

Abstract
Despite the high prevalence of pituitary adenomas they are invariably benign, indicative of unique intrinsic mechanisms controlling pituitary cell proliferation. Cellular senescence is characterized by a largely irreversible cell cycle arrest and constitutes a strong anti-proliferative response, which can be triggered by DNA damage, chromosomal instability and aneuploidy, loss of tumor suppressive signaling or oncogene activation. In vivo senescence is an important protective mechanisms against cancer. Here we discuss prospective mechanisms underlying senescence-associated molecular pathways activated in benign pituitary adenomas. Both deletion and overexpression of pituitary tumor transforming gene (Pttg) promote chromosomal instability and aneuploidy. Pttg deletion abrogates tumor development by activating p53/p21-dependent senescence pathways. Abundant PTTG in GH-secreting pituitary adenomas also triggers p21-dependent senescence. Pituitary p21 may therefore safeguard against further chromosomal instability by constraining pituitary tumor growth. These observations point to senescence as a target for effective therapy for both tumor silencing and growth restraint towards development of pituitary malignancy
The pituitary gland is a key regulator of homeostatic endocrine balance. Five highly differentiated cell types: somatotrophs, gonadotrophs, lactotrophs, thyrotrophs and corticosrophs synthesize and release specific hormones in response to stimulatory and inhibitory central and peripheral signals. Pituitary cells proliferate slowly, apoptosis is low, and high levels of differentiation and specialization occur at the expense of lowered renewal rate. The gland responds in a dynamic and plastic fashion to constantly changing hormonal and metabolic environments. Responding to endogenous and endogenous signals, the gland undergoes reversible changes in cell growth leading to hyperplasia and excess pituitary hormone secretion usually associated with true adenoma formation (Melmed, 2003).
Pituitary adenomas are remarkably common in the general population: sporadic pituitary adenomas are present in 25% of autopsy specimens. Tumors may arise from any of five pituitary cell subtypes (Lania et al., 2006, Fernandez et al., 2009). PRL-producing adenomas are the most common, while approximately 30% of all adenomas are not associated with hormone hypersecretion, and most of these are immunopositive for LH/FSH, ACTH- and GH-producing adenomas each account for 10-15% of all adenomas, while TSH-producing adenomas are extremely rare. Pituitary chromosome instability, and epigenetic changes including methylation of tumor suppressor p16 promoter are considered early markers of pituitary tumorigenesis (Farrell, 2006, Farrell and Clayton, 2003). Pituitary tumors rarely progress to true carcinoma, however some tumors show invasive or recurrent growth. Mitotic activity is low even in aggressive pituitary adenomas, in contrast to tumors arising from more rapidly replicating tissues (Melmed, 2003).
Several factors may account for the biological indolence of these neoplasms. Progression to a malignant phenotype requires multiple oncogenic mutations, which rarely occur. Indeed, with the exception of gsp oncogene observed in a subset of somatotrophinomas (Landis et al., 1989), activating mutations of oncogenes, or mutations resulting in inactivation of tumor suppressor genes have not been found. Remarkably, pituitary adenomas, especially silent microadenomas, often appear to stop growing and prolactinomas can even resolve itself (Levy and Lightman, 2003). It is plausible that intrinsic properties of highly differentiated and specialized pituitary cells limit their ability for uncontrolled proliferation.
Senescence
In 1961 Hayflick and Moorhead described the process of cellular senescence whereby human fibroblasts cultured in vitro had a limited number of divisions before entering a state of irreversible proliferation arrest (Hayflick and Moorhead, 1961). In eukaryotic cells each chromosome shortens telomeres during every round of DNA replication (Harley et al., 1990). The structure of telomeres with replicative sequences functions as a cap to prevent chromosome end fusions and genomic instability. In somatic cells proliferating continuously, loss of telomeres provokes DNA damage response signaling pathway activation leading to irreversible proliferation arrest known as “replicative” age-related senescence(Kim Sh et al., 2002). Proliferative block can also occur without telomere shortening, as a result of oxidative or genetic stress including radiation-, UV- or chemotherapy-induced DNA damage, disrupted chromatin organization, aneuploidy and chromosomal instability (Sharpless and DePinho, 2004). This type of senescence was termed “premature” senescence. Genetic stress triggers DNA damage response followed by accumulation of p53 and p53-mediated induction of the Cdk inhibitor p21 that blocks Rb phosphorylation and enforces G1 cell cycle arrest (Schmitt et al., 2002, Sharpless and DePinho, 2004, Campisi, 2005). Activation of senescence pathways signals cells to exit the cell cycle and cease proliferation. Senescent cells exhibit particular flat morphology and express specific senescence-associated β-galactosidase (SA-β-gal) activity (Dimri et al., 1995, Collado et al., 2007).
