

Abstract
Cyclodextrin (CD) is a well known drug carrier and excipient for enhancing aqueous solubility. CDs themselves are anticipated to have low membrane permeability because of relatively high hydrophilicity and molecular weight. CD derivatization with 17-beta estradiol (E2) was explored extensively using a number of different click chemistries and the cell membrane permeability of synthetic CD–E2 conjugate was explored by cell reporter assays and confocal fluorescence microscopy. In simile with reported dendrimer –E2 conjugates, CD–E2 was found to be a stable, extranuclear receptor selective estrogen that penetrated into the cytoplasm.
Keywords: Cyclodextrin, Estrogen, Bioconjugate, Membrane, Nuclear receptor
1. Introduction
β-Cyclodextrins (CDs) are torus-shaped saccharides consisting of a shallow interior cavity, a primary face of 7 hydroxyls and a secondary face of 14 hydroxyl groups. CDs and modified CDs have many interesting biological properties due to their unique structure, which can be altered most readily by chemical modification of the primary face.1,2 The hydrophobicity of the CD cavity has dominated thinking in terms of CD host–guest chemistry via formation of non-covalent inclusion complexes and native and commercially available CD derivatives, the latter generally heterogeneous compounds, have been used as efficient drug carriers.3 Alternatively, the CD torus can be used as a scaffold to attach drug moieties in order to enhance their effectiveness by facilitating drug delivery or specificity; for example, CD–epoxysuccinyl peptide conjugates were reported as inhibitors of membrane-associated cathepsin B, known to be secreted in several tumor cell lines.4 The synthesized CD–peptide conjugates were full inhibitors of extranuclear cysteine proteases and were reported not to be membrane-permeant. Other examples of CD conjugates as specific ligands of cell-surface receptors include opioid peptides,5 gastrin peptides,6 and mono- and oligosaccharides.7 These approaches rely in part on the assumed membrane impermeability of CD conjugates, based upon the natural hydrophilicity of CD conjugates and a molecular weight (>1500) above the perceived limits of facile, passive membrane transport.
The development of chemically and biologically stable estrogen –macromolecule conjugates has been a challenge of increasing importance given the interest in differentiating non-classical ligand- activated estrogen receptor (ER) actions versus the classical ER function as a ligand-activated nuclear receptor. Estrogens are able to elicit rapid responses that are thought to be mediated by extranuclear receptors that may be the classic ER, splice variants, or G-protein coupled receptors.8–10 The non-classical, extranuclear action of E2 elicit activity via cytosolic kinase cascades, which may have genomic effects via epigenetic actions or transcription factors other than ER.11,12 The classical, genomic actions of ER as a nuclear receptor have been differentiated from membrane-associated ER activity by use of commercially available protein conjugates such as E2–BSA12–14 and E2–peroxidase.15 These conjugates have been widely used, but as remarked by Katzenellenbogen, they have critical limitations such as leakage of free E2 either non-covalently bound or from biological degradation of the conjugate in cell cultures. 8,13,16 In order to overcome these problems and develop efficient E2–macromolecule conjugates, Katzenellenbogen and coworkers utilized a polyamidoamine (PAMAM) dendrimer as core architecture instead of a protein, leading to an E2-conjugate selective for extranuclear ER.
Click chemistry is commonly used today as a synonym for the Huisgen cycloaddition, although originally defined by Sharpless as encompassing simple synthetic steps giving quantitative conversions. 17 Quantitative procedures for formation of CD-conjugates avoid the complex chromatography required to remove by-products of incomplete derivatization. Herein, novel estradiol–CD conjugates were prepared using click chemistry and the estrogenic activity was assayed in order to test the ability of CD derivatives to provide selective probes of estrogenic activity.
