

Abstract
The Bacillus thuringiensis phosphatidylinositol-specific phospholipase C (PI-PLC), an interfacial enzyme associated with prokaryotic infectivity, is activated by binding to zwitterionic surfaces, particularly phosphatidycholine (PC). Two tryptophan residues (Trp47 in the two-turn helix B and Trp242 in a disordered loop) at the rim of the barrel structure are critical for this interaction. The helix B region (Ile43 to Gly48) in wild-type PI-PLC orients the side chains of Ile43 and Trp47 so that they pack together and form a hydrophobic protrusion from the protein surface that likely facilitates initial membrane binding. In previous studies we reported that in the crystal structure of the dimeric W47A/W242A mutant, which is unable to bind to PC, the helix B region has been reorganized by the mutation into an extended loop. Here we report the construction and characterization (catalytic activity, fluorescence and NMR studies) of a series of PI-PLC mutants targeting helix B residues and surrounding regions to explore what is needed to stabilize the ‘membrane active’ conformation of the helix B region. Results strongly suggest that, while hydrophobic groups and presumably an intact helix B are critical for the initial binding of PI-PLC to membranes, disruption of helix B to allow enzyme dimerization is what leads to the activated PI-PLC conformation.
The Bacillus thuringiensis phosphatidylinositol-specific phospholipase C (PI-PLC) exhibits a very specific interfacial activation towards monomer and aggregated substrates upon binding to phosphatidylcholine interfaces (1–3). Although the (βα)8-barrel core of closely related PI-PLC appears relatively rigid, the crystal structure of the enzyme shows hydrophobic residues at the rim of the barrel in helix B (Ile43-Gly48) and the 237–244 loop with significant mobility as measured by B factors (4). Previous work has clearly pointed to the importance of Trp47 and Trp242 in binding to activating PC interfaces (and at least one of them crucial for the protein to bind to anionic phospholipid surfaces) (5–7). The rim Trp residues have been suggested to be part of a binding site for PC molecules (8). Removal of either Trp reduces activation by PC; removal of both renders the enzyme incapable of binding to PC interfaces (5). Indeed, for the rim loop interfacial mutants of PI-PLC examined thus far, the specific activity (in both phosphotransferase and cyclic phosphodiesterase assay systems) correlates with how tightly the PI-PLC binds to activating PC SUVs (Figure 1). However, helix B mutant proteins do not follow this trend. Introduction of a tryptophan at other positions in helix B in the PI-PLC mutant W47A generated proteins that could bind moderately well to PC vesicles. Yet most of those were much less active than wild type PI-PLC (7). Furthermore, the single helix B mutant that regained significant kinetic activation by PC compared to W47A, I43W/W47A, did not bind tightly to PC vesicles (7). I43W/W47A has the tryptophan on the same side of the two-turn helix B as Trp47. The lower activity of I43W/W47A as well as the other helix B mutants, which bind PC well but are relatively inactive (7), suggest most of the mutations can not retain the unusual conformation that is critical for interfacial activity.
Figure 1.

Correlation of apparent Kd, which measures the partitioning of PI-PLC and mutants to POPC SUVs, with enzymatic activity toward (A) 8 mM PI solubilized in 24 mM diC7PC or towards (B) 5 mM cIP with 5 mM diC7PC added. Squares represent W242A and rescue mutants, the open triangle is wild type rPI-PLC, and the filled circles are helix B W47A and rescue mutants (data from (7)).
Recently, the crystal structure of the B. thuringiensis PI-PLC W47A/W242A mutant was solved (9). This interfacially impaired PI-PLC is a dimer in contrast to the highly homologous B. cereus PI-PLC, which crystallizes as a monomer (4). The dimer interface has a hydrophobic core of tyrosine residues that are quite removed from the active site (9). In the dimer, residues Ile43 to Gly48 no longer form a helix but instead adopt an irregular loop with an increased distance between residues 43 and 46 (Figure 2). In the B. cereus PI-PLC, helix B has the Ile43, Val46 and Trp47 side chains packed together to form a plug extending into solution (4). Such a hydrophobic plug could aid in initiating and stabilizing membrane binding. In the W47A/W242A double mutant, the Ile43 side chain is reoriented towards the protein core (9). If attachment of the plug is a part of the initial binding of the protein to a PC interface, then mutations that destabilize the helix in solution would be expected to reduce PC affinity and lead to lower PC activation of cIP hydrolysis. However, there are interactions of residues in and around helix B (e.g., Pro42 and Gln45) that appear to stabilize the dimer interface, and these cannot be accommodated with a helical structure for this segment of the protein (9).
Figure 2.
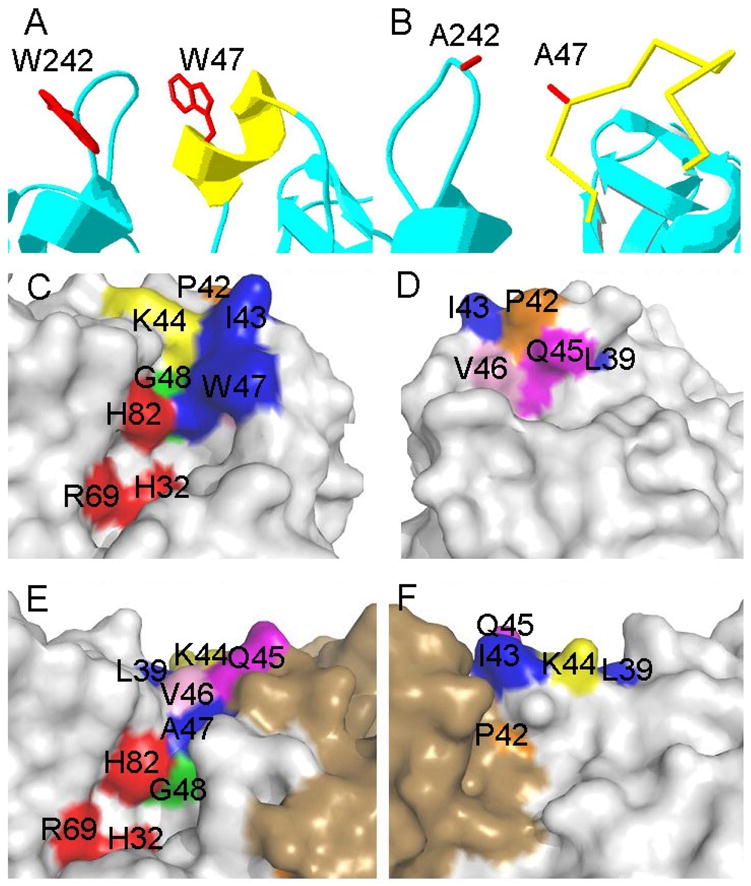
Local structure of helix B in (A) B. cereus PI-PLC and (B) a monomer of B. thuringiensis W47A/W242A PI-PLC. (C–F) Surface representations showing the location of helix B residues (Pro42 orange, Ile43 blue, Lys44 orange, Gln45 magenta, Val46 pink, Trp47 blue, and Gly48 green), and their relation to active site residues (His32, Arg69, and His82 colored in red) in B. cereus PI-PLC (C, D) and B. thuringiensis W47A/W242A (E, F). For each enzyme the left and right side panels represent ‘front’ and ‘back’ (rotated by 180°) views of the helix B, active site, and dimerization interface regions of the protein.