Cellular senescence occurs in vivo in human tumors and in mouse tumor models. Senescence occurs in benign or early stage, but not in advanced tumors, and several lines of evidence show that cell–cycle arrest observed in non-growing benign lesions is triggered by activated oncoproteins like BRAF and RAS (Campisi, 2001, Serrano et al., 1997, Braig et al., 2005, Bartkova et al., 2006) supporting in vitro observations that activation of these pathways leads to an initial burst of proliferation followed by DNA replication stress and cellular senescence. This type of senescence was termed oncogene-induced senescence (OIS). Replicative, premature and oncogene-induced senescence share common characteristics such as increased SA-β-gal activity and condensation of individual chromosomes into distinct heterochromosome bodies, called senescence-associated heterochromatic foci (SAHF)(Narita et al., 2003, Sharpless and DePinho, 2004). Senescence is accompanied by upregulation of Cdk inhibitors including p15, p16 and p21, a set of well-known tumor suppressors that are often inactivated in human cancers, and Rb hypophosphorylation, (Arzt et al., 2009). OIS is specifically associated with the secretory phenotype, activating a plethora of factors including key components of Wnt, IGF1, transforming growth factor-β (TGFβ), plasmin, and IL6/ C/EBP pathways collectively termed senescence-messaging secretome (SMS). SMS allows for signal modulation by intracellular communication, implementing and fine-tuning the senescence response (Kuilman and Peeper, 2009) (Fig.1).
Fig. 1.
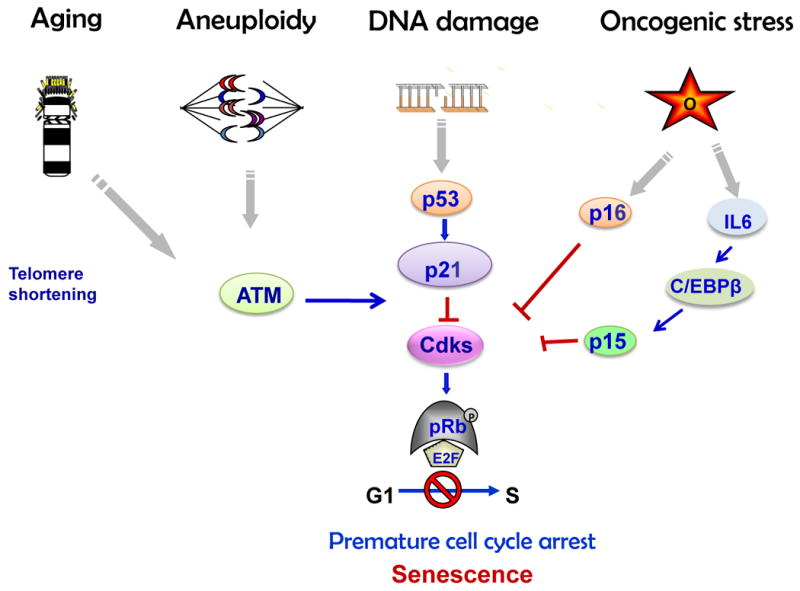
Senescence pathways (Adapted from: Chesnokova et al. Pituitary Today II, Front Horm Res. Basel, Karger 2010, v.38, pp.7-14)
Thus, in addition to cell death programmes such as apoptosis and autophagia, senescence is increasingly recognized as a potent barrier for the development of true malignancies (Collado et al., 2005, Braig et al., 2005, Michaloglou et al., 2005). Mechanisms effecting cell senescence act to buffer the cell from pro-proliferative signals (Michaloglou et al., 2005) and function to protect against oncogenic transformation, thereby suppressing unscheduled proliferation of damaged and early neoplastic cells.
