

Abstract
Human catechol O-methyltransferase (COMT) contains three common polymorphisms (A22S, A52T, V108M), two of which (A22S, V108M) render the protein susceptible to deactivation by temperature or oxidation. We have performed multiple molecular dynamics simulations of the wild-type, A22S, A52T, and V108M COMT proteins to explore the structural consequences of these mutations. In total, we have amassed over 1.4 μs of simulation time representing the largest set of simulations detailing the effects of polymorphisms on a protein system to date. The A52T mutation had no significant effect on COMT structure in accord with experiment, thereby serving as a good negative control for the simulation set. Residues 22 (α2) and 108 (α5) interact with each other throughout the simulations and are located in a polymorphic hotspot ∼20Å from the active site. Introduction of either the larger Ser (22) or Met (108) tightens this interaction, pulling α2 and α5 towards each other and away from the protein core. The V108M polymorphism rearranges active-site residues in α5, β3 and α6, increasing the S-adenosylmethionine site solvent exposure. The A22S mutation reorients α2, moving critical catechol-binding residues away from the substrate-binding pocket. The A22S and V108M polymorphisms evolved independently in Northern European and Asian populations. While the decreased activities of both A22S and V108M COMT are associated with an increased risk for schizophrenia, the V108M induced destabilization is also linked with improved cognitive function. These results suggest that polymorphisms within this hotspot may have evolved to regulate COMT activity and that heterozygosity for either mutation may be advantageous.
Keywords: catechol O-methyltransferase, polymorphism, molecular dynamics, schizophrenia, breast cancer, drug development
Catechol O-methyltransferase (COMT, EC. 2.1.16) catalyzes the transfer of a methyl group from co-substrate S-adenosylmethionine (SAM) to a variety of catechol substrates, thus playing an important role in the metabolism and inactivation of catecholamine neurotransmitters and catechol estrogens (1-3). COMT exists as two isozymes: a ubiquitous 221-residue soluble form (referred to as COMT in this paper) expressed primarily in the liver and kidneys, and a 271-residue membrane-bound form (mb-COMT) found predominantly in the brain and adrenal medulla (4-8). COMT and mb-COMT have different substrate specificities, with mb-COMT methylating neurotransmitters much more efficiently than does the cytosolic protein (9, 10). Inhibitors of COMT are currently included in the primary treatment in the management of Parkinson's disease (11-13).
The human COMT gene contains three common coding polymorphisms: A22S (rs6267), A52T (rs5031015), and V108M (rs4680) (14-16) (Figure 1). 25% of U.S. and Northern European Caucasians are homozygous for the 108M allele (17, 18), while the 22S allele occurs in ∼10 % of Asian populations (www.ncbi.nlm.nih.gov/SNP). There are only limited data regarding the population distribution of the 52T allele, although it appears to occur most frequently in Sub-Saharan African and African American populations (www.ncbi.nlm.nih.gov/SNP). Of the three polymorphisms, the structural and epidemiological effects of the V108M mutation are best characterized. The sidechain of residue 108 is buried within a hydrophobic pocket in a loop between α5 and β3, approximately 16 Å from the protein's active site (Figure 1). While the wild-type and V108M proteins display similar kinetic properties in vitro (10, 19, 20), the V108M polymorphism destabilizes the protein, increasing its susceptibility to thermal (14, 21) and chemical denaturation (21), as well as to oxidative stress (22-24). This destabilization results in decreased protein levels (19, 25, 26), and therefore activity (27-32), in vivo relative to the wild-type protein. The association of the 108M allele with increased risk for breast cancer (20, 33-35) and neuropsychiatric diseases (36-40) has been well documented. Interestingly, recent studies have linked the 108M allele with increased sensitivity to pain (41-43) and with improved prefrontal cognition, especially in working memory (44-46).
Figure 1.
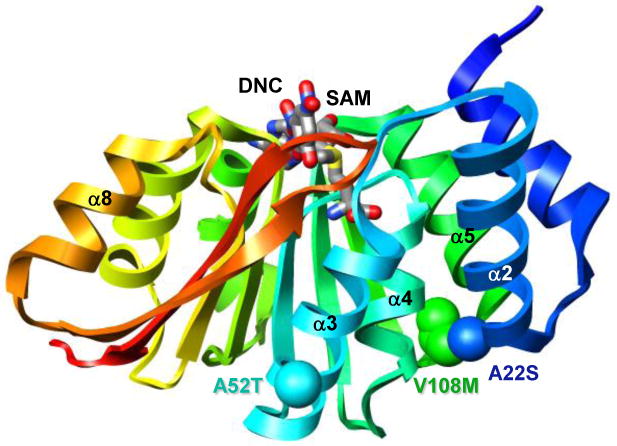
The A22S, A52T, and V108M COMT polymorphisms. Ribbon diagram of wild-type COMT (PDB entry 3BWM (54)) colored from blue (N-terminus) to red (C-terminus). S-adenosylmethionine (SAM) and 3,5-dinitrocatechol (DNC) are shown in stick representation and colored by atom type. Polymorphic residues 22, 52, and 108 are shown in space-filling representation and colored in blue, cyan and green, respectively.
Residue 22 is positioned in a surface loop between α1 and α2 ∼13 Å from the SAM-binding site and ∼20Å from the catechol-binding site (Figure 1). The A22S mutation decreases COMT activity by 30% relative to the wild-type protein, and incubating 22S COMT for 1 hour at 45°C lowers enzymatic activity by 80% compared with the wild-type protein (47). However, the A22S polymorphism significantly increases COMT's affinity for SAM (47). Three studies have been published linking the S22 allele with increased risk for schizophrenia (48, 49) and estrogen-associated cancers (47) in Koreans. However, two studies find no association between the A22S polymorphism and either renal cell (50) or prostate cancer (51) in Japanese men.
Residue 52 is positioned on the protein surface in helix α3 which contains residues critical for binding both SAM and catechol substrates (Figure 1). The wild-type and 52T proteins have T50 (temperatures resulting in 50% inactivation) values of 53.7 ± 0.10 and 52.7 ± 0.34 °C, respectively (14). In addition, Li et al. (2005) showed that while the A52T mutation slightly decreases COMT's affinity for SAM, it has no significant effect on COMT activity (47). No data associating the A52T polymorphism with disease have been published, to our knowledge.
In 2006, we published a molecular dynamics (MD) study describing the destabilizing effects of the V108M polymorphism on the structure of COMT at atomic resolution (52). The mutation distorted the overall structure of COMT, increasing the solvent exposure of both the SAM- and catechol-binding sites. These MD studies were performed using a homology model of human COMT generated from a 2 Å crystal structure of the highly homologous rat protein (1VID, (53)) which shares 81% sequence identity with the human protein. Crystal structures of human wild-type and V108M COMT bound with SAM and a substrate analogue are now available (54). Here we describe MD simulations of the wild-type and V108M COMT crystal structures to validate the results of our previous simulations based on the homology model of human COMT (52). We also expand upon these studies to investigate the effects of the A22S and A52T polymorphisms on COMT structure and dynamics via multiple MD simulations. In total, we have amassed over 1.4 μs of simulation time describing the structural effects of COMT polymorphisms representing the largest set of simulations detailing the effects of polymorphisms on a protein system to date.
Materials and Methods
Homology Model
An initial homology model of human 108V COMT was generated from a 2 Å crystal structure of rat COMT (PDB entry 1VID (53), residues 4-216), which shares 81% sequence identity with the human protein. The side chains of the non-identical rat residues were replaced sequentially using a Monte Carlo procedure that minimized the torsional, electrostatic, and van der Waals energies of each residue (55). Two passes through the sequence were completed so that residues altered early in the sequence could rearrange in response to later modifications. Altogether, 40 residues were changed, including 30 conservative replacements. Models of A22S, A52T, and V108M COMT were generated from the homology model by replacing each wild-type residue with its polymorphic variant and minimizing the torsional, electrostatic, and van der Waals energies of the resultant structure in vacuo (56). The Cα-rmsd between the starting structures of wild-type COMT and all of the variant proteins was ∼0.1 Å.
Protein Preparation
The crystal structures of human 108V and 108M COMT were solved after all of the simulations of the wild-type, A22S, A52T, and V108M COMT homology models described here were completed. We therefore performed simulations using the 108V and 108M COMT crystal structures to validate the simulations of the human homology model described here and in 2006 (52). The 1.98 Å crystal structure of human catechol O-methyltransferase (PDB entry 3BWM (54), residues 2-215) bound with S-adenosylmethionine (SAM), 3,5-dinitrocatechol (DNC), Mg2+, and K+ was the starting structure for the simulation of wild-type COMT. The 1.30 Å crystal structure of human 108M COMT (PDB entry 3BWY (54), residues 2-215) bound with SAM, DNC, Mg2+, and 2-methyl-2,4-pentanediol (MPD) was used as the starting structure for the simulation of 108M COMT. SAM, DNC, Mg2+, K+, and MPD were removed, and the resultant structures underwent 1000 steps of molecular dynamics to minimize the torsional, electrostatic and van der Waals energies of the resultant structures in vacuo (56). One 21 ns control simulation of each of the wild-type and V108M COMT crystal structures was completed at 37°C using the protocol described below.
