

Abstract
Short/branched chain acyl-CoA dehydrogenase (SBCAD) deficiency, also known as 2-methylbutyryl-CoA dehydrogenase deficiency, is a recently described autosomal recessive disorder of isoleucine metabolism. Most patients reported thus far have originated from a founder mutation in the Hmong Chinese population. While the first reported patients had severe disease, most of the affected Hmong have remained asymptomatic. In this study we describe 11 asymptomatic non-Hmong patients brought to medical attention by elevated C5-carnitine found by newborn screening and one discovered because of clinical symptoms. The diagnosis of SBCAD deficiency was determined by metabolite analysis of blood, urine, and fibroblast samples. PCR and bidirectional sequencing were performed on genomic DNA from five of the patients covering the entire SBCAD (ACADSB) gene sequence of 11 exons. Sequence analysis of genomic DNA from each patient identified variations in the SBCAD gene not previously reported. E. coli expression studies revealed that the missense mutations identified lead to inactivation or instability of the mutant SBCAD enzymes. These findings confirm that SBCAD deficiency can be identified through newborn screening by acylcarnitine analysis. Our patients have been well without treatment and call for careful follow-up studies to learn the true clinical impact of this disorder.
Keywords: Branched Chain Acyl Coenzyme A Dehydrogenase, 2-Methylbutyryl Coenzyme A dehydrogenase, organic acidemia, isoleucine metabolism, acyl-CoA dehydrogenase, newborn screening
1.1 INTRODUCTION
The acyl-CoA dehydrogenases (ACADs) are a family of closely related mitochondrial enzymes that catalyzes the removal of hydrogen atoms from the α and β positions (also known as β-oxidation) of straight or branched chain acyl-CoA esters in the metabolism of amino acids and fatty acids, transferring electrons to electron transferring flavoprotein [1–8]. At least 9 members of this family have been described so far. Short chain acyl-CoA dehydrogenase (SCAD), medium chain acyl-CoA dehydrogenase (MCAD), long chain acyl-CoA dehydrogenase (LCAD), very long chain acyl-CoA dehydrogenase (VLCAD), and acyl-CoA dehydrogenase 9 (ACAD9) all catalyze oxidation of acyl-CoAs in mitochondrial fatty acid β-oxidation. Human ACAD9 plays a unique role in the mitochondrial β-oxidation of unsaturated fatty acids [9]. Glutaryl-CoA dehydrogenase is active in the lysine degradation, while isovaleryl-CoA dehydrogenase, short branched chain acyl-CoA dehydrogenase (SBCAD, also known as 2-methylbutyryl-CoA dehydrogenase), and isobutyryl-CoA dehydrogenase (IBD, also designated ACAD8) function in the catabolism of leucine, isoleucine, and valine intermediates, respectively [5, 7, 10–16]. SBCAD, like all ACADs except VLCAD and ACAD9, is a homotetrameric mitochondrial enzyme catalyzing alpha and beta dehydrogenation of acyl-CoA derivatives with greatest activity toward straight/branched short chain acyl-CoA metabolites and specifically toward 2-methybutyryl-CoA, catalyzing the third step of the isoleucine catabolic pathway [13].
Deficiencies in all of these enzymes except LCAD have been associated with human disease [7, 14, 16–24]. SBCAD deficiency (OMIM 600301/610006) has only recently been described. The first two patients were identified due to significant medical problems and were shown to have SBCAD deficiency [16, 25]. Subsequently, this disorder has been shown, through population and newborn screening studies, to be common in individuals of Hmong ancestry [26, 27.] A minority of patients in the Hmong population diagnosed through newborn screening was reported to have transient muscle hypotonia, but the Hmong patients have otherwise been well. Additional non-Hmong patients with SBCAD deficiency have also been identified through newborn screening and remain well, as have others diagnosed through metabolic evaluations for clinical symptoms [26, 28–30]. One patient was reported to have autism and mental retardation, but causation could not be established [31]. Thus, the significance of SBCAD deficiency remains unclear.
In this study we describe 11 patients from various medical centers identified by newborn screening because of elevated C5-acylcarnitine levels. An additional patient who was born before acylcarnitine analysis was added to the respective State’s newborn screening program was identified during an evaluation for clinical symptoms. Further diagnostic testing, including plasma acylcarnitines, plasma amino acids, urine organic acids, urine acylglycines, and fibroblast acylcarnitine profiling, substantiated SBCAD deficiency in all 12 patients. Sequencing of genomic DNA from 5 patient fibroblasts identified mutations predicted to be deleterious in four of the patients identified by newborn screening and in the symptomatic patient. In vitro mutagenesis and expression studies of point mutations in E. coli confirmed that they lead to an inactive or unstable SBCAD protein. Individuals detected through newborn screening programs remain well with the oldest being 4 years old, suggesting that this deficiency may be a biochemical phenotype rather than a disease.
