

Abstract
The isolation and preconcentration of estrogens from new types of biological samples (acellular and protein-free simulated body fluid) by molecularly imprinted solid-phase extraction has been described. In this technique, supramolecular receptors, namely molecularly imprinted polymers (MIPs) are used as a sorbent material. The recognition sites of MIPs were prepared by non-covalent multiple interactions and formed with the target 17β-estradiol as a template molecule. High-performance liquid chromatography with spectroscopic UV, selective, and a sensitive electrochemical CoulArray detector was used for the determination of 17β-estradiol, estrone, and estriol in simulated body fluid which mimicked human plasma.
Keywords: Estrogens, MIPs, SPE, HPLC-UV/EC
Introduction
Attention of many scientists has been drawn to estrogen hormones and their metabolites as substances of both positive and negative clinical significance in the human body. Estrogen synthesis and metabolism take place in men and women in the same way [1]. The proper level of these sexual hormones in the body determines not only fertility and overall condition and functioning of the organism but may also have an effect on development of different diseases including cancer. Therefore, the determination of sexual hormones is very important, and effective analytical procedures must be applied to quantify such analytes in relatively complicated matrices, like tissues or body fluids. Here, the sample preparation methods are of high importance [2]. One of the most important techniques used for the preconcentration of the analytes is solid-phase extraction (SPE) [3–5]. SPE is characterized by a high load, high recovery, enhanced reproducibility, automation capability, and wide spectrum of available stationary phases [6–8]. Typical sorbents for SPE (alkylsilicas, styrene-divinylbenzene, graphitized carbon black, etc.) are not selective, and a large amount of matrix interferents are extracted simultaneously with the target analyte [3, 4]. This factor decreases separation and enrichment efficiency [9, 10]. One of the good solutions is application of “fingerprint adsorbents” like molecularly imprinted polymers. The main task during elaboration of such materials is to mimic the molecular recognition of biological molecules such as receptors, enzymes, antibodies and hormones, for example estrogens. The result of a specially designed synthesis is a three-dimensional network complementary to the structure of a template molecule in the shape and functional group arrangements [11–16]. Two different approaches—covalent and non-covalent—have been developed in molecular imprinting [17]. In both approaches, the functional monomers are chosen to provide interactions with the functional groups of the imprint template. The non-covalent approach is more frequently used due to the following properties: (a) the tedious synthesis of pre-polymerization complex is avoided, (b) removal of the imprint molecule is usually accomplished by a continuous extraction, (c) the multiplicity of functionalities is reached into the molecularly imprinted polymer (MIP) binding sites [18, 19]. MIPs are usually stable at high temperatures and in most of the organic solvents. MIPs present a number of advantages compared to antibodies. For example, immune responses are by nature unpredictable, irreproducible, and can require long periods of time to achieve [20–22]. The bulk polymerization method is often used for the preparation of MIPs. The obtained polymers should be crushed, ground, and sieved to produce packing materials (SPE, HPLC) [23]. Such features of MIPs are also attractive as sorbents for solid-phase extraction as discussed here. A number of MIPs were prepared for steroids such as cholesterol, testosterone [21] 17β-estradiol, [24–26], castasterone [27], 11-α-hydroxyprogesterone and corticosteroids [28, 29]. Only a few imprinted polymers have been applied as separation media. Recent developments in molecularly imprinted solid-phase extraction (MISPE) have been reviewed by several authors [6, 30]. Numerous studies were also devoted to determination of steroids in biological samples by HPLC coupled with various detection techniques [31–36].
In this contribution, we describe the determination of selected estrogens (17β-estradiol, estrone, and estriol) in an acellular and protein-free simulated body fluid with ion concentrations nearly equal to those present in the human plasma. Solid-phase extraction based on MIPs was used for selective isolation and preconcentration of the estrogens from bio-fluids. The MISPE method was followed by HPLC with UV detector as well as a multisensor electrochemical CoulArray detection which was found to be very useful for trace determination and as one of the most sensitive and selective detection technique, especially for biologically active compounds.
Experimental
Apparatus and chromatographic conditions
The HPLC system was an Agilent 1100 Series model (Agilent Technologies, Waldbronn, Germany) equipped with a diode array detector and ChemStation software for data collection. The system was also connected to CoulArray detector model 5600A (ESA Technologies, Chelmsford, USA) and ESA software for data collection and control of the instrument. The acetonitrile–ammonium acetate buffer 25 mM (50:50% v/v) was used as a mobile phase at a flow rate of 1 mL min−1 and injection volume was 10 μL. The column Supelcosil LC-C18-DB (250 mm × 4.6 mm, d p = 5μm) was kept in the column oven (Gynkotek, Germering, Germany) at the temperature of 30 °C. The analytes were detected at the wavelength λ = 280 nm using DAD and at the potential of 790 mV using CoulArray detector. Solid-phase extraction was performed using a 12-port vacuum manifold from Mallinckrodt Baker (Deventer, The Netherlands). MISPE columns were prepared with SPE glass columns equipped with porous PTFE disks used at the top and at the bottom of the polymer bed. Hyperchem Release 7.0 software was used for the molecular modeling (Hypercube Inc., Gainesville, Florida, USA).