Molecular Dynamics Simulations
Molecular dynamics studies of the wild-type and polymorphic COMT proteins were performed with the in lucem molecular mechanics (ilmm) simulation package (57) using protocols and the potential energy function that have been previously described (58-60). The simulations included all hydrogen atoms and explicit flexible three-centered waters (60). The proteins were solvated in a periodic rectangular box with walls extending at least 10 Å from all protein atoms. Solvent densities were set to 0.997 and 0.933 g/ml for simulations performed at 25 and 37°C, respectively (61). Once the densities were set, the box volume was held fixed and the NVE microcanonical ensemble employed. A 10 Å force-shifted, non-bonded cutoff was used for all non-bonded interactions and the interaction pair-lists were updated every two steps (62). Non-bonded interactions between atoms in charge groups separated by three bonds were included and scaled (C-scale) by 0.4 (63, 64); while the inclusion of these interactions is the current working protocol in the group, these interactions were not included in the past (C-scale = 0). Very few protein systems are affected by this change, but some proteins are more stable with C-scale=0.4. In order to facilitate comparisons with our earlier published work (C-scale = 0) (52), we present simulations here using both approaches. COMT is unaffected by the change in C-scale. A time step of 2 fs was used in all calculations, and structures were saved every 1 ps for analysis. One control simulation of each system was performed at 25°C to ensure that the protein fold was maintained at low temperature and that the protein was suitable for study using MD (Supplementary Figure S1A). Three independent simulations at 37°C of between 41 to 80 ns were performed for the wild-type, A22S, A52T, and V108M proteins for a total of 807 ns of simulation time. Combined with the 650 ns of simulation time published in 2006 (52), we have amassed over 1.4 μs of simulation time describing the structural effects of polymorphisms in COMT.
Data Analyses
Statistical analyses was performed using data from the final 5 to 10 ns of each simulation to ensure that the analyses included only structures from equilibrated systems in which the energies and properties had converged. Average values for Cα–rmsd, solvent-accessible surface area (SASA), and contact distances were calculated using structures from the last 5 ns (5,000 structures) of each simulation. A contact was defined as a C-C atom distance of ≤ 5.4 Å, or a heavy atom (O, N, S) distance of ≤ 4.6 Å between two non-contiguous residues. SASA was calculated using in-house software implementing the NACCESS algorithm (65). Cα-rmsf values were calculated relative to the average structure over the last 10 ns of each simulation at 37°C. Cα–rmsf values for the crystal structures of the rat apo (PDB entry 2ZLB (66)) and holoproteins (PDB entry 1VID (53)) were calculated using the crystallographic B-factors (B) using Cα-rmsf = (3B/8π2)1/2 (Figure 3B).
Figure 3.
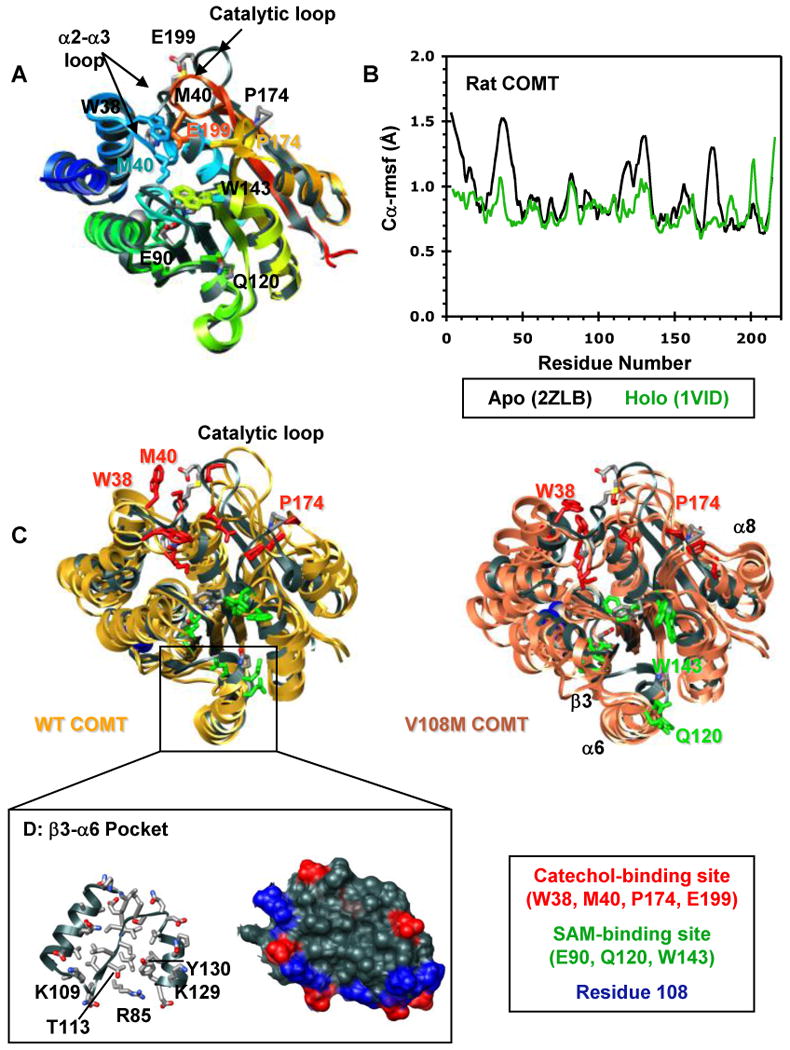
Structural characterization of the COMT apoprotein. (A) Superposed ribbon diagrams of the rat COMT apoprotein (gray, 2ZLB.PDB (66)) and the rat (1VID.PDB (53)) and human (3BWM.PDB (54)) holoproteins bound with SAM and DNC and colored from blue (N-terminus) to red (C-terminus). The overall structures of the rat and human holoproteins are virtually identical. The rat apo and holoproteins differ mainly in the loop regions that define the catechol-binding site. The α2-α3 loop, β5-α8 loop, and the catalytic loop (residues 197-202) pull away from the protein core in the apoprotein exposing the SAM- and catechol-binding sites. This open configuration may facilitate substrate binding. Interestingly, the adenosine pocket of the apo and holoprotein SAM-binding sites are very similar showing only a slight displacement of key active site residues (E90, Q120, W143). Active site residues are shown in stick representation and colored/labeled in either gray (apoprotein) or to match the structure (holoprotein). (B) Cα-rmsf values (Å) per residue for the rat apoprotein (black, 2ZLB.PDB (66)) and holoprotein (green, 1VID.PDB (53)). (C) Structural overlay of the rat apoprotein crystal structure (gray, 2ZLB.PDB (66)) with structures from the final ns of three independent simulations of human WT (gold) and V108M (pink) COMT. The human WT and rat apoproteins behave similarly in that the structure of the adenosine pocket in the SAM-binding site (α5, α6, α7) is maintained, with some flexibility in the orientations of active site residues E90, Q120, and W143, while the α2-α3, β5-α8, and catalytic loops fluctuate greatly opening up the active-site core and moving W38, M40, P174 and E199 out of the substrate-binding pocket. In addition to these motions, both β3 and α6 pull away from the protein core during simulations of V108M COMT. SAM- and catechol-binding residues are shown in stick representation and colored in green and red, respectively. Residue 108 is shown in space-filling representation and colored blue. (D) Ribbon (left) and van der Waals surface (surface) representation of the β3-α6 pocket from human WT COMT. The V108M polymorphism alters the orientations of β3 and α6, possibly destabilizing the protein. Therefore, the β3-α6 pocket may serve as a target site for docking small molecules to stabilize the V108M COMT structure. Positively and negatively charged residues in the surface representation are colored in blue and red.
The data described in Tables 1 and 2 are based on sets of three independent simulations at 37°C for each wild-type and variant protein. The errors are the standard deviations in the average values of the ensembles for each reported property. The statistical significance of the data was determined using the Student's t test (67).
Table 1. Comparison of general properties from MD simulations based on homology models and crystal structures of human wild-type and V108M COMT.
| Property a | Homology Model | Crystal Structure d | ||||
|---|---|---|---|---|---|---|
| 2006b | 2008c | |||||
| WT | V108M | WT | V108M | WT | V108M | |
| Cα-rmsd (Å) | 2.6 ± 0.3 | 3.3 ± 0.6 | 3.1 ± 0.2 | 3.7 ± 0.4i | 2.4 ± 0.2 | 3.9 ± 0.1j |
| Total SASA (Å2)e | 10948 ± 208 | 11148 ± 248 | 11344 ± 245 | 11850 ± 263i | 10944 ± 187 | 12145 ± 143k |
| Active Site | ||||||
| SAM-Binding Site SASA (Å2) f | 1016 ± 56 | 1146 ± 37l | 989 ± 105 | 1150 ± 79 | 983 | 1264 |
| E90 Cγ - Q120 Cγ (Å) | 14.7 ± 1.0 | 15.7 ± 0.9 | 14.0 ± 2.3 | 15.4 ± 1.0 | 16.8 ± 1.4 | 9.2 ± 0.7 |
| Q120 Cγ - W143 Cβ (Å) | 14.9 ± 4.0 | 18.6 ± 5.2 | 13.3 ± 1.8 | 18.1 ± 1.7l | 14.9 ± 1.7 | 15.0 ± 2.2 |
| W143 Cβ - E90 Cγ (Å) | 10.8 ± 0.7 | 12.5 ± 1.8 | 11.5 ± 2.1 | 14.0 ± 3.3 | 11.9 ± 0.5 | 13.8 ± 1.4i |
| Catechol-Binding Site SASA (Å2)g | 723 ± 44 | 812 ± 107 | 937 ± 77 | 851 ± 96 | 754 | 999 |
| W38 Cγ - P174 Cγ (Å) | 20.0 ± 2.3 | 23.7 ± 4.3 | 19.0 ± 1.8 | 23.7 ± 4.8 | 18.8 ± 1.2 | 29.0 ± 1.6k |
| Polymorphic Site | ||||||
| Polymorphic Site SASA (Å2)h | 322 ± 30 | 366 ± 29 | 384 ± 95 | 452 ± 38 | 376 | 362 |
|
V108 Cγ1/ M108 Sδ – A22 Cβ (Å) |
4.1 ± 0.3 | 6.4 ± 3.4 | 6.3 ± 3.0 | 4.9 ± 0.7 | 4.7 ± 0.5 | 6.0 ± 0.6i |
|
V108 Cγ2 /M108 Cε – V74 Cγ1 (Å) |
4.4 ± 0.6 | 8.0 ± 3.3 | 7.6 ± 2.0 | 6.0 ± 1.3 | 5.6 ± 0.8 | 4.8 ± 0.6 |
|
V108 Cγ1 /M108 Sδ – R78 Cβ (Å) |
6.5 ± 0.2 | 6.8 ± 3.3 | 8.4 ± 3.1 | 8.5 ± 2.8 | 8.5 ± 0.6 | 8.6 ± 0.5 |
|
V108 Cγ2 /M108 Cε – V103 Cγ1 (Å) |
4.2 ± 0.3 | 7.5 ± 0.7k | 4.9 ± 0.9 | 6.4 ± 2.0 | 4.5 ± 0.4 | 5.4 ± 0.9 |
All values were calculated using structures from the last 5 ns of each simulation.