1.2 MATERIALS AND METHODS
1.2.1Urine Organic Acid, Acylglycine, and Acylcarnitine, Plasma Acylcarnitine, and Fibroblast In Vitro Probe Analyses
Acylcarnitine analyses from dried blood spots, plasma, and urine samples, urine organic acid and acylglycine analyses, and the in vitro probe assay in fibroblast cultures were performed following established procedures [32].
1.2.2 Molecular Genetic Analysis
Molecular genetic analysis of the ACADSB gene was performed using genomic DNA extracted from fibroblasts samples [16]. All 10 exons and 18 intron-exon junctions were amplified by PCR and DNA analysis was performed by dye-terminator sequencing PCR-amplified fragments on an Applied Biosystems 3730 DNA Analyzer (Foster City, CA) at the University of Pittsburgh Genomics and Proteomics Core Laboratory. Primer sequences for exon amplification and sequencing are available upon request.
1.2.3 Computational Molecular Modeling
Computational modeling and visualization of human SBCAD were performed on a Silicon Graphics Fuel workstation (Mountain View, CA) using the Insight II 2005 software package (Accelrys Technologies, San Diego, CA) and the published atomic coordinates of human SBCAD (PDB code: 2JIF) (Pike, et al, 2007, GenBank entry).
1.2.4 SBCAD Mutagenesis and Expression
Patient mutations c.443C>T and c.1159G>A were introduced into a wild type SBCAD expression vector with QuickChange II mutagenesis Kit (Stratagene, La Jolla, CA) and expressed in E. coli as previously described [33]. The presence of SBCAD cross-reactive material was then determined by western blot analysis of crude fibroblast extracts [34].
1.2.5 Enzyme Activity Assay
Enzyme activity was measured with the anaerobic electron transfer flavoprotein (ETF) fluorescence reduction assay using an LS50B fluorescence spectrophotometer from PerkinElmer Life Sciences (Wellesley, MA) with a heated cuvette block set to 32°C as described [13]. Acyl-CoA substrates were purchased from Sigma (St. Louis, MO). The reaction was started with the addition of the CoA ester substrate to give a final concentration of 25 μM. Activity was calculated with one mU of activity defined as the amount of enzyme necessary to completely reduce one nmol of ETF in one minute.
1.3 RESULTS
1.3.1 Clinical Summary
Eleven patients identified by newborn screening as having elevated C5-carnitine levels have been further studied. All were confirmed to have persistently elevated C5-carnitine levels in blood. Additional testing was variable but all either had elevation of 2-methylburtyrylglycine in urine (with no intermediates consistent with isovaleric acidemia), or an increase in 2-methylbutyrylcarnitine in culture medium from patient fibroblasts incubated with deuterated isoleucine consistent with a diagnosis of SBCAD deficiency. All of the newborn screening patients have been followed for up to three years, and all have remained asymptomatic at the time of the manuscript preparation with the oldest being 4 years old.
Information on the level of “C5-carnitine” reported on the initial newborn screen report was available on 7 of 11 individuals. Because the samples were analyzed in several labs and reported with different cutoffs, we have calculated the fold-increase over the lab-reported maximum normal value to allow comparison. The differences ranged from a low of 1.1 fold to >19 fold increase. C5-carnitine levels decreased with time on all patients with one falling to the normal range. However, since all patients were asymptomatic, no correlations of screening or follow up levels with phenotype were possible. While the asymptomatic patients likely represent all screen positive babies from the respective states of residence of the co-authors of this study, a formal determination of incidence was not made.
One final individual, a 3½-year-old girl, was found to have an elevated plasma C5-carnitine during metabolic evaluation for clinical disease. She was the product of non-consanguineous, European ancestry, born by elective C-section at term with a birth weight of 2,358 g. She had a normal neonatal course and good general health. She was born prior to acylcarnitine analysis being added to her birth state’s newborn screening program. Concerns regarding her development arose when she did not crawl at about eight month of age. Subsequently her gross motor development was noted to be delayed; she walked at the age of two years and climbed stairs at the age of 3 ½ years. Speech and language were delayed. At the age of 3 ½ she was also noted to show balance problems with poor coordination and abnormal wide based gait with ataxia. Since the age of 1 year she demonstrated sleep disturbance and staring episodes as well as behavioural issues including head banging. She was described as an interactive girl with good socializing skills. On examination at 3 ½ years of age she had dysmorphic features including microcephaly (head circumference 46.5 cm; <3), wide set eyes with prominent nasal bridge, prominent but low set ears and micrognathia, and blond and brittle hair. There were no skeletal abnormalities and no organomegaly. Her neurological exam revealed that she was unable to speak and was not able to understand complex commands; cranial nerves were intact. Tone was reduced but muscle bulk was normal and deep tendon reflexes were brisk. MRI scan of the brain showed simplified general pattern with decreased cortical sulci most likely representing brain dysgenesis, MRS was normal. EEG revealed abnormal right temporal inter-ictal discharges consistent with a partial seizure disorder. Chromosome and fragile X testing were normal, as was sequencing of exons 2 and 3 of the MECP2 gene. Her plasma amino acids were normal as were chromosomes, Angelmann syndrome testing and microarray analysis. Urinary organic acid analysis revealed methylbutyryglycine in the urine (amount not quantified). Molecular analysis showed the patient to be homozygous for the intron 3 +1 G>A mutation also identified in one of the asymptomatic infants identified through newborn screening.