PTTG
Pituitary tumor transforming gene (Pttg), the index mammalian securin, is a component of the spindle checkpoint, controlling faithful sister chromatid separation during metaphase(Zou et al., 1999). Pttg exhibits oncogene properties and facilitates cell cycle progression (Pei and Melmed, 1997, Zhang et al., 1999b). Pttg over-expression causes cell transformation in vitro (Yu et al., 2000, Yu et al., 2003), promotes tumor formation in nude mice, and activates angiogenesis (Ishikawa et al., 2001, Heaney et al., 1999). Pttg initially isolated from pituitary tumor cells, is abundantly expressed in pituitary, breast, thyroid, endometrial, oesophageal and colorectal tumors, and levels of PTTG expression are concurrent with tumor invasiveness, recurrence and poor prognosis(Vlotides et al., 2007). Pttg was identified as a key signature genes associated with tumor metastasis (Ramaswamy et al., 2003). PTTG protein is induced early in experimental oestorgen-induced pituitary tumors (Heaney et al., 1999). PTTG is required for pituitary tumorigenesis as pituitary-directed transgenic Pttg over-expression results in focal pituitary hyperplasia and adenoma formation (Abbud R, 2003). Pttg enhances cell proliferation by activating basic fibroblast growth factor (bFGF), c-myc and CCND3 (Zhang et al., 1999a). Both Pttg overexpression and deletion result in dysregulated G2/M checkpoint surveillance, increased aneuploidy and chromosomal instability in vitro and in vivo. (Kim et al., 2007, Kim et al., 2005) highlighting the requirement for intracellular securin equilibrium for maintenance of chromosomal stability (Bernal et al., 2008).
PTTG deletion results in pituitary gland senescence
Mice lacking Pttg are viable and fertile, and exhibit testicular and splenic hypoplasia, thymic hyperplasia, and pancreatic β cell hypoplasia (Wang et al., 2001, Wang et al., 2003). Pttg-deficient pituitary glands are hypoplastic, and Pttg-null multipotent adult progenitor cells (Rubinek et al., 2007) and pituitary cells exhibit intracellular p53 accumulation and p21 induction. High pituitary p21 levels observed in the absence of PTTG were associated with suppressed Cdk2 activity, decreased RB phosphorylation and cyclin A expression, all required for cell cycle progression(Chesnokova et al., 2007, Chesnokova et al., 2005). Pttg deletion also leads to extensive pituitary cell aneuploidy, which activated DNA damage signaling pathways triggering ARF/p53/p21 senescence evidenced by increased SA-β-gal activity in Pttg-/- pituitary gland (Chesnokova et al., 2008) (Fig.2A,B)
Fig.2.
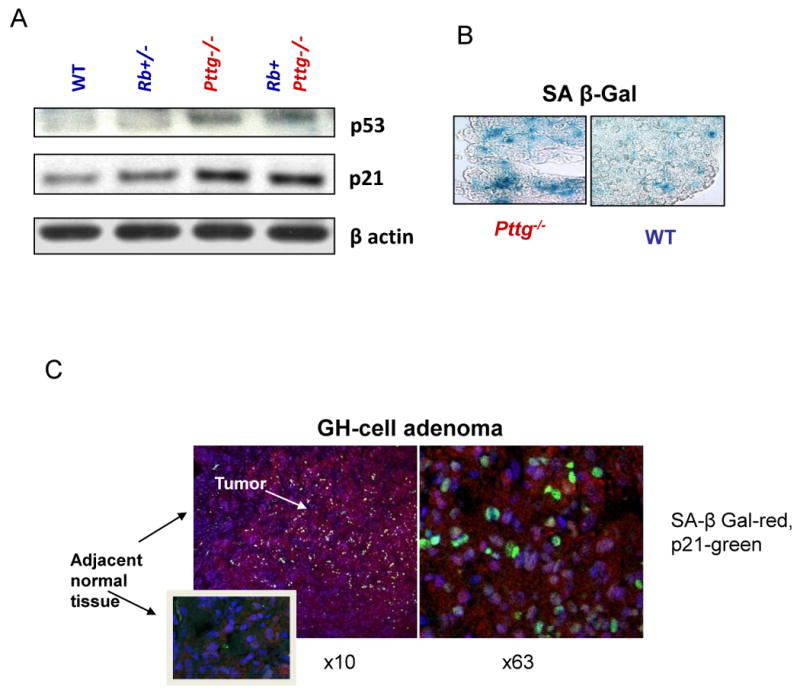
Pttg deletion in murine pituitary gland and Pttg overexpression in human GH-cell adenoma result in activation of p53/p21 senescence pathway and senescence. A) Induced p53 and p21, and B) increased SA-β-gal activity (blue) in Pttg-deficient pituitary glands (Chesnokova, Cancer Res 2007); C) Confocal image of double fluorescence immunohistochemistry of p21 (green) and β-galactosidase (red) proteins co-expression in human pituitary adenomatous but not in normal adjacent tissue. Right panel-high resolution (× 63) image of the same slide (Chesnokova PNAS 2008).