Here we report the characterization of a series of PI-PLC mutant proteins with alterations of helix B residues (as well as regions around this structural feature). These were constructed to explore what residues in this region of the protein are needed to stabilize the ‘membrane active’ conformation of the protein. Alanine mutants at each position in helix B (43–48) as well as Leu39 were constructed and characterized for any changes in secondary structure (CD spectroscopy), PC activation of PI cleavage and cIP hydrolysis, and the ability to bind to PC interfaces (comparing both protein intrinsic fluorescence in the presence of PC vesicles and a vesicle filtration assay). Leu39, a hydrophobic amino acid adjacent to the start of helix B, was altered to an alanine to assess its contribution, if any, to membrane binding. The mutated protein that replaces Lys44, the only charged residue in helix B, with an anionic group (Glu) was also examined. Pro42, the residue immediately before the start of helix B, was also altered to P42G, replacing a conformationally restricted residue with a very flexible one.
All these mutants except K44A exhibited lower activity for one or both steps of PI hydrolysis. However, there were differences among the mutants that allow us to propose a mechanism for PI-PLC activation by PC and to assess the formation of a membrane-induced dimer as a key step for optimal activity. The results are consistent with (i) formation of helix B in solution to prevent premature dimerization of the protein in solution, (ii) initiation of binding to PC-containing membranes by the hydrophobic helix B ‘plug’, and (iii) disruption of helix B once the protein is anchored in preparation for dimer formation with another membrane bound PI-PLC to generate the ‘membrane activated’ form of PI-PLC.
Experimental Procedures
Chemicals
Lipid powders or solutions in organic solvent were purchased from Avanti Polar Lipids, Inc., and used without further purification. These included the long chain lipids 1- palmitoyl-2-oleoyl-phosphatidylcholine (POPC), L-α-phosphatidylinositol (PI) from bovine liver and the synthetic short chain lipid diheptanoyl-PC (diC7PC). Crude soybean PI from Sigma Chemical Co. was used for the enzymatic generation of cIP by PI-PLC as described previously (1). Tryptone and yeast extract were obtained from Fisher, Inc. Most of other chemicals, such as isopropanol (iPrOH), chloroform, ethanol, D2O, Tris, HEPES, glycerol and Triton X-100, were purchased from Sigma. Q Sepharose fast flow anion exchange resin and phenyl sepharose hydrophobic resin were purchased from Amersham Pharmacia Biotech; AG1-X8 (chloride form, 100–200 mesh) was obtained from Bio-Rad Laboratories. All competent cells used in mutagenesis (super blue) and overexpression (BL21 Codonplus) were obtained from Stratagene.
Constructing and overexpression PI-PLC mutants
Single PI-PLC mutants (L39A, P42G, I43A, I43W, K44E, Q45A, V46A, G48A) were constructed from the wild type B. thuringiensis PI-PLC gene using QuikChange methodology (10) with a site-directed mutagenesis kit from Stratagene. The HPLC purified mutagenic primers were purchased from Operon. Mutated genes were sequenced at Sequegen to confirm the identity of the mutation. Also overexpressed were two mutants constructed and reported previously, K44A (6) and W47A/W242A (5). Several double mutant proteins (L39A/V46A, I43W/W47I) were made by introducing the second mutation in the gene coding for a single mutant. A plasmid containing the mutant B. thuringiensis PI-PLC gene was transformed into E. coli BL21-Codonplus (DE3)-RIL cells. Overexpression and purification (via elution from a Q-sepharose fast flow column and then a phenyl sepharose column) of the recombinant proteins followed procedures described preciously for recombinant B. thuringiensis PI-PLC (rPI-PLC) (5). The purity of PI-PLC enzymes was above 90% as monitored by SDS-PAGE. Protein solutions were concentrated using Millipore Centraplus 10 filters; concentrations were usually determined by Lowry assays (11), although absorption at 280 nm was also used.
CD spectroscopy
Recombinant PI-PLC secondary structurewas measured using an AVIV 202 CD spectrophotometer. Comparison of secondary structure for recombinant wild type and mutatedPI-PLC used wavelength scans from 265 to 195 nm with protein(0.3–0.4 mg/ml) in a 0.1-cm cell at 25°C and the program CDNN (12–14).
31P NMR spectroscopy
The specific activity of PI-PLC mutant proteins was measured using 31P NMR (202.3 MHz) spectra obtained on a Varian INOVA 500 using parameters described previously (1, 5, 15). Phosphorylated product formation for either fixed time points or continuous assays was analyzed (7). For both, the buffer was 25 mM HEPES, pH 7.5, and the amount of enzyme added in each kinetic run (50 ng to 3 μg depending on whether the phosphotransferase or the cyclic phosphodiesterase was monitored, respectively) was adjusted to generate less than 20% product within the desired reaction time. PI cleavage and cIP hydrolysis rates were measured from the integrated intensity of cIP and I-1-P resonances (in the absence of 1H decoupling), respectively, as a function of incubation time, using the 31P resonance of glucose-6-phosphate or KH2PO4 of known concentrations (2 or 4 mM) as an internal reference. The PI concentration was 5, 6 or 8 mM; diC7PC was usually 24 or 32 mM but was also increased for surface dilution studies. The assay temperature was 22°C. The hydrolysis of cIP (20 mM) was monitored in the absence or presence of 5 or 8 mM diC7PC. For PI-PLC activity toward PI presented in small unilamellar vesicles (prepared by sonication), the PI concentration was 8 mM with 2 mM POPC.
31 P NMR was also used to monitor the interaction of two specific mutant proteins with the activator diC7PC (2, 8). DiC7PC was titrated into a solution of 3 mg/ml (0.085 mM) enzyme in 0.05 M HEPES, 1 mM EDTA, pH 7. In the presence of rPI-PLC, the linewidth of the diC7PC phosphorus resonance was narrow below the CMC of that short-chain lipid, very broad around the CMC and then narrowed as the concentration of diC7PC increased further and the fraction of enzyme-bound diC7PC decreased (2, 8). The magnitude of the line broadening reflects enzyme binding and diC7PC dynamics and can be used to compare different PI-PLC mutant proteins. In an attempt to quantify the line-broadening, we chose to extrapolate a ‘bound’ linewidth for a 1:1 PI-PLC/diC7PC complex by fitting the increased linewidth for diC7PC (ΔΔδ1/2 = Δν1/2(+PLC) – Δν½ (PC)) for lipid concentrations above the CMC of 1.5 mM with the following expression:ΔΔδ1/2 = Δb [PLC]/[diC7PC] + Δν½ (limit), where Δb is the ‘bound’ linewidth and Δν½ (limit) is the increased diC7PC linewidth caused by particle aggregation. This treatment, which assumes fast exchange for the diC7PC in ‘empty’ micelles, micelles containing PI-PLC, and monomeric diC7PC, is simplistic but useful for assessing differences in mutated PI-PLC/diC7PC interactions where specific activities are essentially the same.