1.3.2 SBCAD Gene Analysis
Bidirectional sequence analysis of genomic DNA from 4 of the newborn screening patients and the one symptomatic individual has identified two variations in the ACADSB gene not previously reported and two previously described mutations (Table 1). The first was homozygous for a c.295C>T mutation in exon 3 that introduces a stop codon in the coding sequence (p.Gln99X). The patient’s parents are first cousins of Indian descent. No further studies were performed on this early nonsense mutation since only a short, unstable piece of the protein is likely to be produced. The second patient identified by newborn screening (of European ancestry) was a compound heterozygote for two previously described mutations, c.443C>T (p.Thr148Ile) in exon 4 and c.1159G>A (p.Glu387Lys) in exon 10 [28, 30]. An additional exon 1 sequence variant (c.50G>A, p.R13K) was detected in individual 2. This is a conservative substitution in the mitochondrial targeting signal and is not predicted to affect function. Moreover, it is polymorphic in the population as determined though the HapMap Project website (www.hapmap.org) with heterozygote G/A frequency in Nigerian Africans of 22.4%, Japanese 22.2% and Chinese 13.3%. One asymptomatic and one symptomatic patient were homozygous for a G>A mutation at the +1 position of the splice donor site for intron 3 that disrupts the canonical splicing signal for that intron. A mutation affecting this intron has previously been described [35]. This is predicted to be a null mutation and so no mutagenesis assay was performed. An additional baby identified through newborn screening had a fibroblast acylcarntine profile consistent with complete SBCAD deficiency, but only one heterozygous nonsense mutation was identified in exon 5 genomic DNA. cDNA was homozygous for the mutation in this region indicating that the mutation on the second allele must either result in loss of transcription of the SBCAD gene, an unidentified intronic mutation that results in abnormal splicing and/or unstable RNA, or deletion of part of the allele containing the normal exon 5 sequence. Unfortunately, samples for gene sequencing were not available for the rest of the patients identified through newborn screening.
Table 1.
Mutations identified in the ACADBS gene in patient cell lines
| Mutation | Presentation | Treatment | Follow up “C5 carnitine” nM/ml (normal range for lab) | Outcome |
|---|---|---|---|---|
| Homozygous c.295 C>T in exon 3 (p.Gln99X, precursor protein) | Detected on NBS, asymptomatic | None | 0.88 (<0.8) | No symptoms |
| Heterozygous c.443C>T in exon 4 (p.Thr148Ile, precursor protein) | ||||
| Heterozygous c.1159 G>A in exon 10 (p.E387K, precursor protein) | Detected on NBS, asymptomatic | None | 1.89 (<0.63) | No symptoms |
| Heterozygous c.50G>A, p.R13K | ||||
| Heterozygous c621G>A in genomic DNA (W207X, precursor protein) | Detected on NBS, asymptomatic | None | 0.25 (0.33) | No symptoms |
| Homozygous for this mutation in cDNA | ||||
| Homozygous IVS3+1G>A | Detected on NBS, asymptomatic | None | 2.58 ((<.63) | No symptoms |
| Homozygous IVS3+1G>A | 4 year old Developmental delay, dysmorphic features | None | 1.47 (<0.38) | Developmental delay, simplified brain sulci pattern, microcephaly |
1.3.3 Computational and Functional Analyses
To examine the possible effects of the identified mutations on SBCAD structure, we modeled each point mutation into the published three dimensional structure of the protein (Figure 1A). The computational molecular modeling suggested that a Thr148Ile substitution might be tolerated, however, the Thr148 residue lies in a hydrophilic pocket, where an Ile replacement could also be disruptive (Figure 1B. In contrast, the Glu387Lys mutation is predicted to be deleterious to enzyme quaternary structure, disrupting a hydrogen bonding conduit that includes residues Arg384:Glu387:Thr411’ (the prime designation for residues from the second subunit) that is likely essential for monomer-monomer binding (Figure 1B). The mutation would also disrupt FAD binding, deleterious to enzyme stability, since the Glu387 backbone oxygen directly binds an FAD hydroxyl group. To investigate these hypothetical effects experimentally, in vitro mutagenesis was used to introduce each mutation into a prokaryotic expression vector for SBCAD. For both mutations, western blot analysis of E. coli cell extract following expression of vectors containing each mutation identified no detectable cross reactive material to SBCAD antigen (Figure 2), and no enzyme activity was measurable in the cellular extract, indicating that the protein folding and/or stability were grossly affected (Table 2).