Rb+/- mice develop pituitary tumors with high penetrance. Crossbreeding Rb+/- mice with Pttg-/- animals abrogated Rb+/- tumor formation with decreased tumor number and size, confirming the Pttg requirement for pituitary tumorigenesis. Abundant intra-nuclear p21 expression underlies decreased pituitary tumor development in Pttg-deficient Rb+/- mice (Chesnokova et al., 2007). Indeed, when p21 was deleted, the number of Rb+/-Pttg-/-p21-/- animals developing pituitary tumors reverted to the high tumor penetrance observed in Rb+/- mice (Chesnokova et al., 2008). p21, a transcriptional target of p53, is induced in response to a broad spectrum of cellular stresses, including DNA damage or oncogene expression, and acts to constrain the cell cycle in unstable or aneuploid cells (Shen et al., 2005, Barboza et al., 2006). p21 induction also triggers senescence, which facilitates Rb hypophosphorylation and tumor growth arrest (Brugarolas et al., 1999, Gartel and Tyner, 1998, Sharpless and DePinho, 2004, Mooi and Peeper, 2006).
PTTG overexpression results in pituitary tumor senescence
Pttg deletion resulted in p21 induction observed mostly in GH-producing mouse Pttg-null pituitary cells(Chesnokova et al., 2007). Aneuploidy, a potent inducer of cellular p21 levels (Shen et al., 2005, Barboza et al., 2006), is induced by and both Pttg deletion and overexpression. We therefore tested effects of Pttg overexpression and silencing on pituitary cell p21 levels. We transiently transfected rat GH3 pituitary cells with plasmids expressing EGFP-PTTG, and immunocytochemistry showed enhanced p21 protein expression in these cells as compared with vector-transfected cells (Chesnokova et al., 2008). Transfected cells were also sorted by flow cytometry, and increased p21 mRNA levels were observed in EGFP-PTTG-positive GH3 cells relative to controls. We measured the protein kinase mutated in ataxia telangiectasia (ATM), essential in sensing aneuploidy and DNA damage. ATM and p21 cooperate to impede aneuploidy and maintain chromosomal stability (Shen et al., 2005). In EGFP-PTTG-positive GH3 cells ATM mRNA levels were induced 9-fold as compared to EGFP-only transfected cells(Chesnokova et al., 2008). Silencing of Pttg in GH3 cells with Pttg siRNA also enhanced p21 mRNA levels thus confirming our in vivo observation showing p21 up-regulation in the Pttg-null pituitary gland(Chesnokova et al., 2008). Thus, both Pttg deficiency and overexpression trigger intranuclear p21 expression.
Pttg is induced in all human pituitary tumor subtypes (Vlotides et al., 2007). When compared to other types of pituitary adenomas, GH-cell and PRL-cell tumors showed both higher expression of Pttg and levels of aneuploidy (Uccella et al., 2005). We (Chesnokova et al., 2008) and others (Neto et al., 2005) demonstrated high intra-nuclear p21 expression in GH-secreting adenomas. In our experiments, of 32 GH-producing adenomas, 25 tested strongly positive, and 7 weakly positive for p21 immunoreactivity, while p21 expression was very low in normal pituitary sections. Intra-nuclear p21 was not detected in four human GH-producing pituitary carcinomas (Chesnokova et al., 2008).