2. Results and discussion
2.1. Synthesis of ACD conjugates
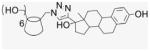
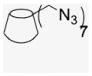
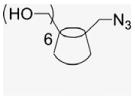
A CD-conjugate persubstituted at the C6 position of the primary face was the initial target. Aminocyclodextrins (ACDs) are a specific subset of persubstituted CDs, which may be readily prepared by direct amination of per-6-bromo-per-6-deoxy-CD or per-6-iodo-per-6-deoxy-CD ACDs possess an annulus of amino groups in place of the ether oxygens of native CD and a corona of pendant side chains; at neutral pH the presence of the positively charged annulus diminishes the ability of the ACD cavity to include hydrophobic small molecules.18 The ACD scaffold provides ACD derivatives with demonstrated, inherent biological activity.19 Synthesis from the more reactive per-6-iodo-per-6-deoxy-CD was revealed by careful chromatography to yield an intramolecular cross-linked impurity, 20 whereas synthesis via the less reactive per-6-bromo-per-6-deoxy-CD is accomplished in quantitative steps requiring no chromatography (Scheme 1).21,22 However, this ‘click’ strategy is limited to reactions with amines in the liquid phase or with high solubility in DMF,21 an alternative click methodology is imine formation from reaction of aldehyde with per-6-amino-per-6-deoxy-β-CD (3), easily obtained from the per-azido derivative 2, and subsequent reduction, ultimately leading to the desired CD–E2 conjugate, 7EACD (Scheme 1).
Scheme 1.

Modifications of β-cyclodextrin.
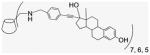
EE2 is a biologically active ER ligand of synthetic utility because of the reactivity of the terminal acetylene group.23 Elaboration of EE2 is readily achieved using palladium catalyzed Sonogashira cross coupling with halide reagents,24 allowing attachment of a benzaldehyde moiety for ACD conjugation. Subsequent reduction of benzaldehyde 4 gave the benzyl alcohol 5 that was used as a control compound. The previously reported heterogeneous dendrimer E2–PAMAM conjugate was obtained by PAMAM condensation with 4 in MeOH at room temperature.8,25 Similarly, condensation of ACD (3) with 4 was attempted, followed by in situ NaBH4 reduction (Scheme 1; Table 1 entry 1). However, reaction did not go to completion, giving a heterogeneous mixture that, from mass spec and NMR examination, contained (EE2)7ACD as major product in addition to smaller amounts of (EE2)6ACD and (EE2)5ACD (Fig. 1). Optimization of reaction conditions failed to yield a homogeneous product, however, semi-preparative RP-HPLC did yield conjugate 7EACD (6) that consists of (EE2)7ACD (>90%) with small amounts of (EE2)6ACD and (EE2)5ACD. Chromatographic and spectroscopic examination revealed no evidence for an inclusion complex between 7EACD and either EE2 or the reaction by-product, 5, compatible with the extensive washing and chromatography employed in synthesis, and the known propensity of ACD to former weak inclusion complexes.18 7EACD is comparable to the heterogeneous E2–dendrimer conjugate described by Katzenellenbogen.
Table 1.
| Entry | CD | Ligand | Reaction condition | Product | |
|---|---|---|---|---|---|
| 1a | 10–70 equiv |
 4 |
60–70 °C, 10 d |
 7EACD,6 |
|
| 2b |
 3 |
8 equiv | 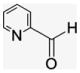 |
rt, 5 h |
 7 |
| 3c | 8 equiv | 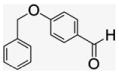 |
rt, 7 d |
8 |
|
| 4c | 8 equiv | 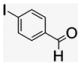 |
60–70 °C, 18 h |
11 |
|
| 5d | 8 equiv |
9 |
60–70 °C, 18 h | S.M recovered | |
| 6d | 10 equiv |
 10 |
60–70 °C, 10 d | S.M recovered | |
| 7d |
 15 |
21 equiv |
 4 |
60–70 °C, 10 d |
16 |
| 8e |
 19 |
1–3 equiv | 60–70 °C, 3 d |
1EACD,20 |
|
| 9f |
 17 |
21 equiv |
 12 |
60–70 °C, 7 d |
21 |
(1) DMF, 60–70 °C, 10 d, (2) NaBH4, MeOH, 2 h.
(1) rt, 5 h, (2) NaBH4, MeOH, 2 h.
(1) DMF, rt, 7 d, (2) NaBH4, MeOH, 2 h.
(1) DMF, 60–70 °C, 18 h, (2) NaBH4, MeOH, 2 h.
(1) DMF, 60–70 °C, 3 d, (2) NaBH4, MeOH, 2 h.
DMF, 60–70 °C, 7 d.
Figure 1.