Data from simulations published in Rutherford et al. (2006) (52). Three independent simulations of each protein were run with a C-scale value of 0.0.
Three independent simulations of each protein were run using a C-scale value of 0.4 (see Methods).
Control simulations were run using either 3BWM (WT COMT) or 3BWY (108M COMT) as a starting structure (see Methods) (54).
The total solvent-accessible surface area (SASA) was determined using the NACCESS algorithm (65).
The following residues were used to calculate the SASA of the SAM-binding site: A39, M40, N41, V42, G66, A67, Y68, S72, E90, I91, N92, G117, A118, S119, Q120, D141, H142, and W143.
The following residues were used to calculate the catechol-binding site SASA: W38, M40, K46, D141, K144, D169, N170, C173, P174, L198, E199, Y200, and D205.
The following residues were used to calculate the polymorphic site SASA: A22, A73, V74, R78, V103, A106, G107, K109, K111, V112 and residue 108.
p < 0.10.
p < 0.005.
p < 0.01.
p < 0.05.
Table 2. Properties from the MD simulations WT, A22S, A52T, and V108M COMT.
| Property a | WT COMT | A52T COMT | A22S COMT | V108M COMT |
|---|---|---|---|---|
| Cα-rmsd (Å) | 3.1 ± 0.2 | 2.9 ± 0.3 | 4.0 ± 0.5 h | 3.7 ± 0.4 h |
| Total SASA (Å2) b | 11344 ± 245 | 11290 ± 329 | 11759 ± 277h | 11850 ± 263 h |
| SAM-Binding Site SASA (Å2) c | 989 ± 105 | 968 ± 180 | 965 ± 180 | 1150 ± 79 h |
| Catechol-Binding Site SASA (Å2) d | 937 ± 77 | 924 ± 30 | 1088 ± 30 i | 851 ± 96 |
| W38 Cγ - P174 Cγ (Å) | 19.0 ± 1.8 | 21.2 ± 2.3 | 22.8 ± 1.6 h | 23.7 ± 4.8 |
| A52T Polymorphic Site | ||||
| Polymorphic Site SASA (Å2) e | 427 ± 49 | 434 ± 41 | 393 ± 15 | 392 ± 20 |
| A/T 52 Cβ – I49 Cγ2 (Å) | 5.8 ± 0.5 | 6.2 ± 0.4 | 6.0 ± 0.4 | 6.0 ± 0.4 |
| A/T 52 Cβ – I54 Cβ (Å) | 7.4 ± 0.2 | 7.4 ± 0.2 | 7.5 ± 0.2 | 7.4 ± 0.2 |
| A/T 52 Cβ – Q55 Cβ (Å) | 5.6 ± 0.4 | 5.7 ± 0.4 | 5.6 ± 0.4 | 6.0 ± 0.5 |
| A52 Cβ /T52 Cγ2 – E56 Cγ (Å) | 6.0 ± 0.7 | 5.2 ± 0.9 | 6.5 ± 0.8 | 5.8 ± 0.9 |
| A/T 52 Cβ – Y194 Cε2 (Å) | 7.5 ± 0.9 | 7.8 ± 1.1 | 6.7 ± 1.1 | 8.1 ± 1.0 |
| A22S Polymorphic Site | ||||
| Polymorphic Site SASA (Å2) f | 318 ± 86 | 299 ± 33 | 386 ± 37 | 392 ± 63 |
| Helix α2 Cα-rmsd (Å) | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 |
| Helix α4 Cα-rmsd (Å) | 0.9 ± 0.3 | 0.7 ± 0.1 | 1.0 ± 0.3 | 0.6 ± 0.2 |
| A/S 22 Cβ – V74 γ1 (Å) | 4.9 ± 0.7 | 4.5 ± 0.5 | 7.0 ± 3.1 | 7.2 ± 1.3 h |
| A/S 22 Cβ – R78 Cγ (Å) | 4.5 ± 0.6 | 5.4 ± 1.3 | 10.5 ± 3.8 h | 8.2 ± 2.5 h |
| A/S 22 Cβ – A106 Cβ (Å) | 6.4 ± 2.4 | 4.9 ± 0.6 | 5.3 ± 0.9 | 4.9 ± 0.8 |
| A22 Cβ / S22 Oγ – G107 Cα (Å) | 8.9 ± 3.0 | 7.2 ± 0.8 | 5.7 ± 1.0 | 6.5 ± 0.7 |
| A/S 22 Cβ – V108 Cγ1/ M108 Sδ (Å) | 6.3 ± 3.0 | 4.6 ± 0.7 | 5.3 ± 1.4 | 4.8 ± 0.8 |
| V108M Polymorphic Site | ||||
| Polymorphic Site SASA (Å2) g | 384 ± 95 | 369 ± 31 | 425 ± 41 | 452 ± 38 |
| V108 Cγ1/ M108 Sδ – A22 Cβ (Å) | 6.3 ± 3.0 | 4.6 ± 0.7 | 5.3 ± 1.4 | 4.9 ± 0.7 |
| V108 Cγ1 – L26 Cδ1/ M108 Cε - L26 Cδ2 (Å) | 11.7 ± 1.7 | 10.2 ± 0.8 | 7.2 ± 2.0 i | 7.0 ± 1.5 i |
| V108 Cγ2 /M108 Sδ – A73 Cβ (Å) | 5.0 ± 1.1 | 4.6 ± 0.5 | 6.5 ± 2.0 | 6.6 ± 2.2 |
| V108 Cγ2 /M108 Cε – V74 Cγ1 (Å) | 7.6 ± 2.5 | 5.2 ± 0.8 | 5.2 ± 0.9 | 6.0 ± 1.3 |
| V108 Cγ1 /M108 Sδ – A77 Cβ (Å) | 5.2 ± 1.3 | 4.2 ± 0.6 | 6.6 ± 3.1 | 5.0 ± 1.4 |
| V108 Cγ1 /M108 Sδ – R78 Cβ (Å) | 8.4 ± 3.1 | 7.6 ± 1.4 | 9.3 ± 1.3 | 8.5 ± 2.0 |
| V108 Cγ2 /M108 Cε – V103 Cγ1 (Å) | 4.9 ± 0.9 | 4.3 ± 0.3 | 4.3 ± 0.3 | 6.4 ± 2.0 |
| V108 Cγ2 /M108 Cε – A106 Cβ (Å) | 4.5 ± 0.4 | 4.2 ± 0.4 | 4.2 ± 0.3 | 4.8 ± 0.8 |
All values were calculated using structures from the last 5 ns of each simulation. All values are expressed as means and standard deviations of the means from three independent simulations at 37°C.
The total solvent-accessible surface area (SASA) was determined using the NACCESS algorithm (65).
The following residues were used to calculate the SASA of the SAM-binding site: A39, M40, N41, V42, G66, A67, Y68, S72, E90, I91, N92, G117, A118, S119, Q120, D141, H142, and W143.
The following residues were used to calculate the catechol-binding site SASA: W38, M40, K46, D141, K144, D169, N170, C173, P174, L198, E199, Y200, and D205.
The following residues were used to calculate the A52T polymorphic site SASA: I49, V53, Q55, E56, Y194, and residue 52.
The following residues were used to calculate the A22S polymorphic site SASA: Q23, V25, L26, V74, A77, R78, A106, residue 108 and residue 22.
The following residues were used to calculate the V108M polymorphic site SASA: residue 22, A73, V74, R78, V103, A106, G107, K109, K111, V112 and residue 108.
p < 0.10.
p < 0.05.
Results and Discussion
V108M Polymorphism: Homology Model vs. Crystal Structure
Our earlier MD study examining the destabilizing effects of the V108M polymorphism utilized a homology model of human COMT generated from a crystal structure of the highly homologous rat protein (1VID, (53)), which shares 81% sequence identity with the human protein (52). Crystal structures of the human wild-type (3BWM, (54)) and V108M (3BWY, (54)) COMT proteins bound with both SAM and dinitrocatechol (DNC) are now available. Following energy minimization, the root-mean-square deviation of the Cα atoms (Cα-rmsd) of the human crystal structures and homology models differed by only 0.2 Å, and the positions of residues within the SAM- and catechol-binding sites were nearly identical suggesting that our homology model was appropriate. It is interesting to note that the crystal structures of wild-type and V108M COMT are virtually identical, probably due to the presence of SAM and DNC. The lack of information from these crystal structures regarding how the V108M polymorphism destabilizes COMT makes MD simulations of this system detailing the structural and dynamic consequences of this mutation all the more important.