Intrinsic fluorescence of PI-PLC
Fluorescence measurements of PI-PLC (2μM) were carried out on Shimadzu RF5000U or Fluorolog R-3 spectrofluorometers. The excitation wavelength was 282 nm. Changes in the fluorescence intensity at 337 nm (the emission maximum) upon the addition of lipids (either diC7PC or POPC SUVs) were expressed as (I- Io)/Io, where Io is the emission intensity of protein alone, and I is the intensity in the presence of an additive. An apparent binding constant to POPC vesicles was obtained by fitting the change in intensity with a hyperbolic function to extract a bulk concentration of PC for half the maximum change. For titrations with diC7PC, the normalized change in fluorescence was fit with a cooperative function ((I-Io)/Io = ΔI [PC]n/(Kdn + [PC]n) where ΔI was the maximum change in fluorescence and the apparent Kd for diC7PC was the concentration of that lipid that led to half the maximum fluorescence change (ΔI/2). The standard errors in Kd were derived from the least- squares fit of the concentration dependence of the fluorescence changes. To estimate an apparent Kd for ‘micellar’ diC7PC, only data above 1.5 mM were used, and the dependence of (I-Io)/I for diC7PC concentrations above the CMC (1.5 mM) of pure lipid was plotted versus micellar diC- 7 PC (estimated as [PC] – CMC).
Centrifugation vesicle binding assay
A centrifugation binding assay was used in addition to the fluorescence assay to characterize the binding of some PI-PLC mutants to interfaces (6). Solutions of PI-PLC incubated with phospholipid vesicles were centrifuged through a filter to separate free from vesicle bound enzyme. Typically, 25 μg of PI-PLC (ET) was added to an aliquot of the vesicle solution (2 ml) in 10 mM Tris HCl, pH 7.5. Vesicle-bound enzyme (EB) was separated from free enzyme (EF) using an Amicon centricon-100 filter (100 kDa molecular mass cutoff). A control containing the same amount of enzyme in Tris HCl, pH 7.5, with no lipid vesicles present indicated that less than 5% of ET was retained on the filters in the control. Filtrates were lyophilized and analyzed by SDS-PAGE. Band intensities, quantified by NIH Image 1.61 software, were used to calculate the concentration of EF by comparing intensities from vesicle-containing samples to the ET value of the control; vesicle-bound enzyme, EB, was calculated as ET – EF.
Results
Helix B alanine mutations – secondary structure
A model (9) that has recently been proposed for the activation of B. thuringiensis PI-PLC upon binding to activating PC surfaces has the following steps. (i) In solution PI-PLC exists primarily as a monomer, although small amounts of dimer have been noted previously (8). In the crystal structure of monomeric B. cereus PI-PLC (4), helix B points the hydrophobic side chains of Trp47 and Ile43 out into solution. (ii) The Trp47 in this hydrophobic plug sticking out from what appears to be a flexible helix B region is instrumental in protein binding to a PC surface. It should be noted that the protein has an electrostatic interaction with pure anionic lipids, but the binding does not involve insertion of the protein, and the protein can easily be removed from such a surface with moderate salt (7). The protein binds to the PC interface by inserting Trp47. (iii) Once bound to the PC surface, PI-PLC can form an interfacial homodimer using a string of Tyr residues (9, 16) near the barrel rim. It is the PLC dimer that is the kinetically activated form of the enzyme, presumably because it enhances processive catalysis and possibly alters some residues in and around the catalytic site leading to an increased kcat. However, the only concrete evidence for PI-PLC dimer formation is from the crystal structure of W47A/W242A, a mutant with little affinity for PC surfaces.
While the crystallographic W47A/W242A dimer is unlikely to be the same as a membrane bound PI-PLC dimer, it serves as a reference point for exploring residues that might be critical for dimerization. A key observation in the W47A/W242A dimer is that helix B no longer exists – rather the residues adopt a loop that is closer to the large rim loop that normally contains Trp242, another key Trp whose fluorescence intensity monitors membrane binding (5). Pro42, right before the start of helix B in the monomer crystal structure, contributes to dimer formation of W47A/W242A through hydrophobic interactions with both Tyr247 and Tyr248 of the opposite chain. In the dimer, Gln45 participates in a hydrogen bond with Gly239 of the opposite chain.
Since the dimer is of an interfacially impaired protein and residues in the helix B region appear to stabilize the dimer, there are two key questions: (i) is the membrane activated structure of this PI-PLC a dimer, and (ii) is helix B intact when the protein binds to interfaces or does it form an extended loop as in W47A/W242A. Recent mutagenesis of a cluster of tyrosines (Tyr246, Tyr247, Tyr248, Tyr251) that form the hydrophobic core of the dimer interface in W47A/W242A strongly supports formation of a dimeric structure of PI-PLC on membranes as necessary for optimal activity (16). To assess the contribution of helix B to activity and binding of activator phospholipids, we have constructed alanine mutations at each position in helix B as well as at Leu39 and introduced a Gly in place of Pro42. All mutant genes overexpressed well in E. coli and generated proteins with similar CD spectra and Tm (as monitored by loss of negative ellipticity at 222 nm (5)) as rPI-PLC indicating that the mutated proteins were well-folded with the same overall structure. I43A, K44A, W47A had the same secondary structure as rPI-PLC. However, L39A, P42G, Q45A, V46A, and G48A exhibited a reduced α-helix content (as much as 3% loss compared to the wild type recombinant protein in solution) in low ionic strength buffers when compared by CD (Figure 3). For all of these but Q45A, the addition of 2 mM diC7PC caused the protein to regain α-helix structure to different degrees. These results illustrate a structural maleability in the helix B region of the protein when it is in solution.
Figure 3.

Difference in α-helix content for each mutant compared to recombinant PI-PLC: protein (~ 0.4 mg/ml in 10 mM borate buffer, pH 8.0, in 1 mm cuvette) in the absence (dark gray;) and presence (light gray) of 2 mM diC7PC. For comparison, the secondary structure of the native protein from CD appears to be 25.5% α-helix, 24.4% β-sheet, 17.8% β-turn, and 32.3 % random (6).