Materials
Methacrylic acid (MAA), trimethylolpropane trimethacrylate (TRIM), 2,2′-azobis(2-methylpropionitrile) (AIBN), 17β-estradiol was purchased from Aldrich (St. Louis, USA). Isooctane (2,2,4-trimethylpentane), toluene, acetonitrile, methanol, 2-propanol, ethyl acetate, ammonium acetate were obtained from J.T. Baker (New Jersey, USA). NaCl (99.5%), NaHCO3 (99.5%), KCl (99.0%), Na2HPO4·2H2O (99.5%), MgCl2·6H2O (99.0%), Na2SO4, Tris (99.5%), CaCl2·H2O (99%) and HCl (37 vol.%) were purchased from POCh (Gliwice, Poland). Water was obtained in our laboratory using a Milli-Q ultrapure water producing system Millipore (Billerica, USA).
Preparation of polymers by non-covalent “bulk” polymerization
The estrogen imprinted polymers were prepared by dissolving the imprint template (17β-estradiol; 0.125 mmol), functional monomer (MAA) the cross-linking monomer (TRIM or EDMA) and the initiator (AIBN; 2 wt.% of total monomer) in porogen solvents (isooctane–toluene 1:99, v/v) or (2-propanol) in a volumetric flask. The composition of polymers is described in Table 1, and the polymerization process is schematically shown in the Fig. 1. Except for the composition of the mixtures mentioned in the Table 1, an initial series of polymers was also prepared to determine the best monomer:crosslinker molar ratio, namely 1:1.15, 1:2.3, 1:4, and 1:9. The polymerization mixture was transferred to 2-mL clear glass vials, then it was purged with nitrogen for 10 min and the vials were immediately closed. The UV polymerization (λ = 365 nm) was performed in a temperature controlled chamber at 20 °C, 4°C, 0 °C, −4 °C for 2–4 h. After the polymerization was complete, the polymers were crushed and the remained unreacted compounds were extracted with acetonitrile using a Soxhlet apparatus for 24 h. Finally, the MIP and non-imprinted polymers (NIP) were dried in a vacuum oven at 40 °C for 24 h.
Table 1.
Composition of the synthesized molecularly imprinted polymers with 17β-estradiol as a template molecule, functional monomer–crosslinker ratio 1:4
| No. | Monomers | Porogen | T (°C) | Time (h) | Usefulness |
|---|---|---|---|---|---|
| P1 | MAA + EDMA | Isooctane–toluene (1:99% v/v) | 20 | 2 | Very small poresa |
| P2 | 4 | Very small poresa | |||
| P3 | 0 | 4 | Non porous | ||
| P4 | −4 | No polymer | |||
| P5 | 2-propanol | 20 | 2 | Very small poresa | |
| P6 | 4 | Very small poresa | |||
| P7 | 0 | 4 | Not homogeneous | ||
| P8 | −4 | No polymer | |||
| P9 | MAA + TRIM | Isooctane–toluene (1:99% v/v) | 20 | 2 | Porousb |
| P10 | 4 | Porousa | |||
| P11 | 0 | 3 | Not homogeneous | ||
| P12 | −4 | 4 | No polymer | ||
| P13 | 2-propanol | 20 | 2 | Porousa | |
| P14 | 4 | Porousa | |||
| P15 | 0 | 3 | Very small poresa | ||
| P16 | −4 | 4 | Not homogeneous |
A rough selection is also presented in a right-hand column
aMicropores (below 2 nm)
bMesopores (in range 2–50 nm)
Fig. 1.

The schematic picture of MIPs synthesis
Preparation of MISPE cartridges
The obtained polymers were ground in a mortar and passed through 63, 90, and 250 μm sieves. The granular fractions of average dimensions of 63 ÷ 90, 90 ÷ 250, and above 250 μm were collected. Twenty or 50 mg of the polymers were packed into 1 or 3 mL (only 50 mg) SPE glass columns.
Acellular and protein-free simulated body fluid samples
Acellular and protein-free simulated body fluid (SBF) used as a biological sample was prepared according to the chemical composition of a human body fluid, with ion concentrations nearly equal to those of the inorganic constituents of human body plasma. SBF is known to be a metastable buffer solution and even a small, undesired variance in both of the preparation steps and storage temperatures, may drastically affect the phase purity and high-temperature stability [37]. We found that the samples could be stored for a month at 5°C without any degradation.
Molecularly imprinted solid-phase extraction procedure
The cartridges were conditioned with methanol and water. The spiked samples of acellular simulated body fluid (0.1 µg mL−1) were applied to the conditioned column. Then the analytes were eluted with different solvents (acetonitrile, acetone, ethyl acetate, and 2-propanol) each applied in 3 mL aliquots. Extracts were evaporated to dryness and the residue dissolved in 100 μL of the mobile phase and injected into the HPLC system. Stock solutions of estrogens were prepared in the mobile phase and stored at approximately −4 °C.