We performed simulations of human wild-type and V108M COMT beginning with the human crystal structures to validate the results obtained from simulations using the homology models. Since the original results were published (52), our simulation package, ilmm, has been updated to include a scaling factor (C-scale) of 0.4 for non-bonded interactions between atoms in charge groups separated by three bonds (63, 64). Therefore, we have performed simulations of both COMT variants using a scaling factor of 0.4 for non-bonded interactions to verify that this change does not affect the previously published results (52), and to ensure consistency with our current simulation protocols, particularly for comparison with other proteins in our Dynameomics database (63, 68).
The overall methyltransferase fold was maintained in all of the simulations (Figure 2). Cα-rmsd plots of structures from the simulations using the human crystal structures and homology models at 25°C are very similar (Figure S1), consistent with experimental data that the WT and V108M behave similarly at low temperatures (21). Our focus here is the behavior of the proteins at physiological pH. Our previous work (52) looked extensively at how the V108M polymorphism affected COMT structure at 25, 37, and 50°C. Those results indicated that the proteins behave similarly at both 25 and 50°C, and that differences in structure and stability are most prevalent at 37°C. Experimental studies describing protein levels in vivo (25, 26), as well as protein stability and aggregation in vitro (21) also show larger differences between the WT and V108M proteins at 37°C.
Figure 2.
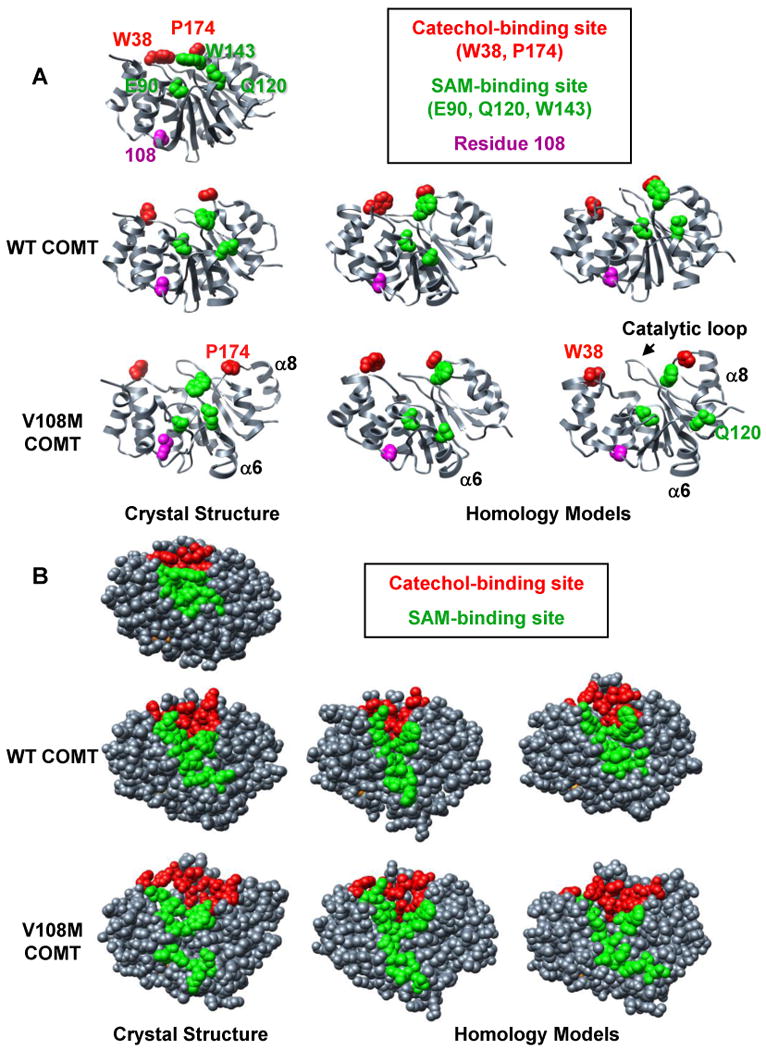
Comparison of MD simulations performed at 37°C using crystal structures and homology models of human wild-type and V108M COMT. Snapshots of structures from the final ns of MD simulations of (left) the crystal structures of human wild-type (3BWM.PDB, (54)) and V108M (3BWY.PDB, (54)) COMT, and (center, right) the homology models of human COMT based on the crystal structure of rat COMT (1VID.PDB, (53)) performed with C-scale values of either 0.0 (center) or 0.4 (right) (see Materials and Methods). (A) The 108M COMT structure is prone to disruption at 37°C. Altered packing around residue M108 reorients helix α6, pulling Q120 away from the SAM-binding site. Helix α8 (and P174) pulls away from the protein core resulting in an expanded conformation. These motions occur during simulations of both the crystal structure and homology models of the V108M COMT protein, but are not observed during the simulations of wild-type COMT. SAM-binding residues (E90, Q120, W143), catechol-binding residues (W38, P174) and residue 108 are shown in space-filling representation and colored in green, red, and magenta, respectively. (B) The 108M COMT active site is more exposed to solvent and distorted at 37°C than that of the wild-type protein. Residues in the SAM- and catechol-binding sites are colored green and red, respectively.
At 37°C, substituting Met for Val at position 108 expands the protein, increasing both the Cα-rmsd and the total solvent accessible surface area of the 108M protein relative to 108V COMT in all of the simulations (Table 1). The larger Met interacted differently than the Val with residues in helices α2 (A22), α4 (V74), and α5 (V103), altering the orientation of secondary structure elements neighboring the polymorphic site (Table 1, Figure 2). 108M COMT was also more prone to distortion in helices α6 (Q120) and α8 (P174), both of which contain active-site residues. These changes resulted in a significant increase in the solvent exposure of the SAM-binding sites of the 108M proteins relative to wild-type COMT (Table 1, Figure 2). While the magnitude of the differences between the 108V and 108M structures varied between simulations, the overall trends describing the destabilizing effects of the V108M polymorphism on COMT structure were consistent under all conditions tested. The results indicate that simulations employing the COMT homology models and the crystal structures provide consistent descriptions of the structural and dynamic effects of the V108M mutation. Also, the addition of a scaling factor for non-bonded interactions does not significantly alter the results of the COMT simulations at 37°C.
A crystal structure of the rat COMT apoprotein (2ZLB.PDB, (66)) is now available. The overall structures of the apo and holo rat proteins are very similar, differing only by a Cα-rmsd of 0.72 Å with all of the secondary structure elements (helices and β-strands) superposeable (Figure 3A). The major differences between the apo and holo structures lie in the loop regions that define the catechol-binding site. The α2-α3 loop (W38, M40), β5-α8 loop (P174), and the catalytic loop (residues 197-202) all pull away from the protein core in the COMT apoprotein exposing the catechol-binding site and a portion of the SAM-binding site (Figure 3A, B). Interestingly, the adenosine-binding pockets of the SAM sites are very similar in both the apo and holoproteins, showing only slight displacements in the positions of key active site residues E90, Q120, W143. COMT first binds SAM and then the catechol substrate in an ordered mechanism (7). The apoprotein structure suggests that the adenosine moiety of SAM directs binding of the co-substrate into a partially formed active-site pocket. It is probable that upon SAM docking, the α2-α3 and β5-α8 loops move in towards the protein core enabling active site residues W38, M40 and N170 to contact SAM. This rearrangement of loops would also bring “gate keeper” residues W38 and P174 closer together to form a binding pocket for the catechol substrate (Figure 3A). Because the rat and human COMT proteins share 81% sequence identity and the crystal structures of the rat and human COMT holoproteins are virtually identical (Figure 3A), it is reasonable to believe that the structure of the wild-type human apoprotein is similar to that of rat COMT.
The starting structure for all of the simulations of human COMT was a COMT holoprotein stripped of both SAM and the catechol substrate. Figure 3C shows the crystal structure of the rat COMT apoprotein overlaid with structures from the final ns of three independent simulations of the human wild-type and V108M COMT apoproteins. The structures from the simulations of human COMT are similar to that of the rat apoprotein structure in that the protein expands while the overall methyltransferase fold and the structure of the binding pocket for the adenosine moiety of SAM are maintained. In addition, the α2-α3, β5-α8 and catalytic loops become highly flexible, revealing the active site to facilitate substrate binding. These similarities suggest that our descriptions of the human apoprotein structures published in 2006 (52) and reported here are correct. These characteristic apoprotein features are also seen during simulations of the V108M variant (Figure 3C). Interestingly, the V108M apoprotein demonstrates an additional large displacement of β3 and α6 that opens up and disorders the SAM-binding pocket (Figure 3C). This displacement is a direct result of altered sidechain packing around the larger Met 108 in the nearby α5-β3 loop (Figures 3C, 5E, F). The β3-α6 motion is quite dynamic at 37°C (Figure 4C), and SAM is still able to bind to the V108M protein. Indeed, several studies have shown that the presence of SAM, which would stabilize α6 through interactions with S119 and Q120, protects both the helical structure and activity of COMT at elevated temperatures (10, 21). However, it is possible that the reorientation of α6 nucleates the destabilization of V108M COMT resulting in the degradation of a portion of the protein pool.
Figure 5.