Figure 1.


Ribbon representation of point mutations identified in SBCAD gene. A. The tetrameric SBCAD crystal structure, PDB code 2JIF, is shown with each subunit colored a different color. FAD is shown as yellow and bound substrate as green. The amino acid substitutions identified in this and other studies are represented as yellow balls with precursor protein numbering given. Leu255Phe and Ser368Pro have previously been reported in Korman, et al, 2005). The former is likely to be important in stabilizing the interaction between two adjacent alpha helices. Ser368 likes in an alpha helix that is likely to have its trajectory altered by substitution of a proline. B. Residues at the 148 and 387 positions (precursor numbering) are illustrated. The Thr148 hydroxlate group lies in a hydrophilic pocket, but does not directly interact with other residues side chains. The carboxylate oxygen molecules of Glu387 lie between the Lys384 side chain amino group and the hydroxyl group of Thr411’ of the second subunit forming a hydrogen bonding conduit. In addition, the Glu387 backbone oxygen is directly involved in FAD binding through hydrogen bonding.
Figure 2.
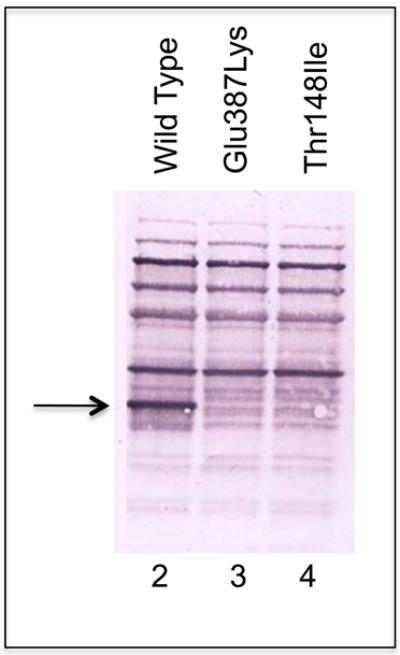
Western blotting of an SDS-PAGE gel of expressed SBCAD patient point mutations. The arrow shows the migration position of purified recombinant SBCAD. Wild type SBCAD expressed in E. coli yields a high level of SBCAD antigen in crude cell extracts. The Glu387Lys and Thr148Ilu are minimally or not detectable.
Table 2.
Activity of Wild type and mutant SBCADH expressed in E. coli
| Vector | Activity (mU/mg cellular protein; % wild type) |
|---|---|
| Wild type | 25; 100% |
| c.443C>T | 0.7; 2.8% |
| c.1159G>A | 0.5; 2.0% |
1.4 DISCUSSION
1.4.1 Clinical Findings in SBCAD Deficiency
In this report, we present 11 non-Hmong infants with SBCAD deficiency identified through newborn screening. To date all have been well, which is in striking contrast to the dramatic phenotype previously reported in the first patients found to have this disorder, as well as the ones identified in this study [16, 25, 31]. The first reported patient presented with acute metabolic acidosis at three days of age after an uncomplicated pregnancy and delivery, and then exhibited chronic seizures, abnormal movements, and developmental delay [16]. His sister was subsequently diagnosed prenatally and has not had any episodes of metabolic decompensation while maintained on a low protein diet. Other individuals identified on evaluation for possible metabolic conditions have presented with a constellation of chronic neurologic symptoms including seizures, developmental delay, hypotonia, recurrent vomiting and attention deficit disorder [25, 29, 30]. In contrast, all reported patients detected through newborn screening with an elevated C5 carnitine, including this study, have remained asymptomatic [26–30]. It is hard to reconcile the clinical differences demonstrated between these groups of patients. SBCAD deficiency may well have predisposed the symptomatic individuals to neurologic damage with or without acute acidosis but the increasing number of asymptomatic patients identified through newborn screening and family studies suggests the original association may have been coincidental. Notably, our one patient identified through an evaluation for significant symptoms had the same SBCAD mutation as an asymptomatic newborn. In addition the symptomatic patient had findings suggestive of a primary neuronal migration defect. Thus, it is likely that the SBCAD deficiency is irrelevant to the patient’s clinical picture.