(I) (A–D) HPLC–UV chromatograms of crude and purified 7EACD product compared to synthetic monomer standards; (E) MALDI-TOF mass spectrum of purified 7EACD. (II) (A) 13C NMR (75 MHz, DMSO-d6) spectrum of 7EqCD (δ ppm) 154.8, 153.6, 137.1, 130.4, 125.9, 124.4, 114.9, 112.6, 101.5, 82.0, 81.0, 72.4, 71.8, 69.5, 49.5, 47.6, 46.8, 43.1, 37.1, 32.6, 30.7, 29.2, 27.2, 26.1, 23.4, 14.3); (B) MALDI-TOF spectrum of 7EqCD; (C) HPLC–UV chromatogram of 7EqCD, detection at 280 nm, m/z = 1261.7 MH3 3+).
The lack of a homogeneous product from the imine synthetic route (Scheme 1) was surprising given that the condensation of 2-pyridinecarboxaldehyde with 3 had been previously demonstrated in our lab to proceed to completion, yielding the persubstituted ACD (7) after reduction in situ (Table 1, entry 2).26 To examine the limitations of this synthetic methodology for persubstituted ACD derivatives, other approaches were attempted. Condensation of 3 with 4-benzyloxybenzaldehyde yielded the desired product 8 (Table 1, entry 3). The possibility that complete reaction of 3 with 4 was hindered by the steric bulk of the ethynyl estradiol group was explored by using a longer linker arm between estradiol and aldehyde groups. However, neither condensation of 10 (Scheme 2) nor the synthon 9 with ACD (3) gave the desired imine products (Table 1, entries 5 and 6), indicating the preference in imine condensation for the more reactive arylaldehydes. Other alternatives were explored, including reaction via the known β-CD derivative, 15 (Scheme 2; Table 1, entry 7).27 however, under all reactions conditions tested, the persubstituted product was the major, but not the sole product observed. The imine condensation reaction of 19 with 4 produced the monosubstituted E2-conjugate 1EACD (20; Table 1, entry 8), and a similar product (21) was obtained by the simple nucleophilic displacement of the tosyl group in mono-6-tosyl-β-CD using EE2 derivative 12 (Table 1, entry 9). Both E2-conjugates 20 and 21 contained small amounts of unmodified β-CD carried through the synthesis as shown by NMR spectra and MALDI-TOF mass spectroscopic analysis.
Scheme 2.

Synthesis of EE2 derivatives and synthons.
2.2. Synthesis of ‘qCD’ conjugates
Cycloaddition click chemistry is currently used extensively for bioconjugation because of the efficient conversion and good yields in aqueous media.28–30 Given the ease with which mono- and persubstituted azido-CD are prepared, this synthetic methodology was explored for E2–CD conjugates. The presence of seven triazole groups in the annulus of such cyclodextrin conjugates is likely to confer inherently different physicochemical properties to CD and ACD conjugates, hence we coin the click-derived conjugates as qCD derivatives.
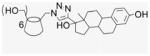
A variety of click reactions were explored based upon literature precedent (Table 2),31,32 monitoring conjugation products by MALDI- TOF spectroscopy. In situ [1,3]-cycloaddition ligation was unsuccessful from 1 (Table 2, entry 10), but was successful from mono-6-tosyl-β-CD giving the desired product 22 in 50% conversion along with unreacted β-CD contaminant 17 (Table 2, entry 11).33–35 The reaction mixture of 2 with seven equivalent of EE2 in the presence of CuI and DIEA changed color from green to brown coincident with the loss of the TLC spot for unreacted EE2. Reaction time was extended for one day beyond this indicator point, yielding quantitative conversion to 7EqCD (23) as a powder in 58% isolated yield after washing (Table 2, entry 12; Fig. 1). The monoazido-β-CD under the same reaction conditions gave 1EqCD (24) in 68% isolated yield (Table 2, entry 13). In all procedures, extensive washing was carried out to remove residual Cu ions. As a control compound for bioassay, a triazole modified estradiol was synthesized (qE2, 25): methyl azide was prepared for in situ click ligation to EE2, resulting in the desired product 25 obtained in 60% yield after chromatography (Table 2, entry 14).
Table 2.