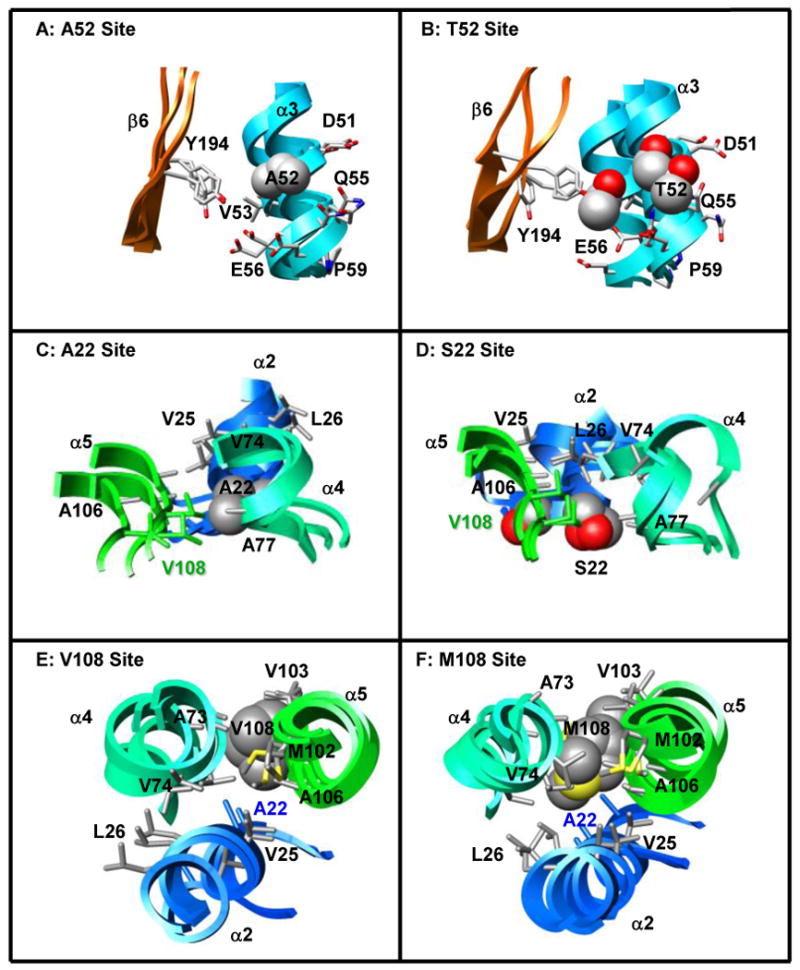
Effects of the A52T, A22S, and V108M COMT polymorphisms on local tertiary structure. (A, B) A52 (α3) forms mainchain hydrogen bonds with K48, I49, and E56 of α3, and sidechain contacts with residues in α3 (I49, V53, Q55, E56) and β6 (Y194). T52 maintains all of these contacts, while T52 OG1 forms additional hydrogen bonds with the backbone carbonyl groups of K48 and I49. (C, D) A22 is positioned in a loop between α1 and α2 where it interacts with residues in α2 (Q23, V25, L26), α4 (V74, A77, R78), and α5 (A106, V108). The larger S22 forms an additional S22 OG – G107 O hydrogen bond enabling closer contact between S22 and residues in α5 (A106, G107, V108). This altered packing moves S22 away from residues V74 and R78 of α4, affecting the orientations of both α2 and α4 relative to their positions in the wild-type protein. (E, F) Residue 108 is located in a loop between α5 and β3. Both V108 and M108 contact residues in α2 (A22), α4 (A73, V74, R78), and α5 (V103, A106). The larger methionine forms additional contacts with V25 and L26 of α2 and M102 of α5, effectively moving helices α2 and α5 closer together. Structures from the final ns of three independent simulations are superposed in A-F and colored to match Figure 1. Sidechains of the polymorphic residues and their contacts are shown in space-filling and stick representations, respectively.
Figure 4.

Mobility of the wild-type, A22S, A52T, and V108M COMT variants at 37°C. Cα-rmsf values (Å) per residue for the wild-type protein (blue) compared with those of the (A) A52T, (B) A22S, and (C) V108M COMT variants (all in red). Cα-rmsf values were calculated relative to the average structure over the last 10 ns of each simulation. Secondary structural elements are depicted as cylinders for α-helices and arrows for β-strands and are colored to match the structure in Figure 1. The polymorphic residues are marked by asterisks.
Competitive substrate-based COMT inhibitors have long been used in the treatment of Parkinson's disease (11, 12, 69-71). Recent studies linking the destabilizing V108M polymorphism with improved prefrontal cognition (45, 46) have led to the use of several of these COMT inhibitors to improve cortical processing in individuals carrying the wild-type COMT allele (72, 73). However, the 108M allele has also been associated with increased risk for breast cancer (20, 34, 35) and neuropsychiatric disease (36-40). Therefore, the development of stabilizing agents to supplement the treatment of schizophrenia and obsessive-compulsive disorder is also desirable. Because β3 and α6 are most readily affected by the V108M polymorphism in simulation and because SAM stabilizes COMT through interactions with residues in the β3-α6 loop, they represent a logical target site for the docking of a stabilizing small molecule that is not competitive with substrate binding. α5, β3 and α6 form a pocket, ∼12 Å in diameter and 6 Å deep, on the protein surface (Figure 3D). The pocket has a hydrophobic core and is bordered by several positively charged residues on α4 (R85), β3 (K109, K111) and α6 (K128, K129). In addition, T113 and Y130 could provide contacts for hydrogen bonding interactions with a stabilizing molecule (Figure 3D). It will be interesting to see if small molecules docked into this site do protect the structure of 108M COMT.
A52T Polymorphism
The A52T polymorphism does not significantly affect either the catalytic activity or thermal stability of COMT (14, 47). The kinetic similarities between the wild-type and A52T proteins are reflected in the simulations. Both proteins reach an overall Cα-rmsd of 3.0 Å and a total solvent-accessible surface area of 11,300 Å2 (Table 2, Figure S1). The average Cα-atom root-mean-square fluctuations (rmsf) values reflecting the degree of motion about the mean structure over all three simulations are shown in Figure 4A. The Cα-rmsf values for both the wild-type and A52T proteins follow similar patterns. The wild-type protein has higher Cα-rmsf values in the catalytic loop, while the 52T variant shows slight spikes in flexibility at residue 52 and in the loop regions containing both SAM- (E90, I91, S119, Q120) and catechol-binding (P174) residues. However, these differences in Cα-rmsf values do not translate to altered active-site structures. The solvent exposures of the SAM and catechol-binding sites are ∼975 and 930 Å2, respectively, in both the wild-type and A52T proteins (Table 2).
Residue 52 is a surface residue found in the third turn of helix α3 (Figure 1). A52 forms main-chain hydrogen bonds with K48, I49, and E56 of α3 and hydrophobic contacts with residues in α3 (I49, V53, Q55, E56) and β6 (Y194) (Table 2, Figure 5A). The larger Thr maintains all of these contacts and the CB atoms of A52 and T52 are positioned similarly throughout the simulations and oriented towards the solvent (Table 2, Figure 5A-B). Instead of interacting with any new nearby residues or solvent molecules, the OG1 atom of T52 forms additional hydrogen bonds with the backbone carbonyl groups of K48 and I49. Phylogenic analyses show conservation of an alanine residue at position 52 in most species. However, bos taurus and sus scrofa have an Arg and a Thr, respectively, at this position suggesting that COMT structure and function are tolerant of several substitutions at this location. The observation that the Ala to Thr polymorphism at residue 52 only minimally affects COMT structure both in simulation and experiment makes the simulations of A52T COMT good negative controls for the effects of computationally introducing mutations into wild-type crystal structures.
A22S Polymorphism
The A22S polymorphism significantly decreases the overall activity of COMT (47). The A22S variant loses ∼80% of its activity upon incubation at 45°C (47). The mechanism for this deactivation is unclear as the A22S mutation decreases the protein's Km for SAM while increasing its Km for hydroxyestradiol relative to the wild-type protein. We performed multiple MD simulations of the A22S COMT variant to investigate the molecular mechanisms underlying its inactivation.
The methyltransferase fold was maintained throughout all of the simulations. Structures of the wild-type protein and A22S variant reached Cα-rmsd values of 3.1 ± 0.2 and 4.0 ± 0.5 Å, respectively, during the simulations at 25 and 37°C (Table 2, Figure S1). The structure of 22S COMT expanded relative to the wild-type protein, with a 400 Å2 increase in total solvent accessible surface area (Table 2). Figure 4 shows Cα-rmsf values for both the wild-type and A22S COMT proteins at 37°C. The A22S variant displayed increased flexibility throughout the protein, with large peaks in Cα-rmsf values for residues in α1, α2 (S22), α5, α6, α7, and the catalytic loop, including SAM- and catechol-binding residues Q120 and E202.
Residue 22 is located in the loop between helices α1 and α2, ∼13 Å from the SAM-binding site and ∼20 Å from the catechol-binding site (Figure 1). No active-site residues are located within α1 or α2. However, the surface loop between α2 and α3 does contain W38, a ‘gate-keeper’ residue that forms a hydrophobic pocket with P174 of α8 to orient the catechol substrate for methylation. Ala 22 forms hydrogen bonds with Q23 OE1 and the backbone amides of V25 and L26. Hydrophobic contacts between A22 and residues in α2 (Q23, V25, L26), α4 (V74, A77, R78) and α5 (A106, V108), are maintained throughout the simulations (Table 2, Figure 5C). S22 maintains many of its contacts with α2 and α5, while forming an additional hydrogen bond with G107 (Table 2, Figure 5D). The S22-G107 interaction packs α2 and α5 more closely together, moving both S22 and α2 away from their contacts with V74, A77, and R78 in α4 (Table 2) and moving W38 away from residues P174 and E199 in the protein's C-terminus (Table 2, Figure 6). This motion moves the opposite (N-terminal) ends of α2 and α4 towards each other enabling salt-bridges between D30-R75-E34 to form. The formation of this network pulls α4 and α3 away from each other, and away from the protein's C-terminus, leading to an expansion of the overall structure and the opening of a large cleft in the protein (Figure 6B). This repacking around the polymorphic site is translated throughout the protein, significantly increasing the solvent exposure of the catechol-binding site ∼ 20 Å away, in agreement with the A22S mutation's detrimental effect on substrate binding (47) (Table 2).
Figure 6.