Results and discussion
Effect of polymerization temperature
To create a strong and selective binding, it is necessary that the template molecule preorganize the functional monomer in a temperature stable configuration prior to polymerization. It is clearly defined that the higher temperature disrupts the balance of template-functional monomer complex resulting in a decrease in the number of strongly binding cavities. Following the literature data, we chose the UV-initiated process to synthesize polymers at lower temperatures. Temperatures of 20, 4, 0, or −4 °C were chosen because 17β-estradiol is not hampered with solubility problems when isooctane–toluene (1:99% v/v) and 2-propanol are used as porogens. The SEM microscopy was used for a rough assessment of the polymers. Our idea was to obtain polymers which morphology is similar to polymeric monoliths, that is the polymer consists of globules (preferably having mesopores) and flow-through pores which allow the liquid sample to flow through. Therefore, we were choosing those polymers which were homogeneous and possessing flow-through pores visible on SEM micrographs. In Table 1, we presented the results of this rough assessment. When the synthesis was performed at temperatures of 20 and 4 °C the obtained polymers were characterized by a uniform and adequate globule size (P5, P6, P9, P10, P13, P14, Table 1). The temperature 0 °C led to a poor globule size or irregular creation of globules in polymers P3, P7, P11, P15 (Table 1). The polymerization process did not run properly at −4 °C (P4, P8, P12, P16, Table 1) which resulted in a soft, wet and gel-like polymeric unusable material. We also adjusted the time of exposition to UV radiation during synthesis at different temperatures to complete the polymerization. It varied from 2 h (20 and 4 °C) through 3–4 h (0 °C) up to 4 h (−4 °C). We can assume that the temperatures above 0 °C and 2 h as exposition time are appropriate for preparation of useful MIPs. These facts also confirmed the comparison of the recovery results performed by 17β-estradiol MISPE analysis illustrated in the Table 2.
Table 2.
Comparison of recovery of 17β-estradiol MISPE analysis using different MIPs as cartridges
| Polymers | R (%)a | RSD (%) |
|---|---|---|
| P1 | 61.96 | 0.63 |
| P2 | 64.81 | 0.62 |
| P5 | 65.77 | 0.89 |
| P6 | 68.93 | 0.78 |
| P9 | 82.15 | 0.64 |
| P10 | 88.96 | 0.49 |
| P13 | 86.53 | 0.58 |
| P14 | 94.19 | 0.86 |
| P15 | 40.61 | 0.56 |
a50 mg cartridge in 1-mL glass column
Non-covalent imprinting with MAA, EDMA, or TRIM
Throughout this research, the MIPs as artificial recognition systems for 17β-estradiol, estrone and estriol were synthesized by bulk polymerization with a non-covalent imprinting approach. The non-covalent manner pioneered by Mosbach and co-workers [38–40], and later developed by Sellergren et al. [41, 42] and Andersson [43] was chosen to provide the appropriate binding of 17β-estradiol template by utilization of H-bonding which is a dominant interaction in the biological systems. In our artificial system the phenolic A-ring with hydroxyl group at the 3-position and D-ring with hydroxyl group at 17-position are capital factors responsible for the affinity and binding of 17β-estradiol to the imprinted and non-imprinted polymers. MAA was chosen as a functional monomer due to the presence of carboxylic groups in a molecule and related potential for creating hydrogen bonding and ionic interactions with 17β-estradiol molecule. The acid carboxylic groups in MAA are responsible for the interactions with −OH group in C3 and C17 position of 17β-estradiol molecule and can lead to the formation of a stable and strong template-functional monomer pre-polymerization complex resulting in the optimal non-covalent imprinting sites in the resulting imprinted polymer. The key problem connected with MIPs is to find out the preparation way for creation of useful and effective polymer with highly selective recognition sites.
Except for the functional monomer, one of the basic factors is choice of crosslinker to assure good supramolecular recognition which may be connected with the nature, solubility of crosslinkers to be used, degree of cross-linking and the length of polymerizable chains. Due these facts, ethylene glycol dimethacrylate (EDMA) and TRIM were used as cross-linking agents in this work. The utilization of EDMA is based on previous findings [17, 44–47]. According to Wulff et al. [48] short alkyl or oxyethylene chain between two methacrylate units in EDMA molecule led to the highest selectivity in the formed polymers. EDMA has so far proven to be the optimum choice, the best compromise between flexibility/rigidity and length [49]. Another study [50] showed that in the optimization of polymer selectivity EDMA turned out to be more universal cross-linking agent employed. In connection with our application of EDMA as a cross-linking agent used to fix MAA-17β-estradiol pre-polymerization complex we can state that it gave ambiguous results. Eight polymers (denoted as P1-P8) were synthesized with EDMA (see Table 1). Only MIPs P1, P2, P5, P6 were useful as sorbents in subsequent MISPE preconcentration. Taking into account these results, we continued syntheses using TRIM which was found to work better. The possible explanation is that TRIM is one carbon bond longer than EDMA and may fix in a better way the created polymer network in the pre-polymerization MAA-17β-estradiol complex. This fact was also confirmed in the previous studies of Shea, Sellergren, and Mosbach [51, 52] who reported that using TRIM resulted in polymers capable of higher loadings in comparison to EDMA supports.