GH-secreting human pituitary adenomas expressed several markers of senescence including high levels of both p21 and ATM. SA-β-gal activity was very low in normal pituitary sections, while GH secreting adenomas were strongly positive for this senescence marker. Accordingly, intra-nuclear p21 expression was observed in paraffin slides derived from corresponding pituitary tumors (Chesnokova et al., 2008). Double-labeling of GH secreting adenoma specimens with antibodies to p21 and β galactosidase showed that all 25 p21-expressing adenomas were positive for β galactosidase, and p21 co-localized with β galactosidase in adenomatose tissue, while adjacent normal tissue did not express either of these proteins (Fig.2C)(Chesnokova et al., 2008).
The results showing induced intra-nuclear p21 in pituitary tumor cells also overexpressing Pttg are seemingly inconsistent with the conclusion that Pttg absence restrains pituitary tumor development by activating p21 senescent pathways. Pttg behaves as an oncogene (Vlotides et al., 2007), and is permissive for pituitary tumor formation (Abbud et al., 2005, Chesnokova et al., 2005). Both loss of Pttg as well as Pttg overexpression result in aneuploidy (Chesnokova et al., 2007, Wang et al., 2001, Yu et al., 2003), and Pttg overexpression triggers genetic instability characterized by DNA breakage (Kim et al., 2007, Kim et al., 2005). High p21 levels were observed in Pttg-null anterior pituitary cells, and also in GH-secreting GH3 cells where Pttg was either silenced or overexpressed, as well as in human GH-secreting adenomas overexpressing Pttg. Pttg-null pituitary cells exhibit activated DNA damage pathway (Chesnokova et al., 2007). In tumors, high PTTG levels may initially mediate excessive proliferation, and lead to defective DNA replication and aneuploidy (Uccella et al., 2005). Accordingly, ATM up-regulation, indicative of aneuploidy or/and DNA damage, is evident in PTTG-overexpressing GH3 cells and GH-secreting human pituitary tumors (Chesnokova et al., 2008) (Fig.3).
Fig.3.
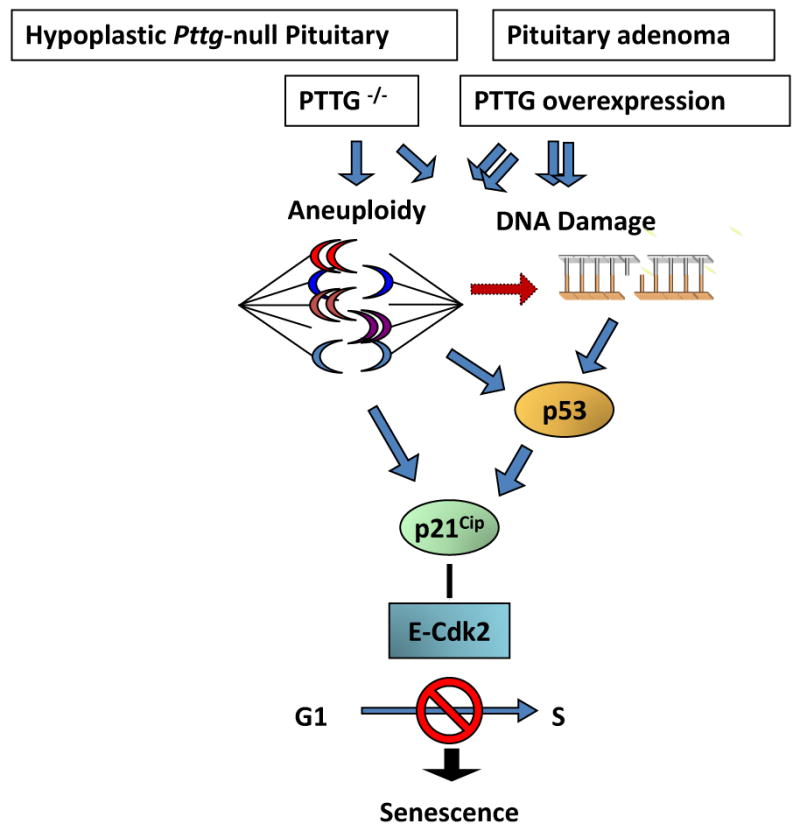
Proposed model for p21-induced senescence in the hypoplastic Pttg-null pituitary gland and Pttg-overexpressing pituitary adenomas (Chesnokova, PNAS 2008).
p21 is strongly induced in aneuploid cells and functions to preserve chromosomal stability in affected cells by suppressing cell cycle progression (Barboza et al., 2006). The evidence thus suggests that pituitary p21 expression is associated with pituitary cell senescence, and leads to restrained cell growth in the hypoplastic Pttg-null pituitary. In pituitary tumors derived from somatotroph lineage, intranuclear p21 acts as a tumor suppressor on the background of genomic instability (Shen et al., 2005) and also triggers senescence.