Phosphotransferase and cyclic phosphodiesterase activities
If formation of a PI-PLC dimer is critical for high interfacial activity, then mutations in the helix B region that disrupt such a dimer should lead to reduced activity toward cIP in the presence of activating diC7PC (but not towards cIP alone). PI cleavage when presented in a PC surface might also be expected to decrease. A comparison of the cIP hydrolysis rates in the absence and presence of diC7PC is shown in Figure 4A for these helix B mutant enzymes. In the absence of any interfaces, all the enzymes had cIP cleavage rates 58 to 96% of rPI-PLC enzyme activity (except for L39A and Q45A which exhibited 10% and 39% rPI-PLC activity). Thus, monomer substrate activity was more or less unaltered with this series of mutations. Addition of diC7PC micelles increased all cIP hydrolysis activities with the activation-fold (ratio of the activity plus PC compared to that in its absence) comparable to rPI-PLC for L39A, I43A and K44A; V46A was reasonably activated as well although not quite to the same extent (Figure 4B). P42G, Q45A, W47A and G48A were all much less responsive to kinetic activation by this nonsubstrate phospholipid suggesting either a reduction in diC7PC affinity or an altered conformation of the protein when bound to these micelles. The first two of these residues with lower PC activation of cIP hydrolysis are ones involved in dimer stabilization in W47A/W242A.
Figure 4.

Activities (μmol min−1 mg−1) of PI-PLC helix B region mutants with Ala or Gly (for P42G) substituted at the position indicated. (A) Specific activity for cleavage of 20 mM cIP in the absence (black) and presence (light gray) of 8 mM diC7PC. Error bars in (A) reflect standard deviations from multiple assays. (B) Extent of diC7PC activation presented as the ratio of average cIP hydrolysis activity in the presence of diC7PC compared to aerage activity in the absence of this nonsubstrate lipid. (C) Specific activity for cleavage of PI (8 mM) solubilized in 32 mM diC7PC; error bars represent standard deviations for multiple assays.
While the cyclic phosphodiesterase rates of P42G, Q45A, W47A and G48A were very sensitive to modification of these helix B residues, the phosphotransferase activities only varied at most three-fold (Figure 4C). Poor PC activation of cIP hydrolysis did not necessarily imply poor phosphotransferase activity (e.g., P42G and G48A show the opposite behavior). Conversely, for some enzymes with high cIP activation, phosphotransferase activity could be low (L39A, I43A). Since PI has acyl chains while cIP does not, these results hint that the presence of substrate with hydrophobic moieties can also lead to the ‘activated’ conformation of PI-PLC.
Binding of mutant proteins to PC SUVs
In the model proposed, binding of PI-PLC to PC surfaces has two phases – an initial interaction and partitioning of Trp47 into the membrane (which presumably is achieved by monomeric protein) and then perhaps enhanced membrane binding once a homodimer is formed on the surface. If helix B need not be intact for initial binding to PC, then most of the mutant proteins would be expected to bind to PC, although affinities might be lower than rPI-PLC if dimerization does not occur to much extent. If helix B must be intact for the initial binding, then interactions that destabilize the helix in solution would be expected to weaken binding of the protein to PC surfaces.
Recombinant PI-PLC bound moderately tightly to POPC SUVs with an apparent Kd = 0.067±0.015 mM (6). Of these helix B region mutant proteins, only K44A and G48A exhibited affinities for PC SUVs comparable to the rPI-PLC protein (Figure 5). Cationic Lys44 does not insert into membranes and the structural change from Gly to Ala would not be expected to alter the Kd for G48A significantly. L39A and W47A had the weakest affinity for PC, with apparent Kd values roughly 50- to 70-fold more than that of rPI-PLC. The calculated Kd values for each protein in Fig. 5 are based on free energy differences for side chains in membranes from the Wimley-White scale (17). The increased apparent Kd for W47A is clearly consistent with the indole ring partitioning into the membrane. However, the apparent Kd for the Leu39 substitution is far greater than expected solely on the basis of membrane partitioning. Although the Leu side chain could partition in the membrane, it is likely that substitution at this position also alters the protein conformation and disposition of other residues involved in membrane partitioning since the observed loss of binding affinity was so severe.
Figure 5.

Apparent dissociation constants for helix B region mutants binding to POPC SUVs in 10 mM Tris HCl, pH 7.5. The binding was monitored by both changes in the intrinsic Trp fluorescence as a function of added POPC SUVs as well as a filtration/centrifugation binding asssay. The Kd in the light gray bars for the nonpolar side chains is what would be predicted for the Kd if that particular residue partitioned into the bilayer.
Alanine substitutions at the other positions in helix B exhibited a 6–12-fold loss of affinity for PC. Here, the loss of binding affinity does not correlate with changing the identity of a group that inserts into the membrane based on free energy changes. Rather, these residues must be involved in other interactions that modulate PC binding. In the crystal structure of B. cereus PI-PLC (4), helix B side chains do not interact significantly with the rest of the protein (Figure 2C and D). If helix B remains intact and acts as a hydrophobic plug for interface binding, only the removal of residues poised on the hydrophobic helix face, Ile43, Val46 and Trp47, should reduce membrane binding. However, in the W47A/W242A crystal dimer, both Pro42 and Gln45 make intersubunit contacts (Figure 2E and F). Therefore, the significantly weakened affinities of P42G and Q45A proteins (compared to rPI-PLC) for PC more likely reflect impaired dimerization on the membrane surface. Reduced dimerization could weaken the binding of the enzyme on the PC surface by increasing the off rate of the protein.
Positional and charge reversal mutant proteins of helix B
If a helical hydrophobic plug (residues Ile43-Gly48) is needed for initial interaction of PI-PLC with PC, it should be stabilized by hydrophobic residues facing outwards on the same side of helix B. Indeed, this is consistent with the observation that only I43W/W47A and no other Trp rescue mutation, could significantly recover the PC interfacial activation of cIP hydrolysis lost in W47A (7). However, full PI cleavage activity was not observed with I43W/W47A, nor was it observed in I43A (Figure 4C). This might suggest that a specific positional arrangement of hydrophobic residues is critical in the helix B region of the protein. Two additional mutant proteins were examined to check for the importance of where the helix B Trp was placed. The ‘swap’ mutation I43W/W47I should be equivalent to rPI-PLC kinetically and structurally if the actual position of Trp on helix B matters less than its orientation in solution and its stabilization by another hydrophobic side chain. I43W, which maintains the critical Trp47 residue in helix B (5) was made to see if the addition of a larger hydrophobic side chain had any effect on PI-PLC kinetic behavior. Both proteins had comparable secondary structure (data not shown) and similar phosphotransferase and cyclic phosphodiesterase specific activities (Table 1). Specific activities were determined for cleavage of PI solubilized in 30% iPrOH, PI in Triton X-100 micelles, and PI in vesicles with 20 mol% PC as well as for the PI/diC7PC mixed micelle system (Table 1). The ‘swap’ mutant enzyme had similar activity to rPI-PLC in all but the iPrOH assay system (18, 19). I43W was not quite as active as rPI-PLC with the additional Trp residue in this region. Nonetheless, these results suggest that the position of the Trp in the helix B region is less important that its packing with another hydrophobic residue (presumably to stabilize helix B in solution).