The amount of the crosslinker is another factor affecting MIP quality. The polymers with functional monomer:crosslinker (M:C) molar ratio of 1:1.5, 1:2.3, 1:4, 1:9 were also prepared. According to the imprinting literature [48, 50] the amount of crosslinker is very important and the addition varied from a few percent up to ∼100%. During our initial investigation, the crosslinker amount also varied and keeping the amount of template constant we observed a surprising effect. At low amounts, 1:1.5, 1:2.3 of M:C ratio, corresponding to 20 % of crosslinker total molar ratio, the obtained polymers were soft and unstable so they did not meet the requirements of the SPE material. On the other hand, the MIPs prepared with M:C =1:9 (crosslinker amount 90%) were hard, non-porous, and smooth without globules. The application of 1:4 monomer to crosslinker ratio resulted in creation of porous, compact and adequate globule-sized polymers (P1, P2, P5, P6, P9, P10, P13, P14) which were used in our further studies. The scanning electron microscope was used for verification of the structural integrity of particles of P1–P16 as well as for a rough assessment of the pore/globule sizes (this can be found in the Table 1). The syntheses of the polymers P4, P8, P12, P16 (Table 1) were not successful and wet, gel-like, and soft materials were obtained in these cases. The SEM observation of the polymers P3, P7, P11, P16 revealed their inhomogeneous nature—parts of the polymers were porous while the other fragments were a hard, non-porous polymer. The polymers P9, P13, P14 with 80% (molar) amount of TRIM (Table 1) were characterized by more compact and regular pore/globule size than polymers P1, P2, P5, P6 prepared with the same amount of EDMA. Despite the negative characteristics of many of the synthesized polymers, most of them were compared with respect to the recovery they provide. Fifty-milligram portions of the P1, P2, P5, P6, P9, P10, P13, P14, and P15 polymers were then packed into the 1-mL glass SPE columns and were used for extraction of 17β-estradiol. The comparison of the results (Table 2) clearly shows the advantage of TRIM over EDMA and indicates the optimal temperature of the polymerization (+4 °C). The P10 and P14 polymers were then used for further investigation. The SEM images and comparison of considerable differences in the morphology of 17β-estradiol imprinted polymer and its non-imprinted control sample are presented in Fig. 2a, b. The NIP (Fig. 2a) has a smoother morphology with more submicrometer-sized globules/pores. This example shows that relatively small amount of the template may have a significant effect on polymer morphology.
Fig. 2.

Exemplary SEM micrographs of non-imprinted polymer (a) and 17β-estradiol imprinted polymer P12 (b)
17β-estradiol as an imprint template
It is generally believed that the amount of the template is crucial for the performance and practical usefulness of MIPs. A current critic of imprinted polymers is the significant amount of template needed to prepare the polymer. For example, to prepare a 5-g batch of MIP, with the template:monomer (T:M) ratios of 1:4 to 1:10 depending on the molecular weight of the template, up to several hundred mg of the template are needed. As a general guideline reports in the imprinting literature, the used amount of template is 1 mmol [21, 22, 24–26]. In our work, smaller 17β-estradiol concentration with respect to the functional monomer was investigated. We tried to synthesize polymers with 0.125 mmol amount of 17β-stradiol. The resulting template:monomer ratio was 1:64 or 1:160 (17β-estradiol:MAA). The main advantages of using only 0.125 mmol amount of 17β-estradiol as imprint molecule were fewer problems with washing out of template in the extraction step and minimizing costs of the prepared polymers. It should also be easier to find a total amount of the template to be sufficient to create selective recognition sites. Such an approach was also reported by several authors, for example Yilmaz, Mosbach, and Haupt [53] who investigated how much template was actually needed to create a sufficient number of binding sites. They examined polymers that were synthesized using different template:monomer ratios ranging from 1:4 to 1:5,000. Surprisingly, even at T:M as high as mentioned above, the MIP still bound significantly more of the analyte than the non-imprinted reference material at the same polymer concentration. Consequently, the preliminary experiments referenced above also confirmed our results that it is possible and absolutely appropriate to reduce the amount of template in the polymer recipe.
Choice of porogen
The selection of an appropriate porogen solvent is a key parameter in designing the properties of MIPs. The porogen affects the complexation strength of non-covalent binding sites between monomers and a template. It serves as a reaction environment and should provide a good miscibility with other constituents; it should not disrupt hydrogen bonds, and it also determines the size, shape, and distribution of pores and the surface area of the resulting material. Hence, the basic problem in MIP synthesis is to find an optimum porogen or porogen mixture which yields the most beneficial porosity for a prepared polymer. In this work, we investigated 2-propanol and isooctane–toluene (1:99% v/v) as porogen agents. The porogens were chosen carefully to maximize the interactions of 17β-estradiol and MAA to form a complex. Normally, during preparation of MIPs non-polar, aprotic solvents, which stabilize hydrogen bonds, e.g., toluene, isooctane (used as porogen mixture), are preferred. On the other hand, the polar protic solvent such as 2-propanol can theoretically enhance a high degree of disruption to hydrogen bonds. This fact can be also confirmed by the dielectric constant (ε) aspect 2-propanol as solvent with higher constant (ε = 18.3) in comparison to isooctane (ε = 2.38) and toluene (ε = 2.1) seems to be obvious to weaken binding strength. Despite of this, we used 2-propanol as a pore forming agent in our non-covalent imprinting polymerization system as we found it was advantageous for a good solubility of the template and the monomers prior to polymerization and because of positive effects on pore formation. The physical characteristics density (ρ s), porosity (P), surface area (S BET) and size of globules of P10 and P14 polymers were determined and can be found in the Table 3. These surprisingly good results obtained with the polymers prepared with 2-propanol triggered us to employ Hyperchem, a molecular modeling software, to see what might happen during creation of a model pre-polymerization complex. To do that we created, a model system of one estradiol molecule surrounded with MAA and in a presence or not of the porogen molecules. 17β-estradiol keeps a flexible arrangement of the −OH functional groups at 3 and 17 positions which allows the supramolecular investigation of their role in combinations with carboxylic group of MAA in molecular recognition process. The undergoing electrostatic or hydrogen bonding interactions of examined MAA-17β-estradiol pre-polymerization complex is shown in the Fig. 3. Apparently, MAA is a significantly superior monomer for interacting with 17β-estradiol via hydrogen bonds. Molecular mechanics of semi-empirical method was also used for illustration of it and for the description of the total-charge density of MAA-17β-estradiol pre-polymerization complex as is shown in the Fig. 3 too. The molecular modeling performed in the presence of the porogens showed that 2-propanol was obviously not repelled from MAA–estradiol complex due to interactions of its OH group, but it was not especially attracted. It rotated and got into the position of pointing its OH group towards MAA–estradiol hydrogen bonds, without having no tendency to disrupt it. Considering the problem more quantitatively, we employed the software to calculate the binding energies using the molecular mechanics (MM+) method and then using the semi-empirical mechanic (PM3) method. The results are shown in Table 4. The data have proved that the H-bond is a dominant interaction between the template (which molecule is relatively poor in the terms of number of polar sites), and the functional monomer. The strength of such interaction was not disrupted by the presence of non-polar porogen (isooctane–toluene) but surprisingly was also not affected by polar 2-propanol. We can also hypothesize here that good projection of the imprint molecule in the polymer matrix may be connected with the smaller size of the alcohol molecule which probably resulted in a higher degree of flexibility of the pre-polymerization complex and easier access of other monomer molecules to the vicinity of a template.