One group of SBCAD deficient patients deserves special mention. The Hmong are a population of ethnic Chinese that migrated first to Southeast Asia and, more recently, to the US. Not surprisingly, a recurrent point mutation has been identified in the SBCAD coding region in this population [27]. Several of these patients initially had mild hypotonia, but this resolved over time. Additional numerous deficient individuals in this population have since been identified through newborn screening or family studies, and they have been essentially asymptomatic with normal development [27].
1.4.2 Newborn Screening and Variable Phenotypes
Organic acidemias were originally recognized as severe, life threatening inborn errors of metabolism that ultimately led to acute episodes of metabolic decompensation with acidosis and often death. This is typified by the classic description of neonatal onset isovaleric acidemia [36]. In contrast, disorders of mitochondrial β-oxidation are more generally recognized as more variable and often conditional [17, 18]. That is, patients may be well for long periods of time, even life long, and only manifest symptoms when under common stress such as intercurrent illness. This distinction is being blurred by the increased recognition of mild forms of organic acidemias by expanded newborn screening. Deficiencies of the acyl-CoA dehydrogenases offer a good example of this phenomenon. Newborn screening has now led to the identification of individuals with increased isovaleryl-CoA derivatives in blood and urine and a common mutation in the IVDH gene leading to reduction of isovaleryl-CoA dehydrogenase activity [15, 37]. All of these patients have remained well through at least 10 years of age. Other individuals with complete IVDH deficiency have remained well with dietary restrictions, albeit some have had episodes of intermittent metabolic acidosis. A very similar story has become evident for short chain acyl-CoA dehydrogenase deficiency [24]. The first reported patient with this latter disorder died of overwhelming neonatal acidosis, however, numerous asymptomatic individuals have now been identified through newborn screening. While the clinical risks related to SBCAD and other ACAD deficiencies now appear to be less than originally thought, it would be imprudent to disregard them completely. Rather, long term studies will be necessary to assess the true risk of acute and chronic disease in these patients.
1.4.3 Diagnosis of SBCAD Deficiency and Follow-up of Abnormal Newborn Screens
Regardless of the clinical significance of SBCAD deficiency, the need to unequivocally identify it is clear since it is now most frequently suspected on the basis of newborn screening results. Under these circumstances, it is crucial to differentiate it from isovaleric acidemia as isovaleryl- and 2-methylbutyrylcarnitine share the same mass/charge ratio (m/z 302 on a precursor of 85 scan) [26, 30, 38]. While fibroblast enzyme and acylcarnitine studies or molecular analysis serve this function, the unavoidable delay in diagnosis would be unacceptable. Fortunately, in analogy to the multiple alternative metabolites that accumulate in isovaleric acidemia, additional metabolites can also be identified in SBCAD deficiency. 2-methylbutyrylglycine and/or 2-methylbutyrylcarnitine may be increased in urine but may also be normal [26, 30, 38]. 2-Ethylhydracrylic acid (2-EHA) present in blood due to catabolism of isoleucine by the alternative R-pathway can be a sensitive though not specific marker for SBCAD deficiency [30]. Three other disorders also accumulate 2-EHA in blood: β-ketothiolase, ethylmalonic encephalopathy, and Barth syndrome, though other metabolites distinguish these disorders from SBCAD deficiency [28, 30, 39–42]. It is important to note that although the C5-carnitine level has been reported to be within the normal range in some SBCAD deficient patients, all of the patients reported in this study had elevated levels of this metabolite on newborn screen. However, in one patient, the level normalized on subsequent evaluation (Table 2). In recognition of these issues, the American College of Medical Genetics ACT sheets for response to the finding of elevated C5-carnitine on a newborn screen advocates urine organic acid and acylglycine determination as the initial test following an abnormal newborn screening result (http://www.acmg.net/resources/policies/ACT/condition-analyte-links.htm).
1.4. 4 Therapy
The need for and type of therapy in SBCAD deficiency is unclear. The therapeutic approach in initial cases included a low protein diet, avoidance of fasting, and carnitine supplementation [29]. However, the Hmong patients with SBCAD deficiency identified through family studies have remained asymptomatic as have all previously reported infants identified by newborn screening [27]. In light of this observation, disruptive dietary manipulations for infants identified by newborn screening are of questionable indication. Rather, it would seem more prudent to continue a normal diet in these infants but also have the family maintain some level of vigilance during intercurrent illnesses. Additional long term studies will be necessary to address this issue
1.5 CONCLUSIONS
We present 11 non-Hmong patients with SBCAD deficiency identified through newborn screening acylcarnitine analysis. The lack of clinical symptoms in these patients in conjunction with similar findings in a high risk population (the Hmong Chinese) emphasizes the need to further evaluate the clinical relevance of this biochemical disorder. Rare symptomatic patients may represent the extreme (and unusual) end of the clinical spectrum or coincidental findings. Thus additional diagnostic testing is required to make a definitive diagnosis in infants who screen positive. Consequently, long term follow up of these patients will be critical to addressing the phenotypic spectrum of SBCAD deficiency.