Table 1.
Relative phosphotransferase (towards PI in different assay systems) and cyclic phosphodiesterase (cIP as substrate) activities of rPI-PLC and selected helix B mutants.a
| Substrate (mM) | PI | cIP | |||||
|---|---|---|---|---|---|---|---|
| (5) | (5) | (5) | (8) | (5)c | Activationd | ||
| Assay system | 30% iPrOH | PI/diC7PC (1:4) | PI/TX100 (1:2) | PI/POPC (4:1) SUVs | diC7PC (5 mM) | ||
| WT | 0.72b | 1.00 | 0.67b | 0.017 | 0.19 | 46 | |
| I43A | 0.32 | 0.36 | 0.31 | 0.006 | 0.15 | 36 | |
| I43W/W47A | 0.13b | 0.54b | 0.42b | 0.008 | 0.056 | 47 | |
| I43W/W47I | 0.39 | 0.90 | 0.60 | 0.016 | 0.09 | 33 | |
| I43W | 0.45 | 0.82 | 0.54 | 0.014 | 0.11 | 55 | |
| K44A | 0.68 | 0.98 | 0.36 | 0.018 | 0.16 | 33 | |
| K44E | 0.44 | 0.024 | 4.5 | ||||
All specific activities, except for K44E, were obtained at 22°C, and are referenced to cleavage of 5 mM PI solubilized in 20 mM diC7PC (955μmol min−1 mg−1 at 22°C). Values are averages of fixed time point assays done in duplicate; errors were < 15% of the indicated value.
From Feng et al. (2002).
Values for cIP in the absence of activating PC were lower than reported previously (5, 7), which were obtained at a higher temperature (28 versus 22°C). The value for rPI-PLC under these conditions was 2.8 μmol min−1 mg−1. The ratio show is for hydrolysis of 5 mM cIP in the presence of 5 mM diC7PC.
The extent of PC activation of the enzyme for cIP hydrolysis was estimated by comparing specific activity in the presence of diC7PC to the activity towards cIP alone.
I43W/W47I had about the same affinity for POPC SUVs as rPI-PLC (Table 2). I43A, where the Ile that stabilizes outward facing Trp47 in PI-PLC has been removed, had a weakened affinity for PC vesicles compared to rPI-PLC protein that correlates with its reduced phosphotransferase and PC activation of cIP hydrolysis rates. Changing Ile to Trp (I43W) enhanced enzyme partitioning to SUVs, but not enough to imply that both Trp in helix B are fully inserted into the PC membrane (the predicted Kd if both Trp insert would be 0.005 mM versus the experimental value of 0.026 mM).
Table 2.
Apparent binding constants for recombinant PI-PLC and selected helix B mutants interacting with PC aggregates.
| Assay system | app Kd (mM) | calc. Kd (mM) | K0.5 (mM)a | K0.5-CMC (mM)b |
|---|---|---|---|---|
| POPC SUVs | diC7PC | micelles | ||
| WT | 0.067±0.010 | 1.86±0.08 | 0.36±0.06 | |
| I43A | 0.37±0.07 | 0.15 | 2.28±0.10 | 0.75±0.06 |
| I43W/W47A | 0.85±0.17d | 0.15 | 2.48±0.39d | 1.08±0.33d |
| I43W/W47I | 0.064±0.008 | 0.067 | 1.67±0.07 | 0.18±0.07 |
| I43W | 0.026±0.06 | 0.005 | 1.65±0.07 | 0.17±0.09 |
| K44A | 0.087±0.01 | 1.59±0.07 | 0.08±0.04 | |
| K44E | 0.50±0.20e | |||
K0.5 is estimated as the bulk concentration of diC7PC that leads to 50% of the maximum change in fluorescence intensity upon binding to that short-chain PC; the value for WT PI-PLC is the average of three separate experiments.
K0.5-CMC is the value extracted for 50% of the fluorescence increase if only data above the CMC is used and fit with a hyperbolic function that reflects micellar diC7PC only. Micellar PC is calculated as the concentration of diC7PC minus the CMC (1.5mM).
Extracted from data presented in (5).
Extracted from Feng et al. (7).
We also examined in greater detail changes at residue 44. Lys44 represents the only charged residue in the helix B region. Replacement of the cationic side chain by a methyl group had no significant effect on enzyme activity in assay systems with PC or iPrOH or binding to PC vesicles (6). Specific phosphotransferase activity was only reduced with PI dispersed in Triton X-100. Clearly, removal of the cationic side chain in this region and replacement with a small neutral side chain does not contribute to enzyme binding to PC or to specific activity. However, replacing Lys44 with an anionic group (Glu) reverses the net charge on helix B, and leads to an enzyme, K44E, with both reduced PI cleavage and cIP hydrolysis in the presence of activating PC (Table 1) and reduced affinity for POPC vesicles (Table 2). Introduction of a negative charge at this position also weakens binding of the enzyme to PC interfaces. The K44E protein also had an unusual CD spectrum in that it had 7.4% less α-helix than rPI-PLC, suggesting less stable helical conformations in solution. However, the addition of 2 mM diC7PC led to an increase in α-helix at the expense of β-sheet and random coil (similar to what was observed for P42G). The selective loss of PC activation, most noticeably for cIP hydrolysis, with the charge reversal of the lone charged group in helix B might suggest that it is very close to a PC binding site, possibly near the phosphate rather than the trimethylammonium moiety.
Previous work suggested that Trp242 is the main fluorophore responsible for the intrinsic fluorescence change when PI-PLC binds to activator PC micelles (5). As shown in Figure 6A, the change in fluorescence intensity of rPI-PLC upon micelle formation showed a steeper increase than that of I43A, consistent with the weaker binding of I43A to POPC surfaces. The mutant proteins I43W/W47I and I43W exhibited fluorescence behavior comparable to rPI-PLC. The K0.5 values, which represent total diC7PC, range from 1.59 to 2.48 mM. If the CMC for diC7PC (1.5 mM) is subtracted from the bulk PC concentrations, and the resultant curves fit (except for I43A) with a hyperbolic function (Figure 6B), we obtain apparent micelle Kd values that show a more striking change in micellar PC affinity (Table 2). Removal of the hydrophobic side chain of Ile43 or placing a Trp at this position and an Ala at 47 generates enzyme with weaker affinity for diC7PC micelles. The sigmoidal nature of I43A strongly suggests that its binding is much weaker than the other mutated proteins examined since a higher amount of micellar diC7PC must be present to observe binding (the sigmoidal behavior may result because the protein needs a certain concentration of micelles prior to binding rather than recruiting monomers to form a complex). The swapped mutant protein (I43W/W47I), I43W, and K44A all bind slightly more tightly to PC micelles than rPI-PLC protein (Table 2). Interestingly, K44A shows less than half the fluorescence change of rPI-PLC (Figure 6), indicting a difference in the environment of the dominant fluorophore, previously shown to be Trp242 (5), in this mutant protein. In monomeric PI-PLC structures (4, 16), residue 44 is not close to Trp242. However, in the W47A/W242A dimer, the residues in the helix B region are much closer to the Trp242 in the rim loop (9).