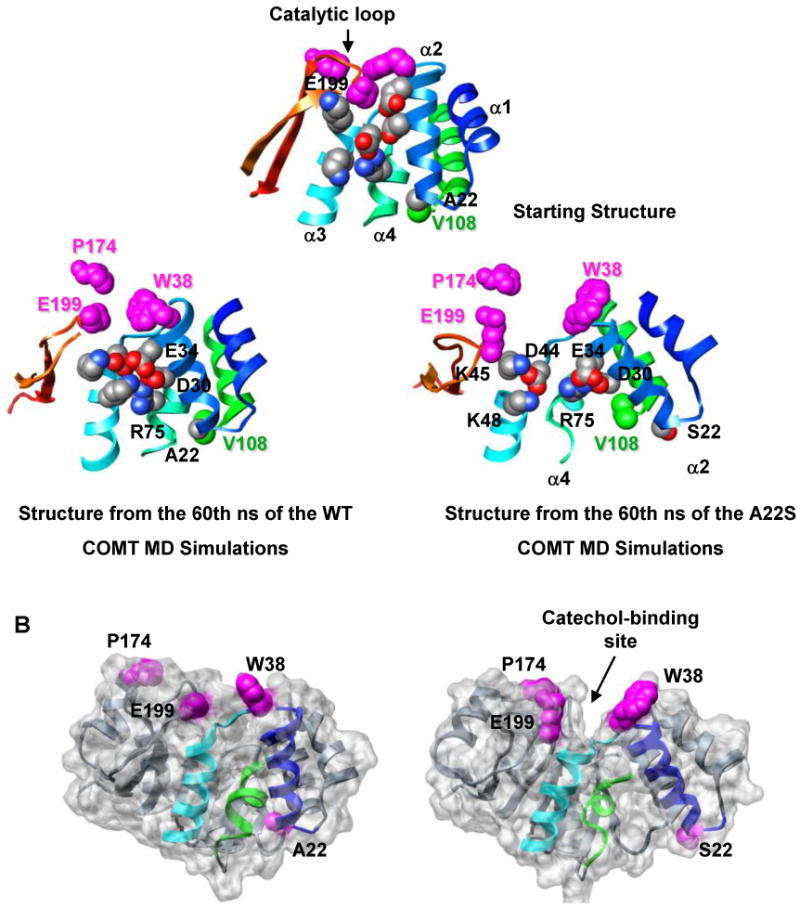
The A22S polymorphism disrupts the catechol-binding site. (A) The altered packing around residue S22 causes α2 and α5 to move towards each other while the last turn of α4 pulls away from the polymorphic site. This rearrangement results in the formation of new salt bridges between D30 (α2) – R75 (α4), E34 (α2) – R75 (α4), and E34 (α2) – K48 (α3). The altered packing around the polymorphic residue is propagated through α3 towards the protein's C-terminus leading to an overall expansion of the protein and the separation of key catechol-binding residues W38, P174, and E199. Residue 22, and residues involved in salt bridges are shown in space-filling representation and colored by atom type. V108 and catechol-binding residues are colored in green and magenta, respectively. Structures are colored as in Figure 1. (B) The rearrangement of α2 and α3 both increases the mobility of several residues in the α2-α3 loop including W38, a gate-keeping residue involved in catechol binding, and pulls these residues away from the protein's C-terminus. This movement opens a large cleft in the protein, which greatly increases the solvent exposure of the catechol-binding site, relative to the wild-type protein. Residue 22 and the catechol-binding residues (W38, P174, E199) are shown in space-filling representation and colored magenta. α2, α3, and α4 are colored as in Figure 1.
The A22S and V108M polymorphic hotspot
The A22S and V108M polymorphisms have been shown experimentally to decrease COMT stability and activity. The A22S mutation alters catechol binding (47), while the V108M mutation distorts the SAM-binding site (10, 21, 52). Both A22 (α1-α2) and V108 (α5-β3) are located in surface loops 16-20 Å from the active sites. However, the polymorphic residues pack against secondary structure elements that contain substrate-binding residues on their distal ends. Interestingly, polymorphic residues 22 and 108 contact each other throughout all of the simulations (Figure 5, Table 2), marking this region as a hotspot for mutations that regulate, but do not abolish, COMT activity. Substitution of the larger Ser and Met residues for Ala and Val similarly distort interactions and packing within the polymorphic sites of both residues 22 and 108 (Table 2). These packing differences are translated throughout both proteins resulting in large increases in the overall solvent-accessible surface area relative to the wild-type protein. However, the two polymorphisms have very different effects on the active sites. The A22S mutation results in the formation of a large cleft in the protein, distorting and significantly increasing the solvent exposure of the catechol-binding site. The introduction of S22 has little effect on the solvent exposure or structure of the SAM-binding site. The opposite is true for the V108M polymorphism, which distorts the SAM-binding site through the rearrangement of active-site residues positioned on α5 (E90, N92), β3 (A116) and α6 (S119, Q120). This difference could depend simply on whether SAM- or catechol-binding residues are on the distal ends of the reoriented helices (α2, α5 and α6).
It is interesting to note that the A22S and V108M polymorphisms developed independently in Northern European and Asian populations, possibly as different ways of destabilizing and deactivating the COMT protein. Both the 22S and 108M alleles have been associated with increased risk of cancer (20, 33-35, 47) and neuropsychiatric dysfunction (36-40, 48, 49). However, the 108M allele also confers improved prefrontal cognitive function, especially in working memory (44-46). These results suggest that individuals heterozygous for the V108M polymorphism may have an advantage, in that intermediate COMT activity may minimize the individual's risk for neuropsychiatric disease while slightly improving cortical processing. A similar phenomenon is seen with protein isoaspartate O-methyltransferase (PIMT), an enzyme critical for protein repair, where heterozygosity for a V119I polymorphism promotes successful aging, due in part to the complementary substrate affinities of the 119V and 119I proteins (74). Both the V108M and V119I mutations in COMT and PIMT, respectively, are present at high frequencies in Caucasian populations, with ∼50% of individuals heterozygous for either polymorphism (17, 74, 75). Further studies are necessary to determine if the A22S polymorphism is associated with any ‘beneficial’ phenotypes.
Supplementary Material
Figure 7.
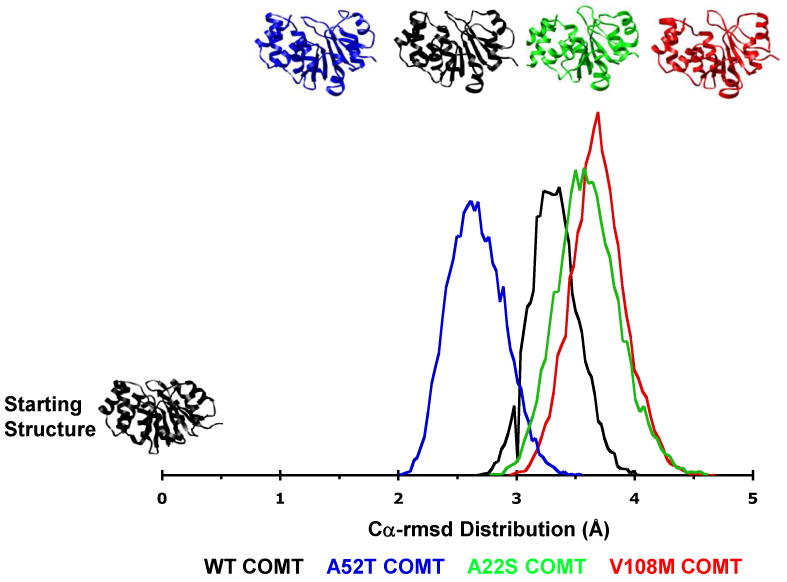
Total Cα–rmsd distributions of structures from MD simulations of wild-type, A22S, A52T, and V108M COMT. Structures from the WT COMT simulations fall into a narrow distribution centered at 3.3 Å Cα-rmsd from the starting structure. The 52T distribution is centered at 2.6 Å Cα-rmsd indicating that the A52T polymorphism somewhat stabilizes the protein structure. However, structures from the 52T COMT simulations fall into a broader distribution than those of the WT protein. Both the 22S and 108M COMT variants exist as ensembles of structures that are more distorted than those of the WT protein. The vertical scale is arbitrary.
Acknowledgments
Most simulations in this study were performed using processor time from Microsoft Research through Computing @ Microsoft (www.microsoft.com/science). Figures were produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081) (76). K.R. thanks Dr. William Parson for helpful discussions, and Amanda Jonsson for running the simulations of the COMT homology models.
- COMT
catechol O-methyltransferase
- Cα-rmsd
Cα-root-mean-square deviation from the starting structure
- Cα-rmsf
Cα-root-mean-square fluctuation about the mean structure
- DNC
3,5-dinitrocatechol
- MD
molecular dynamics
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SASA
solvent accessible surface area
- Tm
temperature resulting in 50% unfolding
- T50
temperature resulting in 50% loss of enzymatic activity
- WT
wild-type (22A, 52A, 108V) protein
Footnotes
Financial support for this work was provided by the National Institutes of Health (GM50789 to VD) and the Canadian Institutes of Health Research (DRA MD-75910 to KR).