Acknowledgments
JV was supported in part by NIH grant R01 DK54936.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ikeda Y, Dabrowski C, Tanaka K. Separation and properties of five distinct acyl-CoA dehydrogenases from rat liver mitochondria. J Biol Chem. 1983;258:1066–1076. [PubMed] [Google Scholar]
- 2.Ikeda Y, Okamura-Ikeda K, Tanaka K. Purification and characterization of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases from rat liver mitochondria. Isolation of the holo- and apoenzymes and conversion of the apoenzyme to the holoenzyme. J Biol Chem. 1985;260:1311–1325. [PubMed] [Google Scholar]
- 3.Ikeda Y, Tanaka K. Purification and Characterization of 2-Methyl-Branched Chain Acyl Coenzyme A Dehydrogenase, an Enzyme Involved in Isoleucine and Valine Metabolism, from Rat Liver Mitochondria. J Biol Chem. 1983;258:9477–9487. [PubMed] [Google Scholar]
- 4.Izai K, Uchida Y, Orii T, Yamamoto S, Hashimoto T. Novel fatty acid β-oxidation enzymes in rat liver mitochondria. 1. Purification and properties of very-long-chain acyl-coenzyme A dehydrogenase. J Biol Chem. 1992;267:1027–1033. [PubMed] [Google Scholar]
- 5.Rozen R, Vockley J, Zhou L, Milos R, Willard J, Fu K, Vicanek C, Low-Nang L, Torban E, Fournier B. Isolation and expression of a cDNA encoding the precursor for a novel member (ACADSB) of the acyl-CoA dehydrogenase gene family. Genomics. 1994;24:280–287. doi: 10.1006/geno.1994.1617. [DOI] [PubMed] [Google Scholar]
- 6.Willard J, Vicanek C, Battaile KP, Vanveldhoven PP, Fauq AH, Rozen R, Vockley J. Cloning of a cDNA for short/branched chain acyl-coenzyme A dehydrogenase from rat and characterization of its tissue expression and substrate specificity. Archives of Biochemistry & Biophysics. 1996;331:127–133. doi: 10.1006/abbi.1996.0290. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen TV, Andresen BS, Corydon TJ, Ghisla S, Abd-El Razik N, Mohsen AW, Cederbaum SD, Roe DS, Roe CR, Lench NJ, Vockley J. Identification of isobutyryl-CoA dehydrogenase and its deficiency in humans. Mol Genet Metab. 2002;77:68–79. doi: 10.1016/s1096-7192(02)00152-x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Zhang W, Zou D, Chen G, Wan T, Zhang M, Cao X. Cloning and functional characterization of ACAD-9, a novel member of human acyl-CoA dehydrogenase family. Biochem Biophys Res Commun. 2002;297:1033–1042. doi: 10.1016/s0006-291x(02)02336-7. [DOI] [PubMed] [Google Scholar]
- 9.Ensenauer R, He M, Willard JM, Goetzman ES, Corydon TJ, Vandahl BB, Mohsen AW, Isaya G, Vockley J. Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J Biol Chem. 2005;280:32309–32316. doi: 10.1074/jbc.M504460200. [DOI] [PubMed] [Google Scholar]
- 10.Goodman SI, Kratz LE, DiGiulio KA, Biery BJ, Goodman KE, Isaya G, Frerman FE. Cloning of Glutaryl-CoA Dehydrogenase cDNA, and Expression of Wild Type and Mutant Enzymes in Escherichia coli. Hum Mol Genet. 1995;4:1493–1498. doi: 10.1093/hmg/4.9.1493. [DOI] [PubMed] [Google Scholar]
- 11.Battaile KP, Nguyen TV, Vockley J, Kim JJ. Structures of isobutyryl-CoA dehydrogenase and enzyme-product complex: comparison with isovaleryl- and short-chain acyl-CoA dehydrogenases. J Biol Chem. 2004;279:16526–16534. doi: 10.1074/jbc.M400034200. [DOI] [PubMed] [Google Scholar]
- 12.Tiffany KA, Roberts DL, Wang M, Paschke R, Mohsen AWA, Vockley J, Kim JJP. Structure of human isovaleryl-coA dehydrogenase at 2.6 angstrom resolution - basis for substrate specificity. Biochemistry. 1997;36:8455–8464. doi: 10.1021/bi970422u. [DOI] [PubMed] [Google Scholar]
- 13.Vockley J, Mohsen AW, Binzak B, Willard J, Fauq A. Mammalian branched-chain acyl-CoA dehydrogenases: molecular cloning and characterization of recombinant enzymes. Methods Enzymol. 2000;324:241–258. doi: 10.1016/s0076-6879(00)24236-5. [DOI] [PubMed] [Google Scholar]