Mechanisms underlying specific endocrine cell sensitivity to globally disrupted PTTG levels are unclear. Of note, the evolutionarily conserved GH-IGF1 growth pathway is critical for lifespan regulation, and when DNA damage and genomic instability accumulate, this axis is attenuated (Schumacher et al., 2008, Niedernhofer et al., 2006). These observations undersore the sensitivity of GH-secreting cells to PTTG-induced genomic instability, and resultant p21 induction and senescence may constrain tumor growth. This postulate is consistent with experimental and clinical observations that somatotrophs are more sensitive to aging, radiation damage or compressive damage than other pituitary cell types (Darzy and Shalet, 2008, Chandrashekar et al., 2007). High levels of Pttg behaving as a securin protein, cause defective metaphase-anaphase progression and chromosomal instability and promote pituitary tumor formation. Activation of pituitary DNA damage pathways triggers p21, a barrier to tumor growth (Halazonetis et al., 2008), which in turn may restrain further growth and malignant transformation of pituitary tumors.
Pituitary cell growth regulation by IL-6 also underlies the involvement of cytokines as factors controlling pituitary cell division (Arzt, 2001, Arzt et al., 2009). New findings of the role of cytokines, and particular of IL-6 in OIS(Kuilman and Peeper, 2009), suggest the possible contribution of endogenous IL-6 in development of pituitary adenoma senescence, which, as well as its induction by aberrant intracellular PTTG levels, may explain the benign nature of these abundant tumors(Arzt et al., 2009).
Thus, pituitary adenomas constitute faithful in vivo models of senescence. Pituitary cells are one of the few epithelial cell types that do not readily undergo malignant transformation. Pituitary cells undergo premature senescence enabling escape from the proliferative pressure of oncogenes, hormones and transformation factors. Senescence restrains proliferation but allows the cell to remain viable and perform its physiological function invaluable for homeostasis control. As senescence is considered an important tumor protection barrier, (Campisi, 2005, Collado et al., 2005, Mooi and Peeper, 2006, Collado et al., 2007, Cichowski and Hahn, 2008), understanding mechanisms underlying the ability of pituitary cells to escape aggressive growth and malignant transformation may provide important insights into cancer-restraining pathways and present new opportunities for sub-cellular therapeutic approaches.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbud RCV, Heney T, Takuni I, Valeria-Castro A, Yu R, Melmed S. Molecular Pathogenesis. Abstracts, 8th International Pituitary Congress; June 22-25,2003; New York. 2003. [Google Scholar]
- Abbud RA, Takumi I, Barker EM, Ren SG, Chen DY, Wawrowsky K, Melmed S. Early multipotential pituitary focal hyperplasia in the alpha-subunit of glycoprotein hormone-driven pituitary tumor-transforming gene transgenic mice. Mol Endocrinol. 2005;19:1383–91. doi: 10.1210/me.2004-0403. [DOI] [PubMed] [Google Scholar]
- Arzt E. gp130 cytokine signaling in the pituitary gland: a paradigm for cytokine-neuro-endocrine pathways. J Clin Invest. 2001;108:1729–33. doi: 10.1172/JCI14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzt E, Chesnokova V, Stalla GK, Melmed S. Pituitary adenoma growth: a model for cellular senescence and cytokine action. Cell Cycle. 2009;8:677–8. doi: 10.4161/cc.8.5.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G. p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci U S A. 2006;103:19842–7. doi: 10.1073/pnas.0606343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bernal JA, Roche M, Mendez-Vidal C, Espina A, Tortolero M, Pintor-Toro JA. Proliferative potential after DNA damage and non-homologous end joining are affected by loss of securin. Cell Death Differ. 2008;15:202–12. doi: 10.1038/sj.cdd.4402254. [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Moberg K, Boyd SD, Taya Y, Jacks T, Lees JA. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc Natl Acad Sci U S A. 1999;96:1002–7. doi: 10.1073/pnas.96.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11:S27–31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309:886–7. doi: 10.1126/science.1116801. [DOI] [PubMed] [Google Scholar]