Supporting Information Available
Evolution of Cα-rmsd (Å) with time for simulations of the WT, A22S, A52T, and V108M COMT proteins performed at 25°C and 37°C (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Axelrod J, Szara S. Enzymic conversion of metanephrine to epinephrine. Biochim Biophys Acta. 1958;30:188–189. doi: 10.1016/0006-3002(58)90259-2. [DOI] [PubMed] [Google Scholar]
- 2.Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- 3.Weinshilboum RM, Otterness DM, Szumlanski CL. Methylation pharmacogenetics: catechol O-methyltransferase, thiopurine methyltransferase, and histamine N-methyltransferase. Annu Rev Pharmacol Toxicol. 1999;39:19–52. doi: 10.1146/annurev.pharmtox.39.1.19. [DOI] [PubMed] [Google Scholar]
- 4.Tenhunen J, Salminen M, Jalanko A, Ukkonen S, Ulmanen I. Structure of the rat catechol-O-methyltransferase gene: separate promoters are used to produce mRNAs for soluble and membrane-bound forms of the enzyme. DNA Cell Biol. 1993;12:253–263. doi: 10.1089/dna.1993.12.253. [DOI] [PubMed] [Google Scholar]
- 5.Tenhunen J, Salminen M, Lundstrom K, Kiviluoto T, Savolainen R, Ulmanen I. Genomic organization of the human catechol O-methyltransferase gene and its expression from two distinct promoters. Eur J Biochem. 1994;223:1049–1059. doi: 10.1111/j.1432-1033.1994.tb19083.x. [DOI] [PubMed] [Google Scholar]
- 6.Rivett AJ, Francis A, Roth JA. Localization of membrane-bound catechol-O-methyltransferase. J Neurochem. 1983;40:1494–1496. doi: 10.1111/j.1471-4159.1983.tb13597.x. [DOI] [PubMed] [Google Scholar]
- 7.Rivett AJ, Roth JA. Kinetic studies on the O-methylation of dopamine by human brain membrane-bound catechol O-methyltransferase. Biochemistry. 1982;21:1740–1742. doi: 10.1021/bi00537a006. [DOI] [PubMed] [Google Scholar]
- 8.Huh MM, Friedhoff AJ. Multiple molecular forms of catechol-O-methyltransferase. Evidence for two distinct forms, and their purification and physical characterization. J Biol Chem. 1979;254:299–308. [PubMed] [Google Scholar]
- 9.Bonifati V, Meco G. New, selective catechol-O-methyltransferase inhibitors as therapeutic agents in Parkinson's disease. Pharmacol Ther. 1999;81:1–36. doi: 10.1016/s0163-7258(98)00032-1. [DOI] [PubMed] [Google Scholar]
- 10.Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melen K, Julkunen I, Taskinen J. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–4210. doi: 10.1021/bi00013a008. [DOI] [PubMed] [Google Scholar]
- 11.Lees AJ. Evidence-based efficacy comparison of tolcapone and entacapone as adjunctive therapy in Parkinson's disease. CNS Neurosci Ther. 2008;14:83–93. doi: 10.1111/j.1527-3458.2007.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan DK, Cordato DJ, O'Rourke F. Management for motor and non-motor complications in late Parkinson's disease. Geriatrics. 2008;63:22–27. [PubMed] [Google Scholar]
- 13.Factor SA. Current status of symptomatic medical therapy in Parkinson's disease. Neurotherapeutics. 2008;5:164–180. doi: 10.1016/j.nurt.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shield AJ, Thomae BA, Eckloff BW, Wieben ED, Weinshilboum RM. Human catechol O-methyltransferase genetic variation: gene resequencing and functional characterization of variant allozymes. Mol Psychiatry. 2004;9:151–160. doi: 10.1038/sj.mp.4001386. [DOI] [PubMed] [Google Scholar]
- 15.Saito S, Iida A, Sekine A, Miura Y, Sakamoto T, Ogawa C, Kawauchi S, Higuchi S, Nakamura Y. Identification of 197 genetic variations in six human methyltranferase genes in the Japanese population. J Hum Genet. 2001;46:529–537. doi: 10.1007/s100380170035. [DOI] [PubMed] [Google Scholar]
- 16.Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 17.Palmatier MA, Kang AM, Kidd KK. Global variation in the frequencies of functionally different catechol-O-methyltransferase alleles. Biol Psychiatry. 1999;46:557–567. doi: 10.1016/s0006-3223(99)00098-0. [DOI] [PubMed] [Google Scholar]
- 18.Weinshilboum RM, Raymond FA. Inheritance of low erythrocyte catechol-o-methyltransferase activity in man. Am J Hum Genet. 1977;29:125–135. [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, Kolachana BS, Hyde TM, Herman MM, Apud J, Egan MF, Kleinman JE, Weinberger DR. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman JE, Jensen LT, He P, Yager JD. Characterization of human soluble high and low activity catechol-O-methyltransferase catalyzed catechol estrogen methylation. Pharmacogenetics. 2002;12:517–528. doi: 10.1097/00008571-200210000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Rutherford K, Alphandery E, McMillan A, Daggett V, Parson WW. The V108M mutation decreases the structural stability of catechol O-methyltransferase. Biochim Biophys Acta. 2008;1784:1098–1105. doi: 10.1016/j.bbapap.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Cotton NJ, Stoddard B, Parson WW. Oxidative inhibition of human soluble catechol-O-methyltransferase. J Biol Chem. 2004;279:23710–23718. doi: 10.1074/jbc.M401086200. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Yao J, Chang M, Nikolic D, Yu L, Yager JD, Mesecar AD, van Breemen RB, Bolton JL. Equine catechol estrogen 4-hydroxyequilenin is a more potent inhibitor of the variant form of catechol-O-methyltransferase. Chem Res Toxicol. 2004;17:512–520. doi: 10.1021/tx0342464. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Yang X, Chang M, Yager JD, van Breemen RB, Bolton JL. Functional and structural comparisons of cysteine residues in the Val108 wild type and Met108 variant of human soluble catechol O-methyltransferase. Chem Biol Interact. 2005;152:151–163. doi: 10.1016/j.cbi.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Doyle AE, Goodman JE, Silber PM, Yager JD. Catechol-O-methyltransferase low activity genotype (COMTLL) is associated with low levels of COMT protein in human hepatocytes. Cancer Lett. 2004;214:189–195. doi: 10.1016/j.canlet.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 26.Doyle AE, Yager JD. Catechol-O-methyltransferase: effects of the val108met polymorphism on protein turnover in human cells. Biochim Biophys Acta. 2008;1780:27–33. doi: 10.1016/j.bbagen.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aksoy S, Klener J, Weinshilboum RM. Catechol O-methyltransferase pharmacogenetics: photoaffinity labelling and western blot analysis of human liver samples. Pharmacogenetics. 1993;3:116–122. doi: 10.1097/00008571-199304000-00008. [DOI] [PubMed] [Google Scholar]
- 28.Boudikova B, Szumlanski C, Maidak B, Weinshilboum R. Human liver catechol-O-methyltransferase pharmacogenetics. Clin Pharmacol Ther. 1990;48:381–389. doi: 10.1038/clpt.1990.166. [DOI] [PubMed] [Google Scholar]
- 29.Grossman MH, Szumlanski C, Littrell JB, Weinstein R, Weinshilboum RM. Electrophoretic analysis of low and high activity forms of catechol-O-methyltransferase in human erythrocytes. Life Sci. 1992;50:473–480. doi: 10.1016/0024-3205(92)90386-4. [DOI] [PubMed] [Google Scholar]
- 30.Scanlon PD, Raymond FA, Weinshilboum RM. Catechol-O-methyltransferase: thermolabile enzyme in erythrocytes of subjects homozygous for allele for low activity. Science. 1979;203:63–65. doi: 10.1126/science.758679. [DOI] [PubMed] [Google Scholar]
- 31.Spielman RS, Weinshilboum RM. Genetics of red cell COMT activity: analysis of thermal stability and family data. Am J Med Genet. 1981;10:279–290. doi: 10.1002/ajmg.1320100311. [DOI] [PubMed] [Google Scholar]
- 32.Weinshilboum RM, Dunnette J. Thermal stability and the biochemical genetics of erythrocyte catechol-O-methyltransferase and plasma dopamine-beta-hydroxylase. Clinical Genetics. 1981;19:426–437. doi: 10.1111/j.1399-0004.1981.tb00740.x. [DOI] [PubMed] [Google Scholar]
- 33.Dawling S, Roodi N, Mernaugh RL, Wang X, Parl FF. Catechol-O-methyltransferase (COMT)-mediated metabolism of catechol estrogens: comparison of wild-type and variant COMT isoforms. Cancer Res. 2001;61:6716–6722. [PubMed] [Google Scholar]
- 34.Wedren S, Rudqvist TR, Granath F, Weiderpass E, Ingelman-Sundberg M, Persson I, Magnusson C. Catechol-O-methyltransferase gene polymorphism and post-menopausal breast cancer risk. Carcinogenesis. 2003;24:681–687. doi: 10.1093/carcin/bgg022. [DOI] [PubMed] [Google Scholar]
- 35.Yim DS, Parkb SK, Yoo KY, Yoon KS, Chung HH, Kang HL, Ahn SH, Noh DY, Choe KJ, Jang IJ, Shin SG, Strickland PT, Hirvonen A, Kang D. Relationship between the Val158Met polymorphism of catechol O-methyl transferase and breast cancer. Pharmacogenetics. 2001;11:279–286. doi: 10.1097/00008571-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Lachman HM, Morrow B, Shprintzen R, Veit S, Parsia SS, Faedda G, Goldberg R, Kucherlapati R, Papolos DF. Association of codon 108/158 catechol-O-methyltransferase gene polymorphism with the psychiatric manifestations of velo-cardio-facial syndrome. Am J Med Genet. 1996;67:468–472. doi: 10.1002/(SICI)1096-8628(19960920)67:5<468::AID-AJMG5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 37.Lachman HM, Nolan KA, Mohr P, Saito T, Volavka J. Association between catechol O-methyltransferase genotype and violence in schizophrenia and schizoaffective disorder. Am J Psychiatry. 1998;155:835–837. doi: 10.1176/ajp.155.6.835. [DOI] [PubMed] [Google Scholar]