- 14.Vockley J, Parimoo B, Tanaka K. Molecular characterization of four different classes of mutations in the isovaleryl-CoA dehydrogenase gene responsible for isovaleric acidemia. Am J Hum Genet. 1991;40:147–157. [PMC free article] [PubMed] [Google Scholar]
- 15.Vockley J, Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C:95–103. doi: 10.1002/ajmg.c.30089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibson KM, Burlingame TG, Hogema B, Jakobs C, Schutgens RBH, Millington D, Roe CR, Roe DS, Sweetman L, Steiner RD, Linck L, Pohowalla P, Sacks M, Kiss D, Rinaldo P, Vockley J. 2-Methylbutyryl-coenzyme A dehydrogenase deficiency: A new inborn error of L-isoleucine metabolism. Pediatric Research. 2000;47:830–833. doi: 10.1203/00006450-200006000-00025. [DOI] [PubMed] [Google Scholar]
- 17.Vockley J, Singh RH, Whiteman DA. Diagnosis and management of defects of mitochondrial beta-oxidation. Curr Opin Clin Nutr Metab Care. 2002;5:601–609. doi: 10.1097/00075197-200211000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Vockley J, Whiteman DA. Defects of mitochondrial beta-oxidation: a growing group of disorders. Neuromuscul Disord. 2002;12:235–246. doi: 10.1016/s0960-8966(01)00308-x. [DOI] [PubMed] [Google Scholar]
- 19.Shih VE, Mandell R, Tanaka K. Diagnosis of isovaleric acidemia in cultured fibroblasts. Clinica Chimica Acta. 1973;48:437–439. doi: 10.1016/0009-8981(73)90425-7. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka K, Yokota I, Coates PM, Strauss AW, Kelly DP, Zhang Z, Gregersen N, Andresen BS, Matsubara Y, Curtis D, et al. Mutations in the medium chain acyl-CoA dehydrogenase (MCAD) gene. Hum Mutat. 1992;1:271–279. doi: 10.1002/humu.1380010402. [DOI] [PubMed] [Google Scholar]
- 21.Yokota I, Indo Y, Coates PM, Tanaka K. Molecular basis of medium chain acyl-Coenzyme A dehydrogenase deficiency: An A to G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J Clin Invest. 1990;86:1000–1003. doi: 10.1172/JCI114761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aoyama T, Uchida Y, Kelley RI, Marble M, Hofman K, Tonsgard JH, Rhead WJ, Hashimoto T. A novel disease with deficiency of mitochondrial very-long-chain acyl-CoA dehydrogenase. Biochem Biophys Res Commun. 1993;191:1369–1372. doi: 10.1006/bbrc.1993.1368. [DOI] [PubMed] [Google Scholar]
- 23.He M, Rutledge S, Kelly D, Palmer C, Murdoch G, Majumder N, Nicholls R, Pei Z, Watkins PA, Vockley J. A new genetic disorder in mitochondrial fatty acid b-oxidation, ACAD9 deficiency. Am J Hum Genet. 2007;81:87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jethva R, Bennett MJ, Vockley J. Short-chain acyl-coenzyme A dehydrogenase deficiency. Mol Genet Metab. 2008;95:195–200. doi: 10.1016/j.ymgme.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andresen B, Christensen E, Corydon T, Bross P, Pilgaard B, Wanders R, Ruiter J, Simonsen H, Winter V, Knudsen I, Schroeder L, Gregersen N, Skovby F. Isolated 2-methylbutyrylglycinuria caused short/branched-chain acyl-CoA dehydrogenase deficiency: Identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism. Am J Hum Genet. 2000;67:1095–1103. doi: 10.1086/303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matern D, He M, Berry SA, Rinaldo P, Whitley CB, Madsen PP, van Calcar SC, Lussky RC, Andresen BS, Wolff JA, Vockley J. Prospective diagnosis of 2-methylbutyryl-CoA dehydrogenase deficiency in the Hmong population by newborn screening using tandem mass spectrometry. Pediatrics. 2003;112:74–78. doi: 10.1542/peds.112.1.74. [DOI] [PubMed] [Google Scholar]
- 27.van Calcar SC, Gleason LA, Lindh H, Hoffman G, Rhead W, Vockley G, Wolff JA, Durkin MS. 2-methylbutyryl-CoA dehydrogenase deficiency in Hmong infants identified by expanded newborn screen. Wisconsin Medical Journal. 2007;106:12–15. [PubMed] [Google Scholar]