Figure 6.

Increase in fluorescence intensities of B. thuringiensis PI-PLC proteins (WT (filled circle), I43A (), I43W (square), I43W/W47I (triangle), K44A (X)) at 337 nm as a function of diC7PC (A) total concentration and (B) micellar diC7PC.
PI-PLC induced aggregation of diC7PC
PI-PLC interaction with diC7PC micelles can also be monitored by the increase in the 31P linewidth of diC7PC when the protein is present (2). The width at half height, Δν1/2, for the phosphorus resonance is broad in the presence of rPI-PLC protein as soon as the diC7PC concentration is greater than the CMC (Figure 7). The very large increase in 31P linewidth when recombinant wild type protein binds to micellar diC7PC reflects a combination of aggregation of the protein/diC7PC system (20) and slower exchange between monomer, micelle, and enzyme-bound diC7PC such that the exchange is intermediate on the NMR time-scale (2, 8). At high concentrations of diC7PC (~20 mM) where the ratio of diC7PC/rPI-PLC is around 200, the limiting linewidth of the phosphorus in the presence of rPI-PLC is much larger than that for pure diC7PC micelles. At 20 mM diC7PC, if the diC7PC molecules were in fast exchange between monomer, micelles of the same size as those in the absence of protein, and enzyme-sites, the weight-averaged linewidth should approach the linewidth of those pure diC7PC micelles (~3 Hz). That it does not with rPI-PLC present is consistent with formation of an rPI-PLC/diC7PC complex that is considerably larger than pure diC7PC micelles and that likely alters the size distribution of diC7PC micelles (16). Although the detailed contribution of different species and exchange rates to this observed linewidth has not been determined, the increased linewidth has been interpreted as formation of an activated PI-PLC aggregate (20). Interestingly, the same titration experiments carried out for I43W and I43W/W47I show much smaller increases in 31P linewidth right above the CMC and much smaller limiting linewidths (Figure 7). Both mutant proteins exhibit K0.5 values for diC7PC similar to rPI-PLC as measured by intrinsic fluorescence changes. From the difference in diC7PC 31P linewidths in the absence and presence of protein at PC concentrations above the CMC, a ‘bound’ linewidth, Δb, (extrapolated broadening of one diC7PC molecule caused by one PLC) can be estimated as well as an excess limiting particle linewidth, which must reflect increases in the average protein/lipid mixed micelle particle size. For rPI-PLC, Δb = 373±41 Hz; this value is reduced to 36.1±5.7 Hz for I43W/W47I and 14.1±6.6 Hz for I43W. The limiting excess particle linewidth is 5.9±1.6 Hz for rPI-PLC, 3.2±0.2 Hz for the double mutant and essentially zero (0.30±0.27 Hz) for I43W. Since all three species are quite active, this suggests that the micellar aggregate structure per se is not responsible for diC7PC activation of cIP hydrolysis. Rather the aggregation upon interaction with diC7PC micelles is likely to reflect placement of the hydrophobic Trp residues at the rim of the PI-PLC barrel and the size and orientation of the hydrophobic plug this produces.
Figure 7.

(A) 31P linewidth (Hz) of the diC7PC phosphorus resonance in the presence of 3 mg/ml WT (filled circle), I43W (open circle), or I43W/W47I (square) PI-PLC. The top scale on the figure also shows the corresponding diC7PC/PI-PLC ratio for these diC7PC titrations. For comparison the linewidth for diC7PC as a function of concentration without enzyme is shown (bold line). (B) Difference in diC7PC linewidth (ΔΔν1/2) in the presence of PI-PLC for the three PI-PLC enzymes.
Discussion
Interfacial activation of water-soluble phospholipases can occur by diverse mechanisms. In the case of the secreted bacterial PI-PLC from Bacillus sp., there is a very specific kinetic activation towards both phosphotransferase and phosphodiesterase substrates when a PC interface is present (1–3, 21). In the absence of PC and with moderate salt, the protein does not bind to vesicles composed of anionic phospholipids (e.g., phosphatidylmethanol (PMe), phosphatidylglycerol (PG)) that are competitive inhibitors of PI (3, 22), while it binds well to PC vesicles. Enhanced PI cleavage in the presence of PC can then be explained by PC anchoring the protein to the interface allowing it to access substrate. More problematic is how to explain activation (increased kcat and decreased Km) of diC4PI cleavage or cIP hdyolysis (1, 6). Activation towards these monomeric substrates strongly indicates binding to a PC surface alters the protein conformation to make it a better catalyst. However, determination of a detailed molecular picture of the mechanism for this activation has been difficult to achieve since the activation occurs on PC surfaces.
Based on the crystal structure of the monomeric B. cereus PI-PLC (4), the helix B region (Ile43 to Gly48) in wild-type PI-PLC, whose Trp residue has been proposed to partially insert into the membrane (5, 7), orients the side chains of Ile43 and Trp47 (and possibly Val46 as well) so that they pack together and form a hydrophobic protrusion that extends outwards toward solvent. This would suggest that an intact helix B is critical for the initial interaction of PI-PLC with PC containing membranes. However, a recent crystal structure (9) of an interfacially impaired mutant, W47A/W242A, showed that the helical secondary structure of the Ile43 to Gly48 segment has been disrupted to form an extended loop. The rim loop that would contain Trp242 has also been displaced. The removal of the two Trp residues thought to insert into membranes could explain the loss of PC binding and low phosphotransferase and cyclic phosphodiesterase activities. However, an unexpected and very interesting feature of this double mutation protein was that PI-PLC W47A/W242A exists in the crystal as a homodimer. Other crystal structures of this bacterial PI-PLC show it is a monomer, and various sizing techniques showed the protein to have a predominantly monomer structure in solution (2). Residues in the helix B region, notably Pro42 and Gln45, play important roles in stabilizing the W47A/W242A dimer. The key question becomes whether a dimer forms with rPI-PLC on membrane surfaces and, if so, how close the structure of W47A/W242A is to the ‘active’ dimer proposed to form on PC interfaces. Formation of a dimer on the membrane surface is appealing because it could provide the protein with an easier means of processive catalysis – release of water-soluble products by one subunit could occur with the other still tightly anchored to the membrane. Recent studies that examined mutations of three or more of the tyrosine residues that form the hydrophobic core of the dimer interface in W47A/W242A, showed a dramatic loss of PI-PLC phosphotransferase and phosphodiesterase activities when these tyrosine residues, not close to the active site or directly around the barrel rim, were replaced with serine (16). Those results strongly support a dimer as the membrane-bound form of this PI-PLC. However, whether or not helix B is intact in the membrane-bound form still needs to be addressed.