Table 3.
The density, porosity, SBET values and globules size of selected polymers
| MIP/NIP | ρ s (g cm−3) | P (%) | S BET (g m−2) | Size of globules (µm) |
|---|---|---|---|---|
| P10 | 0.79/0.76 | 28/20 | 16.76 | 4.38 |
| P14 | 0.81/0.79 | 30/22 | 81.27 | 5.14 |
Fig. 3.

The Hyperchem-derived structure of the complex formed between the 17β-estradiol molecule and two molecules of MAA. The presence of hydrogen bond is indicated by the dashed lines
Table 4.
Calculated energies of the methacrylic acid-17β-estradiol preassembly in a vacuum
| Molecules | Energy (KJ mol−1) |
|---|---|
| Complex (MAA + 17β-estradiol) | −36.56 |
| Complex (MAA + 17β-estradiol + 2-propanol) | −48.31 |
| Complex (MAA + 17β-estradiol + isooctane–toluene (1:99% v/v)) | −45.56 |
Validation parameters
RP-HPLC method with DAD and electrochemical CoulArray (ED) detection was developed and validated for quantification of estriol, 17β-estradiol, and estrone from acellular simulated body fluid. Calibration curves 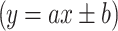 were linear within the range about 10–1,000 ng mL−1 for the both DAD and ED detectors. The selected validation parameters were evaluated and compared by RP/HPLC-DAD/ED (Table 5).
were linear within the range about 10–1,000 ng mL−1 for the both DAD and ED detectors. The selected validation parameters were evaluated and compared by RP/HPLC-DAD/ED (Table 5).
Table 5.
Selected validation characteristics of estrogens
| Compound | Detector |
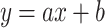
|
r | LODa (ng mL−1) | LOQb (ng mL−1) |
|---|---|---|---|---|---|
| 17β-estradiol | DAD | 0.3141x – 2.3508 | 0.9998 | 1.34 | 4.3 |
| Estrone | 0.2638x – 2.0138 | 0.9996 | 1.39 | 4.47 | |
| Estriol | 0.2921x – 2.5312 | 0.9997 | 0.82 | 2.7 | |
| 17β-estradiol | ED | 0.3141x – 0.5615 | 0.9998 | 0.43 | 1.45 |
| Estrone | 0.2413x + 1.0895 | 0.9999 | 0.44 | 1.5 | |
| Estriol | 0.2921x + 1.5615 | 0.9997 | 0.22 | 1.12 |
a3 S/N
b10 S/N
The basic chromatographic characteristics of estrogens have also been performed. As it is shown in the Table 6, 17β-estradiol imprinted polymers exhibited relatively high retention and good resolution. Uracil was used as a t 0 marker. The chromatographic separation of estrogens (Fig. 4) which was performed after MISPE analysis using imprinted and non-imprinted materials revealed that the utilization of the MIP cartridge in the sample preparation step led to a substantial reduction of matrix interferences (chromatogram drawn with a solid line). It can be also noticed in the Table 5 that the reproducibility of the MISPE-HPLC-DAD/ED system was relatively high and exhibited RSD values below 2%. Taking into account our results, we can state that the developed MISPE-HPLC-DAD/ED technique can be effectively applied in routine analysis.
Table 6.
Basic chromatographic characteristics of the analyzed estrogens

k was calculated from uracil, α was calculated from estriol
Fig. 4.