- 28.Sass JO, Ensenauer R, Roschinger W, Reich H, Steuerwald U, Schirrmacher O, Engel K, Haberle J, Andresen BS, Megarbane A, Lehnert W, Zschocke J. 2-Methylbutyryl-coenzyme A dehydrogenase deficiency: functional and molecular studies on a defect in isoleucine catabolism. Mol Genet Metab. 2008;93:30–35. doi: 10.1016/j.ymgme.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Korman SH. Inborn errors of isoleucine degradation: a review. Molecular Genetics & Metabolism. 2006;89:289–299. doi: 10.1016/j.ymgme.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Korman SH, Andresen BS, Zeharia A, Gutman A, Boneh A, Pitt JJ. 2-ethylhydracrylic aciduria in short/branched-chain acyl-CoA dehydrogenase deficiency: application to diagnosis and implications for the R-pathway of isoleucine oxidation. Clinical Chemistry. 2005;51:610–617. doi: 10.1373/clinchem.2004.043265. [DOI] [PubMed] [Google Scholar]
- 31.Kanavin OJ, Woldseth B, Jellum E, Tvedt B, Andresen BS, Stromme P. 2-methylbutyryl-CoA dehydrogenase deficiency associated with autism and mental retardation: a case report. J Med Case Reports. 2007;1:98. doi: 10.1186/1752-1947-1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rinaldo P, Cowan TM, Matern D. Acylcarnitine profile analysis. Genet Med. 2008;10:151–156. doi: 10.1097/GIM.0b013e3181614289. [DOI] [PubMed] [Google Scholar]
- 33.Goetzman ES, Wang Y, He M, Mohsen AW, Ninness BK, Vockley J. Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system. Mol Genet Metab. 2007;91:138–147. doi: 10.1016/j.ymgme.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Binzak B, Willard J, Vockley J. Identification of the Catalytic Residue Of Human Short/Branched Chain Acyl-CoA Dehydrogenase ByIn Vitro Mutagenesis. Biochim Biophys Acta. 1998;1382:137–142. doi: 10.1016/s0167-4838(97)00161-1. [DOI] [PubMed] [Google Scholar]
- 35.Madsen PP, Kibaek M, Roca X, Sachidanandam R, Krainer AR, Christensen E, Steiner RD, Gibson KM, Corydon TJ, Knudsen I, Wanders RJ, Ruiter JP, Gregersen N, Andresen BS. Short/branched-chain acyl-CoA dehydrogenase deficiency due to an IVS3+3A>G mutation that causes exon skipping. Hum Genet. 2006;118:680–690. doi: 10.1007/s00439-005-0070-4. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka K, Budd MA, Efron ML, Isselbacher KJ. Isovaleric acidemia: a new genetic defect of leucine metabolism. Proc Natl Acad Sci USA. 1966;56:236–242. doi: 10.1073/pnas.56.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ensenauer R, Vockley J, Willard JM, Huey JC, Sass JO, Edland SD, Burton BK, Berry SA, Santer R, Grunert S, Koch HG, Marquardt I, Rinaldo P, Hahn S, Matern D. A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am J Hum Genet. 2004;75:1136–1142. doi: 10.1086/426318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rinaldo P, Tortorelli S, Matern D. Recent developments and new applications of tandem mass spectrometry in newborn screening. Curr Opin Pediatr. 2004;16:427–433. doi: 10.1097/01.mop.0000133635.79661.84. [DOI] [PubMed] [Google Scholar]
- 39.Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 40.Mamer OA, Tjoa SS. 2-Ethylhydracrylic acid: a newly described urinary organic acid. Clin Chim Acta. 1974;55:199–204. doi: 10.1016/0009-8981(74)90295-2. [DOI] [PubMed] [Google Scholar]
- 41.Tiranti V, Briem E, Lamantea E, Mineri R, Papaleo E, De Gioia L, Forlani F, Rinaldo P, Dickson P, Abu-Libdeh B, Cindro-Heberle L, Owaidha M, Jack RM, Christensen E, Burlina A, Zeviani M. ETHE1 mutations are specific to ethylmalonic encephalopathy. J Med Genet. 2006;43:340–346. doi: 10.1136/jmg.2005.036210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowaczyk MJ, Lehotay DC, Platt BA, Fisher L, Tan R, Phillips H, Clarke JT. Ethylmalonic and methylsuccinic aciduria in ethylmalonic encephalopathy arise from abnormal isoleucine metabolism. Metabolism. 1998;47:836–839. doi: 10.1016/s0026-0495(98)90122-6. [DOI] [PubMed] [Google Scholar]