The results in this study of mutating residues in the helix B region of PI-PLC are also consistent with dimer formation on PC interfaces. Q45A has reduced affinity for PC interfaces. Yet if helix B remains intact when the protein is bound to PC, the residue at position 45 could not contribute to membrane binding (or dimer formation) since it is on the opposite face of helix B (Figure 2D). With an intact helix B, both Ile43 and Trp47 would dominate binding of the helix to the membrane. The reduced PC activation of Q45A indicates that the membrane-active conformation of the enzyme is less favorable with this mutation, consistent with the hypothesis that helix B is unlikely to remain intact at the membrane surface during catalysis. In the W47A/W242A dimer Gln45 forms a hydrogen bond with Gly239 of the other subunit (Figure 2E). If this interaction contributes to rPI-PLC dimer stabilization of at the membrane, Gln45 removal would be expected to disfavor dimer formation thereby reducing PC affinity and enzyme activities. Indeed both cIP and PI cleavage were reduced with the Q45A mutation. Pro42 is also involved in dimer stabilization in W47A/W242A. However, P42G could also alter helix B formation and orientation – the introduction of a very flexible residue allows the enzyme to have much greater variability in the helix B region. Interestingly, diC7PC activation of cIP hydrolysis was reduced with only small effects on PI cleavage. If helix B in solution has been destabilized by introduction of a glycine at position 42, then the initial interaction of the protein with PC would be compromised. These results could also suggest that the presence of acyl chains on the bound PI substrate also contribute to dimerization of PI-PLC.
Leu39 provides an interesting wrinkle on membrane binding and dimer formation of B. thuringiensis PI-PLC. Like W47A, L39A binds very poorly to PC vesicles. Furthermore, the magnitude of the drop in affinity is much greater than would be predicted if an alanine rather than a leucine side chain partitioned in the membrane. In the crystal structure of B. cereus PI-PLC (4) as well as the W47A/W242A dimer (9), the side chain of Leu39 is embedded in the structure and would not be expected to have a significant effect on PC binding. The significantly weaker binding of L39A to POPC vesicles suggests the membrane-active dimer is not exactly the same as the W47A/W242A dimer and that there is a conformational change in this region of the barrel rim that either orients Leu39 outward so that it can partition directly into the PC surface and/or positions this residue to interact with the initial portions of the hydrophobic acyl chains of the substrate.
A proposed model for activation of B. thuringiensis PI-PLC is shown in Figure 8. PI-PLC in solution has an intact helix B (shown in green) that keeps the enzyme as a monomer and prevents premature dimerization (conformation A in Figure 8). Solution dimerization, were it to occur, might prevent tight binding of the protein to membranes since the resultant dimer (based on W47A/W242A) has a highly concave surface (9). An intact helix B also provides the hydrophobic surface for the initial binding of the protein to a PC membrane (or a PC-enriched region in mixed substrate/PC bilayers). Once contact occurs, Trp47 is inserted into the bilayer/interface. The insertion of Trp47 triggers a rearrangement of helix B residues altering both the position and structure to an extended loop that could provide several points of contact, Ile43 and Val46 as well as Trp47, with the membrane (conformation B in Figure 8). Membrane anchored protein monomers can now dimerize on the membrane surface via Tyr246, Tyr247, Tyr248, and Tyr251 residues (16) with both Pro42 and Gln45 part of the dimerization network. Enhanced PI cleavage to cIP would in part reflect the increased lifetime of the protein dimer on the membrane. However, other changes must occur for cIP hydrolysis to also be activated by PC interfaces. Work by us (1–3, 5–8, 21) and others (15, 20) has suggested there is a conformational change in the Bacillus protein (as well as in a similar PI-PLC from Listeria monocytogenes (23)) when it binds to activating interfaces that increases kcat and decreases the apparent Kd for membranes. This is reflected in a tighter affinity for activating interfaces (6, 8) and an increased inhibition of cIP hydrolysis by substrate analogues such as phosphatidic acid (PA) or PMe (21). In a W47A/W242A-like dimer, the accessibility of active site residues is altered to present a more extended surface (9) that should increase interactions of substrate molecules with the enzyme.
Figure 8.
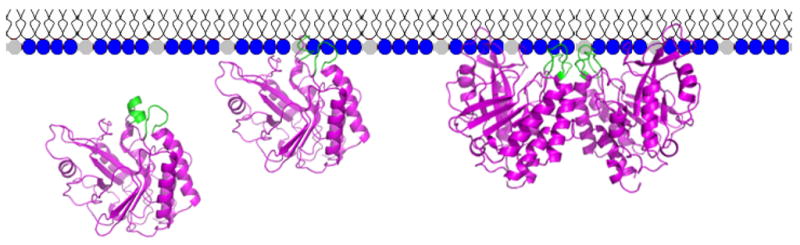
Model for the role of helix B residues in PI-PLC activation by PC interfaces.
There are also likely to be more subtle changes in the active sites of the dimer that modulate the catalytic step. The transformation of PI to cIP occurs by His32 acting as a general base to deprotonate the inositol C(2)-OH so it can attack the phosphate group. His82 then acts as the general acid to protonate the DAG leaving group. In the second step (cIP hydrolysis to I-1-P), water, deprotonated by His82 acting as a general base, acts as a nucleophile to attack the phosphorus while protonated His32 contributes a proton to the inositol C2 oxygen. It has been suggested (24) that the catalytic triad of His82-Asp33-Arg69 is not optimally arranged to catalyze the hydrolysis of cIP. Rather, hydrophobic interactions of the rim and perhaps the hydrocarbon chains of the PI substrate facilitate the changes necessary for optimal catalysis (hence PI cleavage will be much faster than cIP hydrolysis). For cIP in solution, both His32 and His82 would have the ‘wrong’ initial ionization states. Enhancing catalysis, particularly for the second step, would be provided by an allosteric pathway between helix B residues and catalytic residues that shifts local pKa values so that enhanced bond cleavage occurs. Binding to a PC interface provides this local change while not binding to the active site in place of cIP. Gly48, at the C-terminal end of helix B, would appear to be a key residue in this process. In structures of the B. cereus PI-PLC and W47A/W242A, Gly48 is close to His82 (< 4 Å separation). While the binding of G48A to PC is very similar to rPI-PLC, the activities for both PI cleavage and cIP hydrolysis are lower than rPI-PLC. Furthermore, the activation by PC for cIP hydrolysis is dramatically decreased for this mutant enzyme relative to rPI-PLC (3-fold versus 22-fold activation). Since binding to PC induces a conformational change of the enzyme, Gly48 might be a very important site for communicating with the active site His82 about the conformational change of the helix B region.