Comparison of resulting HPLC-ED chromatograms of endogenous estrogens separated after MISPE analysis using imprinted and non-imprinted polymers from spiked acellular and protein-free simulated fluid body samples. HPLC conditions: acetonitrile–ammonium acetate 25 mM (50:50% v/v) as mobile phase, flow rate 1 mL min−1, temperature 30 °C, injection volume 10 µl
Selective extraction of estrogens from synthetic body fluid
In order to investigate the potential of MIPs for the selective entrapment of target analytes (estrone, 17β-estradiol and estriol) the samples of spiked (0.1 µg mL−1) acellular and protein-free simulated body plasma were applied onto P14 sorbent using the optimum MISPE protocol. Due to the nature of estrogens (estriol, 17β-estradiol, and estrone) acetonitrile, acetone, ethyl acetate, and 2-propanol were chosen as eluents in the MISPE procedure. Here, acetone turned out to be an optimal eluent which provided high recoveries (93 ÷ 96%) with very good reproducibility (RSD values below 2%; Fig. 5). Acetonitrile provided slightly worse but still acceptable and very reproducible results (recovery between 89% and 93%, RSD < 1.5%). The worst recoveries (79 ÷ 82%, RSD below 2%) were observed for 2-propanol and ethyl acetate (recovery 81 ÷ 83%, RSD < 2%; Fig. 5, Table 7). The effect of MIP sorbent amount in several MISPE columns was also investigated. Different amounts (20 or 50 mg) of the P14 polymer were packed into 1 or 3 mL glass columns and the efficiency of this material was examined using acetone in the elution step. The 50 mg of P14 polymer which particle size ranged from 90 to 250 µm and which was placed in a 1 mL glass column showed the best values of recovery in MISPE procedure of isolation and preconcentration of the above-mentioned analytes from the SBF samples. The 50 mg of MIP sorbent in 1 mL volume of MISPE column showed the highest recovery rates (93 ÷ 96%, RSD < 2%), while the lowest recovery rates (78 ÷ 83 %, RSD < 1.5%) were observed for the 1-mL column containing 20 mg of the MIP. Slightly higher values (85–89%) were recorded for 50 mg of sorbent in 3 mL column. The reason of the differences in the recoveries obtained for the same amount of sorbent is the thickness of the bed. In a 1-mL glass column, the sorbent bed is higher which makes the passage time of the analytes longer and thus increasing the possibility for the analytes of being trapped by the active sites of the MIP. The same experiments were performed using the NIPs sorbents. The efficiency of synthesized P14 MIP expressed by values of recovery is shown in the Table 7.
Fig. 5.

Comparison of recovery in imprinted and non-imprinted polymers using different solvents in the elution step during MISPE procedure performed with the P14 polymer. Measurement was performed on 50 mg mass of sorbent in a 1-mL glass column
Table 7.
Results of the MISPE and HPLC analysis from synthetic body fluids
| Estrogens | Solvent | 20 mg of MISPEa | 50 mg of MISPEa | 50 mg of MISPEb | |||
|---|---|---|---|---|---|---|---|
| R (%) | RSDc (%) | R (%) | RSDc (%) | R (%) | RSDc (%) | ||
| Estriol | Acetonitrile | 77.44 | ±0.88 | 92.7 | ±1.25 | 86.05 | ±0.56 |
| 17β-estradiol | 75.82 | ±1.06 | 90.62 | ±0.98 | 83.35 | ±0.78 | |
| Estrone | 74.73 | ±0.92 | 89.13 | ±1.18 | 81.99 | ±0.93 | |
| Estriol | Acetone | 83.46 | ±0.98 | 96 | ±1.62 | 89.06 | ±0.47 |
| 17β-estradiol | 81.07 | ±0.63 | 94.55 | ±1.21 | 87.58 | ±0.82 | |
| Estrone | 78.25 | ±1.19 | 92.65 | ±0.99 | 85.83 | ±0.93 | |
| Estriol | Ethyl acetate | 69.68 | ±0.81 | 83.2 | ±1.72 | 78.95 | ±0.76 |
| 17β-estradiol | 68.35 | ±0.75 | 82.93 | ±1.73 | 77.22 | ±0.59 | |
| Estrone | 67.83 | ±0.94 | 81.15 | ±1.36 | 76.23 | ±0.28 | |
| Estriol | 2-propanol | 67.22 | ±0.57 | 81.88 | ±0.67 | 76.9 | ±0.83 |
| 17β-estradiol | 64.29 | ±0.53 | 80.82 | ±0.56 | 74.84 | ±0.46 | |
| Estrone | 62.85 | ±0.94 | 78.65 | ±0.41 | 72.56 | ±0.78 | |
a1 mL glass column
b3 mL glass column
cStandard deviation
R mean recoveries for n = 3
Concluding remarks
The 17β-estradiol imprinted polymers were synthesized by utilizing UV-initiated bulk polymerization based on non-covalent approach. 17β-estradiol MIPs with template–monomer ratio 1:160 and 1:64 were prepared which we found to be really practical and economical. We surprisingly found that MIPs prepared in 2-propanol as a porogen solvent were more porous, compact and better in the terms of recovery values than MIPs synthesized in the isooctane–toluene (1:99% v/v) mixture. The MIPs were utilized in MISPE preconcentration of estradiol, estrone and estriol from acellular and protein-free synthetic body fluid and evaluated by HPLC followed by electrochemical CoulArray detection using acetonitrile–ammonium acetate buffer (25 mM; 50:50% v/v) as a mobile phase. A good analytical performance was attained, including a suitable precision of low detection limits. At the moment, the above-mentioned results make the MISPE-HPLC-ED technique comparable to more sensitive and selective mass spectrometry detection.
