, Prince A. Role of Pseudomonas aeruginosa pili in acute pulmonary infection. Infect Immun. 1995;63:1278–1285. doi: 10.1128/iai.63.4.1278-1285.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. Bacterial adhesion to target cells enhanced by shear force. Cell. 2002;109:913–923. doi: 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- Turner LR, Lara JC, Nunn DN, Lory S. Mutations in the consensus ATP-binding sites of XcpR and PilB eliminate extracellular protein secretion and pilus biogenesis in Pseudomonas aeruginosa. J Bacteriol. 1993;175:4962–4969. doi: 10.1128/jb.175.16.4962-4969.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallis AJ, Yahr TL, Barbieri JT, Frank DW. Regulation of ExoS production and secretion by Pseudomonas aeruginosa in response to tissue culture conditions. Infect Immun. 1999;67:914–920. doi: 10.1128/iai.67.2.914-920.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Alphen LB, Bleumink-Pluym NM, Rochat KD, van Balkom BW, Wosten MM, van Putten JP. Active migration into the subcellular space precedes Campylobacter jejuni invasion of epithelial cells. Cell Microbiol. 2008;10:53–66. doi: 10.1111/j.1462-5822.2007.01014.x. [DOI] [PubMed] [Google Scholar]
- Whitchurch CB, Hobbs M, Livingston SP, Krishnapillai V, Mattick JS. Characterisation of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialised protein export system widespread in eubacteria. Gene. 1991;101:33–44. doi: 10.1016/0378-1119(91)90221-v. [DOI] [PubMed] [Google Scholar]
- Winther-Larsen HC, Wolfgang M, Dunham S, van Putten JP, Dorward D, Lovold C, et al. A conserved set of pilin-like molecules controls type IV pilus dynamics and organelle-associated functions in Neisseria gonorrhoeae. Mol Microbiol. 2005;56:903–917. doi: 10.1111/j.1365-2958.2005.04591.x. [DOI] [PubMed] [Google Scholar]
- Winther-Larsen HC, Wolfgang MC, van Putten JP, Roos N, Aas FE, Egge-Jacobsen WM, et al. Pseudomonas aeruginosa Type IV pilus expression in Neisseria gonorrhoeae: effects of pilin subunit composition on function and organelle dynamics. J Bacteriol. 2007;189:6676–6685. doi: 10.1128/JB.00407-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang M, Park HS, Hayes SF, van Putten JP, Koomey M. Suppression of an absolute defect in type IV pilus biogenesis by loss-of-function mutations in pilT, a twitching motility gene in Neisseria gonorrhoeae. Proc Natl Acad Sci U S A. 1998;95:14973–14978. doi: 10.1073/pnas.95.25.14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang M, van Putten JP, Hayes SF, Dorward D, Koomey M. Components and dynamics of fiber formation define a ubiquitous biogenesis pathway for bacterial pili. Embo J. 2000;19:6408–6418. doi: 10.1093/emboj/19.23.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang MC, Lee VT, Gilmore ME, Lory S. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell. 2003;4:253–263. doi: 10.1016/s1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- Yu L, Lee KK, Hodges RS, Paranchych W, Irvin RT. Adherence of Pseudomonas aeruginosa and Candida albicans to glycosphingolipid (Asialo-GM1) receptors is achieved by a conserved receptor-binding domain present on their adhesins. Infect Immun. 1994;62:5213–5219. doi: 10.1128/iai.62.12.5213-5219.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolfaghar I, Evans DJ, Fleiszig SM. Twitching motility contributes to the role of pili in corneal infection caused by Pseudomonas aeruginosa. Infect Immun. 2003;71:5389–5393. doi: 10.1128/IAI.71.9.5389-5393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Subsurface twitching motility measured as the zone of bacterial expansion at the agar-plastic interface. Mutants lacking TFP (pilA, pilY1 and fimU-pilE) or expressing non-retractile TFP due to deletion of the pilus retraction gene pilT, show an absolute defect in twitching motility. Data are represented as mean +/− SEM (n=3).