Abbreviations
- cIP
1,2-cyclic inositol phosphate
- CMC
critical micelle concentration
- DAG
diacylglycerol
- HEPES
- I-1-P
D-inositol-1-phosphate
- iPrOH
isopropanol
- PI
phosphatidylinositol
- PI-PLC
phosphatidylinositol-specific phospholipase C
- rPI-PLC
recombinant PI-PLC
- PC
phosphatidylcholine
- POPC
1-palmitoyl-2-oleoyl-PC
- diC7PC
diheptanoylphosphatidylcholine
- SUV
small unilamellar vesicle
Footnotes
This research was supported by National Institutes of Health GM60418.
References
- 1.Zhou C, Wu Y, Roberts MF. Activation of phosphatidylinositol-specific phospholipase C toward inositol 1,2-(cyclic)-phosphate. Biochemistry. 1997;36:347–355. doi: 10.1021/bi960601w. [DOI] [PubMed] [Google Scholar]
- 2.Zhou C, Qian X, Roberts MF. Allosteric activation of phosphatidylinositol-specific phospholipase C: specific phospholipid binding anchors the enzyme to the interface. Biochemistry. 1997;36:10089–10097. doi: 10.1021/bi970846o. [DOI] [PubMed] [Google Scholar]
- 3.Qian X, Zhou C, Roberts MF. Phosphatidylcholine activation of bacterial phosphatidylinositol-specific phospholipase C toward PI vesicles. Biochemistry. 1998;37:6513–6522. doi: 10.1021/bi972650u. [DOI] [PubMed] [Google Scholar]
- 4.Heinz DW, Ryan M, Bullock TL, Griffith OH. Crystal structure of the phosphatidylinositol-specific phospholipase C from Bacillus cereus in complex with myo-inositol. EMBO J. 1995;14:3855–3863. doi: 10.1002/j.1460-2075.1995.tb00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng J, Webhi H, Roberts MF. Role of tryptophan residues in interfacial binding of phosphatidylinositol-specific phospholipase C. J Biol Chem. 2002;277:19867–19875. doi: 10.1074/jbc.M200938200. [DOI] [PubMed] [Google Scholar]
- 6.Wehbi H, Feng J, Kolbeck J, Ananthanarayanan B, Cho W, Roberts MF. Investigating the interfacial binding of bacterial phosphatidylinositol-specific phospholipase C. Biochemistry. 2003;42:9374–9382. doi: 10.1021/bi034195+. [DOI] [PubMed] [Google Scholar]
- 7.Feng J, Bradley W, Roberts MF. Optimizing the interfacial binding and activity of a bacterial phosphatidylinositol-specific phospholipase C. J Biol Chem. 2003;278:24651–24657. doi: 10.1074/jbc.M301207200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Wehbi H, Roberts MF. Cross-linking phosphatidylinositol-specific phospholipase C traps two activating phosphatidylcholine molecules on the enzyme. J Biol Chem. 2004;279:20490–20500. doi: 10.1074/jbc.M401016200. [DOI] [PubMed] [Google Scholar]
- 9.Shao C, Shi X, Wehbi H, Zambonelli C, Head JF, Seaton BA, Roberts MF. Dimer structure of an interfacially impaired phosphatidylinositol-specific phospholipase C. J Biol Chem. 2007;282:9228–9235. doi: 10.1074/jbc.M610918200. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. BioTechniques. 1999;26:680–682. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- 11.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 12.Braman J, Papworth C, Greener A. Site-directed mutagenesis using double-stranded plasmid DNA templates. Meth Mol Biol. 1996;57:31–44. doi: 10.1385/0-89603-332-5:31. [DOI] [PubMed] [Google Scholar]
- 13.Bohm G, Muhr R, Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- 14.Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 15.Volwerk JJ, Shashidhar MS, Kuppe A, Griffith OH. Phosphatidylinositol-specific phospholipase C from Bacillus cereus combines intrinsic phosphotransferase and cyclic phosphodiesterase activities: a 31P NMR study. Biochemistry. 1990;29:8056–8062. doi: 10.1021/bi00487a010. [DOI] [PubMed] [Google Scholar]
- 16.Shi X, Shao C, Zhang X, Zambonelli C, Redfield AG, Head JF, Seaton BA, Roberts MF. Modulation of Bacillus thuringiensis phosphatidylinositol-specific phospholipase C activity by mutations in the putative dimerization interface. J Biol Chem. 2007 doi: 10.1074/jbc.M901601200. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 18.Wu Y, Roberts MF. Phosphatidylinositol-specific phospholipase C cyclic phosphodiesterase activity depends on solvent polarity. Biochemistry. 1997;36:8514–8521. doi: 10.1021/bi970560j. [DOI] [PubMed] [Google Scholar]
- 19.Wehbi H, Feng J, Roberts MF. Water-miscible organic cosolvents enhance phosphatidylinositol-specific phospholipase C phosphotransferase as well as phosphodiesterase activity. Biochim Biophys Acta. 2003;1613:15–27. doi: 10.1016/s0005-2736(03)00134-2. [DOI] [PubMed] [Google Scholar]
- 20.Berg OG, Yu B-Z, Apitz-Castro RJ, Jain MK. Phosphatidylinositol-specific phospholipase C forms different complexes with monodisperse and micellar phosphatidylcholine. Biochemistry. 2004;43:2080–2090. doi: 10.1021/bi035063j. [DOI] [PubMed] [Google Scholar]
- 21.Zhou C, Roberts MF. Nonessential activation and competitive inhibition of bacterial phosphatidylinositol-specific phospholipase C by short-chain phospholipids and analogues. Biochemistry. 1998;37:16430–16439. doi: 10.1021/bi980601r. [DOI] [PubMed] [Google Scholar]
- 22.Roberts MF, Redfield AG. High-resolution 31P field cycling NMR as a probe of phospholipid dynamics. J Am Chem Soc. 2004;126:13765–13777. doi: 10.1021/ja046658k. [DOI] [PubMed] [Google Scholar]
- 23.Ryan M, Zaikova TO, Keana JF, Goldfine H, Griffith OH. Listeria monocytogenes phosphatidylinositol-specific phospholipase C: activation and allostery. Biophys Chem. 2002;101–102:347–358. doi: 10.1016/s0301-4622(02)00158-8. [DOI] [PubMed] [Google Scholar]
- 24.Kubiak RJ, Yue X, Hondal RJ, Mihai C, Tsai MD, Bruzik KS. Involvement of the Arg-Asp-His catalytic triad in enzymatic cleavage of the phosphodiester bond. Biochemistry. 2001;40:5422–5432. doi: 10.1021/bi002371y. [DOI] [PubMed] [Google Scholar]