Acknowledgment
This work was supported by grant (project No R05 037 03) from the Ministry of Science and Higher Education (Warsaw, Poland) and Ceepus II, Socrates Erasmus Grant and VEGA 1/4461/07.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Lippert TH, Seeger H, Mueck AO (1999) In: Oettel M (ed) Metabolism of endogenous estrogens, Berlin, Springer, 243–271
- 2.Ricanyova J, Gadzala-Kopciuch R, Reiffova K, Buszewski B. Crit Rev Anal Chem. 2009;39:13–31. doi: 10.1080/10408340802569506. [DOI] [Google Scholar]
- 3.Buszewski B, Šebeková K, Božek P, Štibrányi L, Novak I, Berek D. Chromatographia. 1986;22:299–302. doi: 10.1007/BF02268778. [DOI] [Google Scholar]
- 4.Buszewski B. J Pharm Biomed Anal. 1990;8:645–649. doi: 10.1016/0731-7085(90)80095-7. [DOI] [PubMed] [Google Scholar]
- 5.Thurman EM, Mills MS (1998) Solid-Phase Extraction: Principles and Practice, Wiley, 9–97
- 6.Shi Y, Zhang JH, Shi D, Jiang M, Zhu Y-X, Mei S-R, Zhou Y-K, Dai K, Lu B. J Pharm Biomed Anal. 2006;42:549–555. doi: 10.1016/j.jpba.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 7.Hoving EB. J Chromatogr B. 1995;671:341–362. doi: 10.1016/0378-4347(95)00223-6. [DOI] [PubMed] [Google Scholar]
- 8.Johnson JH, McIntyre P, Zdunek J. J Chromatogr A. 1995;718:371–381. doi: 10.1016/0021-9673(95)00679-6. [DOI] [PubMed] [Google Scholar]
- 9.Ferrer I, Barcelo D. TrAC Trends Anal Chem. 1999;18:180–192. doi: 10.1016/S0165-9936(98)00108-3. [DOI] [Google Scholar]
- 10.Hennion MC. J Chromatogr A. 1999;856:3–54. doi: 10.1016/S0021-9673(99)00832-8. [DOI] [PubMed] [Google Scholar]
- 11.Brüggemann O, Haupt K, Ye L, Yilmaz E, Mosbach K. J Chromatogr A. 2000;889:15–24. doi: 10.1016/S0021-9673(00)00350-2. [DOI] [PubMed] [Google Scholar]
- 12.Andersson LI. J Chromatogr B. 2000;745:3–13. doi: 10.1016/S0378-4347(00)00135-3. [DOI] [PubMed] [Google Scholar]
- 13.Ansell RJ, Ramström O, Mosbach K. Clin Chem. 1996;42:1506–1512. [PubMed] [Google Scholar]
- 14.Katz A, Davis ME. Macromolecules. 1999;32:4113–4121. doi: 10.1021/ma981445z. [DOI] [Google Scholar]
- 15.Matsui J, Kato J, Takeuchi T, Suzuki M, Yokoyama K, Tamiya E, Karube I. Anal Chem. 1993;65:2223–2225. doi: 10.1021/ac00065a009. [DOI] [Google Scholar]
- 16.Matsui J, Doblhoff-Dier O, Takeuchi T. Anal Chim Acta. 1997;343:1–4. doi: 10.1016/S0003-2670(96)00596-X. [DOI] [Google Scholar]
- 17.Sreenivasan K. J Appl Polym Sci. 1998;68:1863–1866. doi: 10.1002/(SICI)1097-4628(19980613)68:11<1863::AID-APP18>3.0.CO;2-V. [DOI] [Google Scholar]
- 18.Takeda K, Kobayashi T. Sci Technol Adv Mater. 2005;6:165–171. doi: 10.1016/j.stam.2004.11.008. [DOI] [Google Scholar]
- 19.Yan H, Row KH. Int J Mol Sci. 2006;7:155–178. doi: 10.3390/i7050155. [DOI] [Google Scholar]
- 20.Sellergren B. J Chromatogr A. 2001;906:227–252. doi: 10.1016/S0021-9673(00)00929-8. [DOI] [PubMed] [Google Scholar]
- 21.Cheong SH, McNiven S, Rachkov A, Levi R, Yano K, Karube I. Macromolecules. 1997;30:1317–1322. doi: 10.1021/ma961014l. [DOI] [Google Scholar]
- 22.Sanbe H, Haginaka J. J Pharm Biomed Anal. 2002;30:1835–1844. doi: 10.1016/S0731-7085(02)00526-5. [DOI] [PubMed] [Google Scholar]
- 23.Darryl YS, Sasaki D, Rush D, Daitch C, Alam T, Assink R, Ashley C, Brinker C, Shea K (1998) In: Bartsch RA, Maeda M (ed) Molecular imprinted receptors in sol–gel materials for aqueous phase recognition of phosphates and phosphonates, American Chemical Society Symposium Series 703, Washington 314–323
- 24.Rachkov A, McNiven S, Cheong SH, El’skaya AV, Yano K, Karube I. Supramol Chem. 1998;9:317–322. doi: 10.1080/10610279808035001. [DOI] [Google Scholar]