- 38.Nolan KA, Volavka J, Czobor P, Cseh A, Lachman H, Saito T, Tiihonen J, Putkonen A, Hallikainen T, Kotilainen I, Rasanen P, Isohanni M, Jarvelin MR, Karvonen MK. Suicidal behavior in patients with schizophrenia is related to COMT polymorphism. Psychiatr Genet. 2000;10:117–124. doi: 10.1097/00041444-200010030-00003. [DOI] [PubMed] [Google Scholar]
- 39.Strous RD, Bark N, Parsia SS, Volavka J, Lachman HM. Analysis of a functional catechol-O-methyltransferase gene polymorphism in schizophrenia: evidence for association with aggressive and antisocial behavior. Psychiatry Res. 1997;69:71–77. doi: 10.1016/s0165-1781(96)03111-3. [DOI] [PubMed] [Google Scholar]
- 40.Strous RD, Nolan KA, Lapidus R, Diaz L, Saito T, Lachman HM. Aggressive behavior in schizophrenia is associated with the low enzyme activity COMT polymorphism: a replication study. Am J Med Genet B Neuropsychiatr Genet. 2003;120B:29–34. doi: 10.1002/ajmg.b.20021. [DOI] [PubMed] [Google Scholar]
- 41.Diatchenko L, Nackley AG, Slade GD, Bhalang K, Belfer I, Max MB, Goldman D, Maixner W. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain. 2006;125:216–224. doi: 10.1016/j.pain.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 42.Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]
- 43.Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both beta2- and beta3-adrenergic receptors. Pain. 2007;128:199–208. doi: 10.1016/j.pain.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheldrick AJ, Krug A, Markov V, Leube D, Michel TM, Zerres K, Eggermann T, Kircher T. Effect of COMT val158met genotype on cognition and personality. Eur Psychiatry. 2008;23:385–389. doi: 10.1016/j.eurpsy.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 45.Goldberg TE, Egan MF, Gscheidle T, Coppola R, Weickert T, Kolachana BS, Goldman D, Weinberger DR. Executive subprocesses in working memory: relationship to catechol-O-methyltransferase Val158Met genotype and schizophrenia. Arch Gen Psychiatry. 2003;60:889–896. doi: 10.1001/archpsyc.60.9.889. [DOI] [PubMed] [Google Scholar]
- 46.Meyer-Lindenberg A, Kohn PD, Kolachana B, Kippenhan S, McInerney-Leo A, Nussbaum R, Weinberger DR, Berman KF. Midbrain dopamine and prefrontal function in humans: interaction and modulation by COMT genotype. Nat Neurosci. 2005;8:594–596. doi: 10.1038/nn1438. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Yang X, van Breemen RB, Bolton JL. Characterization of two new variants of human catechol O-methyltransferase in vitro. Cancer Lett. 2005;230:81–89. doi: 10.1016/j.canlet.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 48.Hong JP, Lee JS, Chung S, Jung J, Yoo HK, Chang SM, Kim CY. New functional single nucleotide polymorphism (Ala72Ser) in the COMT gene is associated with aggressive behavior in male schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:658–660. doi: 10.1002/ajmg.b.30649. [DOI] [PubMed] [Google Scholar]
- 49.Lee SG, Joo Y, Kim B, Chung S, Kim HL, Lee I, Choi B, Kim C, Song K. Association of Ala72Ser polymorphism with COMT enzyme activity and the risk of schizophrenia in Koreans. Hum Genet. 2005;116:319–328. doi: 10.1007/s00439-004-1239-y. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka Y, Hirata H, Chen Z, Kikuno N, Kawamoto K, Majid S, Tokizane T, Urakami S, Shiina H, Nakajima K, Dhir R, Dahiya R. Polymorphisms of catechol-O-methyltransferase in men with renal cell cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:92–97. doi: 10.1158/1055-9965.EPI-06-0605. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka Y, Sasaki M, Shiina H, Tokizane T, Deguchi M, Hirata H, Hinoda Y, Okayama N, Suehiro Y, Urakami S, Kawakami T, Kaneuchi M, Pookot D, Igawa M, Okuyama A, Ishii N, Dahiya R. Catechol-O-methyltransferase gene polymorphisms in benign prostatic hyperplasia and sporadic prostate cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:238–244. doi: 10.1158/1055-9965.EPI-05-0550. [DOI] [PubMed] [Google Scholar]
- 52.Rutherford K, Bennion BJ, Parson WW, Daggett V. The 108M polymorph of human catechol O-methyltransferase is prone to deformation at physiological temperatures. Biochemistry. 2006;45:2178–2188. doi: 10.1021/bi051988i. [DOI] [PubMed] [Google Scholar]
- 53.Vidgren J, Svensson LA, Liljas A. Crystal structure of catechol O-methyltransferase. Nature. 1994;368:354–358. doi: 10.1038/368354a0. [DOI] [PubMed] [Google Scholar]
- 54.Rutherford K, Le Trong I, Stenkamp RE, Parson WW. Crystal structures of human 108V and 108M catechol O-methyltransferase. J Mol Biol. 2008;380:120–130. doi: 10.1016/j.jmb.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 55.Alden RG, Parson WW, Chu ZT, Warshel A. Orientation of the OH dipole of tyrosine (M)210 and its effect on electrostatic energies in photosynthetic bacterial reaction centers. Journal Physical Chemistry. 1996;100:16761–16770. [Google Scholar]
- 56.Levitt M. ENCAD, Energy Calculations and Dynamics. Molecular Applications Group; Palo Alto, CA, and Yeda, Rehovot, Israel: 1990. [Google Scholar]
- 57.Beck DAC, Alonso DOV, Daggett V. ilmm, in lucem Molecular Mechanics. University of Washington; Seattle, WA: 2004-2008. [Google Scholar]
- 58.Beck DA, Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 59.Levitt M, Hirshberg M, Sharon R, Daggett V. Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Computer Physics Communications. 1995;91:215–231. [Google Scholar]
- 60.Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. Journal of Physical Chemistry B. 1997;101:5051–5061. [Google Scholar]
- 61.Kell GS. Precise representation of volume properties of water at one atmosphere. Journal of Chemical Engineering Data. 1967;12:66–69. [Google Scholar]
- 62.Beck DA, Armen RS, Daggett V. Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochemistry. 2005;44:609–616. doi: 10.1021/bi0486381. [DOI] [PubMed] [Google Scholar]
- 63.Beck DA, Jonsson AL, Schaeffer RD, Scott KA, Day R, Toofanny RD, Alonso DO, Daggett V. Dynameomics: mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations. Protein Eng Des Sel. 2008;21:353–368. doi: 10.1093/protein/gzn011. [DOI] [PubMed] [Google Scholar]
- 64.Armen RS, Bernard BM, Day R, Alonso DO, Daggett V. Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative diseases. Proc Natl Acad Sci U S A. 2005;102:13433–13438. doi: 10.1073/pnas.0502068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hubbard SJ, Thorton JM. NACCESS, Department of Biochemistry and Molecular Biology. University College; London: 1993. [Google Scholar]
- 66.Tsuji E, Okazaki K, Isaji M, Takeda K. Crystal structures of the Apo and Holo form of rat catechol-O-methyltransferase. J Struct Biol. 2009;165:133–139. doi: 10.1016/j.jsb.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 67.Hoel PG. Introduction to Mathematical Statistics. 2nd. John Wiley; New York: 1954. [Google Scholar]
- 68.Benson N, Daggett V. Dynameomics: Large-Scale Assessment of Native Protein Flexibility. Protein Sci. 2008 doi: 10.1110/ps.037473.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Masjost B, Ballmer P, Borroni E, Zurcher G, Winkler FK, Jakob-Roetne R, Diederich F. Structure-based design, synthesis, and in vitro evaluation of bisubstrate inhibitors for catechol O-methyltransferase (COMT) Chemistry. 2000;6:971–982. doi: 10.1002/(sici)1521-3765(20000317)6:6<971::aid-chem971>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 70.Kaakkola S, Gordin A, Mannisto PT. General properties and clinical possibilities of new selective inhibitors of catechol O-methyltransferase. Gen Pharmacol. 1994;25:813–824. doi: 10.1016/0306-3623(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 71.Kaakkola S, Teravainen H, Ahtila S, Rita H, Gordin A. Effect of entacapone, a COMT inhibitor, on clinical disability and levodopa metabolism in parkinsonian patients. Neurology. 1994;44:77–80. doi: 10.1212/wnl.44.1.77. [DOI] [PubMed] [Google Scholar]
- 72.Apud JA, Mattay V, Chen J, Kolachana BS, Callicott JH, Rasetti R, Alce G, Iudicello JE, Akbar N, Egan MF, Goldberg TE, Weinberger DR. Tolcapone improves cognition and cortical information processing in normal human subjects. Neuropsychopharmacology. 2007;32:1011–1020. doi: 10.1038/sj.npp.1301227. [DOI] [PubMed] [Google Scholar]
- 73.Apud JA, Weinberger DR. Treatment of cognitive deficits associated with schizophrenia: potential role of catechol-O-methyltransferase inhibitors. CNS Drugs. 2007;21:535–557. doi: 10.2165/00023210-200721070-00002. [DOI] [PubMed] [Google Scholar]
- 74.DeVry CG, Clarke S. Polymorphic forms of the protein L-isoaspartate (D-aspartate) O-methyltransferase involved in the repair of age-damaged proteins. J Hum Genet. 1999;44:275–288. doi: 10.1007/s100380050161. [DOI] [PubMed] [Google Scholar]
- 75.Tsai W, Clarke S. Amino acid polymorphisms of the human L-isoaspartyl/D-aspartyl methyltransferase involved in protein repair. Biochem Biophys Res Commun. 1994;203:491–497. doi: 10.1006/bbrc.1994.2209. [DOI] [PubMed] [Google Scholar]
- 76.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.