- Chandrashekar V, Dawson CR, Martin ER, Rocha JS, Bartke A, Kopchick JJ. Age-related alterations in pituitary and testicular functions in long-lived growth hormone receptor gene-disrupted mice. Endocrinology. 2007;148:6019–25. doi: 10.1210/en.2007-0837. [DOI] [PubMed] [Google Scholar]
- Chesnokova V, Kovacs K, Castro AV, Zonis S, Melmed S. Pituitary hypoplasia in Pttg-/- mice is protective for Rb+/- pituitary tumorigenesis. Mol Endocrinol. 2005;19:2371–9. doi: 10.1210/me.2005-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova V, Zonis S, Kovacs K, Ben-Shlomo A, Wawrowsky K, Bannykh S, Melmed S. p21(Cip1) restrains pituitary tumor growth. Proc Natl Acad Sci U S A. 2008;105:17498–503. doi: 10.1073/pnas.0804810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova V, Zonis S, Rubinek T, Yu R, Ben-Shlomo A, Kovacs K, Wawrowsky K, Melmed S. Senescence mediates pituitary hypoplasia and restrains pituitary tumor growth. Cancer Res. 2007;67:10564–72. doi: 10.1158/0008-5472.CAN-07-0974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichowski K, Hahn WC. Unexpected pieces to the senescence puzzle. Cell. 2008;133:958–61. doi: 10.1016/j.cell.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, Beach D, Serrano M. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- Darzy KH, Shalet SM. Hypopituitarism following radiotherapy. Pituitary. 2008 doi: 10.1007/s11102-008-0088-4. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell WE. Pituitary tumours: findings from whole genome analyses. Endocr Relat Cancer. 2006;13:707–16. doi: 10.1677/erc.1.01131. [DOI] [PubMed] [Google Scholar]
- Farrell WE, Clayton RN. Epigenetic change in pituitary tumorigenesis. Endocr Relat Cancer. 2003;10:323–30. doi: 10.1677/erc.0.0100323. [DOI] [PubMed] [Google Scholar]
- Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK) Clin Endocrinol (Oxf) 2009 doi: 10.1111/j.1365-2265.2009.03667.x. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. The growth-regulatory role of p21 (WAF1/CIP1) Prog Mol Subcell Biol. 1998;20:43–71. doi: 10.1007/978-3-642-72149-6_4. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Heaney AP, Horwitz GA, Wang Z, Singson R, Melmed S. Early involvement of estrogen-induced pituitary tumor transforming gene and fibroblast growth factor expression in prolactinoma pathogenesis. Nat Med. 1999;5:1317–21. doi: 10.1038/15275. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Heaney AP, Yu R, Horwitz GA, Melmed S. Human pituitary tumor-transforming gene induces angiogenesis. J Clin Endocrinol Metab. 2001;86:867–74. doi: 10.1210/jcem.86.2.7184. [DOI] [PubMed] [Google Scholar]
- Kim D, Pemberton H, Stratford AL, Buelaert K, Watkinson JC, Lopes V, Franklyn JA, McCabe CJ. Pituitary tumour transforming gene (PTTG) induces genetic instability in thyroid cells. Oncogene. 2005;24:4861–6. doi: 10.1038/sj.onc.1208659. [DOI] [PubMed] [Google Scholar]
- Kim DS, Franklyn JA, Smith VE, Stratford AL, Pemberton HN, Warfield A, Watkinson JC, Ishmail T, Wakelam MJ, McCabe CJ. Securin induces genetic instability in colorectal cancer by inhibiting double-stranded DNA repair activity. Carcinogenesis. 2007;28:749–59. doi: 10.1093/carcin/bgl202. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kaminker P, Campisi J. Telomeres, aging and cancer: in search of a happy ending. Oncogene. 2002;21:503–11. doi: 10.1038/sj.onc.1205077. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–6. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- Lania AG, Mantovani G, Spada A. Mechanisms of disease: Mutations of G proteins and G-protein-coupled receptors in endocrine diseases. Nat Clin Pract Endocrinol Metab. 2006;2:681–93. doi: 10.1038/ncpendmet0324. [DOI] [PubMed] [Google Scholar]