- 25.Rachkov AE, Cheong SH, El’skaya AV, Yano K, Karube I. Polym Adv Technol. 1998;9:511–519. doi: 10.1002/(SICI)1099-1581(199808)9:8<511::AID-PAT790>3.0.CO;2-H. [DOI] [Google Scholar]
- 26.Haginaka J, Sanbe H. Chem Lett. 1998;11:1089–1090. doi: 10.1246/cl.1998.1089. [DOI] [Google Scholar]
- 27.Kugimiya A, Matsui J, Abe H, Aburatani M, Takeuchi T. Anal Chim Acta. 1998;365:75–79. doi: 10.1016/S0003-2670(98)00095-6. [DOI] [Google Scholar]
- 28.Ramström O, Ye L, Mosbach K. Chem Biol. 1996;3:471–477. doi: 10.1016/S1074-5521(96)90095-2. [DOI] [PubMed] [Google Scholar]
- 29.Ramström O, Ye L, Krook M, Mosbach K. Anal Commun. 1998;35:9–11. doi: 10.1039/a707876e. [DOI] [Google Scholar]
- 30.Qiujin Z, Liping W, Shengfang W, Wasswa J, Xiaohong G, Jian T. Food Chem. 2009;113:608–615. doi: 10.1016/j.foodchem.2008.07.044. [DOI] [Google Scholar]
- 31.Buszewski B, Jezierska-Switala M, Kowalska S. J Chromatogr B. 2003;792:279–286. doi: 10.1016/S1570-0232(03)00283-6. [DOI] [PubMed] [Google Scholar]
- 32.Özkan SA. Chromatographia. 2007;66:3–13. doi: 10.1365/s10337-007-0223-9. [DOI] [Google Scholar]
- 33.Yamada H, Yoshizawa K, Hayase T. J Chromatogr B. 2002;775:209–213. doi: 10.1016/S1570-0232(02)00292-1. [DOI] [PubMed] [Google Scholar]
- 34.Kuronen P, Volin P, Laitalainen T. J Chromatogr B. 1998;718:211–224. doi: 10.1016/S0378-4347(98)00351-X. [DOI] [PubMed] [Google Scholar]
- 35.Giese R. J Chromatogr A. 2003;1000:401–412. doi: 10.1016/S0021-9673(03)00306-6. [DOI] [PubMed] [Google Scholar]
- 36.Gadzała-Kopciuch R, Ričanyová J, Buszewski B. J Chromatogr B. 2009;877:1177–1184. doi: 10.1016/j.jchromb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Cüneyt AT. Biomaterials. 2000;21:1429–1438. doi: 10.1016/S0142-9612(00)00019-3. [DOI] [PubMed] [Google Scholar]
- 38.Ye L, Yu Y, Mosbach K. Analyst. 2001;126:760–765. doi: 10.1039/b009048o. [DOI] [PubMed] [Google Scholar]
- 39.Ye L, Weiss R, Mosbach K. Macromolecules. 2000;33:8239–8245. doi: 10.1021/ma000825t. [DOI] [Google Scholar]
- 40.Sellergren B, Lepistö M, Mosbach K. J Am Chem Soc. 1988;110:5853–5860. doi: 10.1021/ja00225a041. [DOI] [Google Scholar]
- 41.Arshady R, Mosbach K. Makromol Chem. 1981;182:687–692. doi: 10.1002/macp.1981.021820240. [DOI] [Google Scholar]
- 42.Sellergren B. Macromol Chem Phys. 1989;190:2703–2711. doi: 10.1002/macp.1989.021901104. [DOI] [Google Scholar]
- 43.Andersson L. React Polym. 1988;9:29–41. [Google Scholar]
- 44.Rachkov AE, Cheong S-H, Eĺskaya AV, Yano K, Karube I. Polym Adv Technol. 1998;9:511–519. doi: 10.1002/(SICI)1099-1581(199808)9:8<511::AID-PAT790>3.0.CO;2-H. [DOI] [Google Scholar]
- 45.Szumski M, Buszewski B. J Sep Sci. 2004;27:837–842. doi: 10.1002/jssc.200401799. [DOI] [PubMed] [Google Scholar]
- 46.Wei S, Mizaikoff B. Biosens Bioelectron. 2007;23:201–209. doi: 10.1016/j.bios.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 47.Le Noir M, Lepeule A-S, Guieysse B, Mattiasson B. Water Res. 2007;41:2825–2831. doi: 10.1016/j.watres.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 48.Wulff G, Vietmeier J, Poll H-G. Makromol Chem. 1987;188:731–740. doi: 10.1002/macp.1987.021880407. [DOI] [Google Scholar]
- 49.Wulff G, Vesper W. J Chromatogr. 1978;167:171–186. doi: 10.1016/S0021-9673(00)91156-7. [DOI] [Google Scholar]
- 50.Wulff G (1986) In: Ford WT (ed) Polymeric reagents and catalysts, American Chemical Society Symposium Series 308, Washington, 186
- 51.Sellergren B, Shea K. J Chromatogr. 1993;635:31–49. doi: 10.1016/0021-9673(93)83112-6. [DOI] [PubMed] [Google Scholar]
- 52.Kempe M, Mosbach K. J Chromatogr A. 1995;691:317–323. doi: 10.1016/0021-9673(94)00820-Y. [DOI] [PubMed] [Google Scholar]
- 53.Yilmaz E, Mosbach K, Haupt K. Anal Commun. 1999;36:167–170. doi: 10.1039/a901339c. [DOI] [Google Scholar]