- Levy A, Lightman S. Molecular defects in the pathogenesis of pituitary tumours. Front Neuroendocrinol. 2003;24:94–127. doi: 10.1016/s0091-3022(03)00012-8. [DOI] [PubMed] [Google Scholar]
- Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112:1603–18. doi: 10.1172/JCI20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, Van Der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Mooi WJ, Peeper DS. Oncogene-induced cell senescence--halting on the road to cancer. N Engl J Med. 2006;355:1037–46. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Neto AG, McCutcheon IE, Vang R, Spencer ML, Zhang W, Fuller GN. Elevated expression of p21 (WAF1/Cip1) in hormonally active pituitary adenomas. Ann Diagn Pathol. 2005;9:6–10. doi: 10.1053/j.anndiagpath.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, Van Leeuwen W, Theil AF, Vermeulen W, Van Der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–43. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG) Mol Endocrinol. 1997;11:433–41. doi: 10.1210/mend.11.4.9911. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Rubinek T, Chesnokova V, Wolf I, Wawrowsky K, Vlotides G, Melmed S. Discordant proliferation and differentiation in pituitary tumor-transforming gene-null bone marrow stem cells. Am J Physiol Cell Physiol. 2007;293:C1082–92. doi: 10.1152/ajpcell.00145.2007. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–46. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- Schumacher B, Garinis GA, Hoeijmakers JH. Age to survive: DNA damage and aging. Trends Genet. 2008 doi: 10.1016/j.tig.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Depinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004;113:160–8. doi: 10.1172/JCI20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KC, Heng H, Wang Y, Lu S, Liu G, Deng CX, Brooks SC, Wang YA. ATM and p21 cooperate to suppress aneuploidy and subsequent tumor development. Cancer Res. 2005;65:8747–53. doi: 10.1158/0008-5472.CAN-05-1471. [DOI] [PubMed] [Google Scholar]
- Uccella S, Tibiletti MG, Bernasconi B, Finzi G, Oldrini R, Capella C. Aneuploidy, centrosome alteration and securin overexpression as features of pituitary somatotroph and lactotroph adenomas. Anal Quant Cytol Histol. 2005;27:241–52. [PubMed] [Google Scholar]
- Vlotides G, Eigler T, Melmed S. Pituitary tumor-transforming gene: physiology and implications for tumorigenesis. Endocr Rev. 2007;28:165–86. doi: 10.1210/er.2006-0042. [DOI] [PubMed] [Google Scholar]
- Wang Z, Moro E, Kovacs K, Yu R, Melmed S. Pituitary tumor transforming gene-null male mice exhibit impaired pancreatic beta cell proliferation and diabetes. Proc Natl Acad Sci U S A. 2003;100:3428–32. doi: 10.1073/pnas.0638052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yu R, Melmed S. Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol Endocrinol. 2001;15:1870–9. doi: 10.1210/mend.15.11.0729. [DOI] [PubMed] [Google Scholar]
- Yu R, Heaney AP, Lu W, Chen J, Melmed S. Pituitary tumor transforming gene causes aneuploidy and p53-dependent and p53-independent apoptosis. J Biol Chem. 2000;275:36502–5. doi: 10.1074/jbc.C000546200. [DOI] [PubMed] [Google Scholar]
- Yu R, Lu W, Chen J, McCabe CJ, Melmed S. Overexpressed pituitary tumor-transforming gene causes aneuploidy in live human cells. Endocrinology. 2003;144:4991–8. doi: 10.1210/en.2003-0305. [DOI] [PubMed] [Google Scholar]
- Zhang X, Horwitz GA, Heaney AP, Nakashima M, Prezant TR, Bronstein MD, Melmed S. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab. 1999a;84:761–7. doi: 10.1210/jcem.84.2.5432. [DOI] [PubMed] [Google Scholar]
- Zhang X, Horwitz GA, Prezant TR, Valentini A, Nakashima M, Bronstein MD, Melmed S. Structure, expression, and function of human pituitary tumor-transforming gene (PTTG) Mol Endocrinol. 1999b;13:156–66. doi: 10.1210/mend.13.1.0225. [DOI] [PubMed] [Google Scholar]
- Zou H, Mcgarry TJ, Bernal T, Kirschner MW. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science. 1999;285:418–22. doi: 10.1126/science.285.5426.418. [DOI] [PubMed] [Google Scholar]