Click chemistry routes to qCD derivatization
| Entry | CD | Ligand | Reaction condition | Product |
|---|---|---|---|---|
| 10g |
 1 |
 |
CuSO4–5H2O, NaN3 sodium ascorbate | EE2 recovered |
| 11h |
 17 |
 |
CuSO4–5H2O, NaN3 sodium ascorbate |
 22 |
| 12i |
 2 |
7 equiv 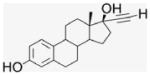
|
CuI, DIEA |
 7Eqcd,23 |
| 13i |
 18 |
1 equiv 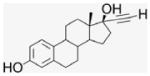
|
CuI, DIEA |
 1Eqcd,24 |
| 14j | 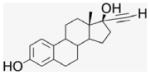 |
Dimethylsulfate, NaN3CuSO4–5H2O, sodium ascorbate |
 qE2,25 |
|
| 15k |
 7EqCD,23 |
NHS–rhodamine |  |
 |
| Rhodamine–perchlorate | 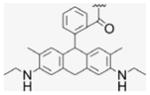 |
|||
| 16i |
 2 |
CuI, DIEA |
 26 |
|
| 17k |
 26 |
NHS–rhodamine | 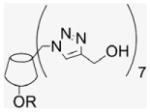 |
 |
| Rhodamine–perchlorate |  |
DMF/H2O (3:1), 60 °C, 4 d.
DMF/H2O (3:1), 60 °C, 4 d.
DMF/MeOH, rt, 1 d.
t-BuOH/H2O (3:1), rt, 1 d.
DMAP/DMF, EDC–HCl/0 °C.
In order to estimate the membrane permeability of 7EqCD, rhodamine was coupled to the secondary face of 7EqCD (26; Table 2, entry 15). Non-covalent inclusion of rhodamine would be difficult to rule out in the rhodamine–7EqCD conjugates, therefore the triazole- CD derivative (26) was prepared and the rhodamine–qCD conjugate synthesized for comparison (Table 2, entries 16 and 17). Both compounds 23 and 26 were coupled with 1 mol equiv of fluorophore, either as the free carboxylic acid (rhodamine-19-perchlorate, yielding conjugates 23R′ and 26R′, respectively) or as the succinimidyl ester (NHS–rhodamine, yielding products 23R and 26R, respectively). In each case, the presence of the monosubstituted rhodamine conjugate was demonstrated by MALDI-TOF spectroscopy.
2.3. Summary of synthetic strategies
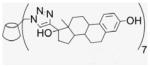
Elaboration of the ACD scaffold via imine formation with aryl aldehydes is efficient when the aldehyde can be used as a co-solvent or in high concentration, however other aldehydes do not give quantitative reaction. Monitoring of reactions by MALDI-TOF is useful to identify qualitatively the extent of multivalent substitution of CD conjugates, however, the intensity of signals in MALDI-TOF is not directly related to relative quantities of each multivalent ACD conjugate. The problematic ability of mass spec to provide quantitation has been previously described for CD derivatives. 36 Furthermore, only perfacially substituted CD derivatives give simple NMR spectra; incompletely substituted β-CD derivatives have 42 chemically and spectroscopically non-equivalent glucose carbons. From HPLC–UV chromatography, 7EACD was estimated predominantly to contain the desired persubstituted product, (EE2)7ACD, however the MALDI-TOF spectrum indicated the presence of both (EE2)6ACD and (EE2)5ACD isomers (Fig. 1). Cycloaddition click chemistry provided a more efficient route to perfacial modification, yielding qCD conjugates perfacially derivatized with EE2 on the primary face (Figs. 1 and 2). Four biocompatible E2–CD conjugates were obtained for bioassay: 7EACD, 1EACD, 7EqCD, and 1EqCD.
Figure 2.

Structure of 7EqCD and one representative conformer optimized at the AM1level in Spartan 04 (Wavefunction Inc., CA) showing top and side views with small cavity (yellow), annulus of triazole groups (cyan), and large flexible corona of pendant estradiol groups visible.
2.4. Estrogenic activity of CD conjugates
The ERα-positive human endometrial Ishikawa cell line is routinely used to quantify estrogenic and antiestrogenic activity of test compounds.37 In this cell line, the induction of alkaline phosphatase is an ER-mediated estrogenic response, allowing measurement of EC50 values for estrogenic and IC50 for antiestrogenic potency by enzyme assay. The measured EC50 values for 1EACD, 1EqCD, 7EACD, and 7EqCD were 4.0 μM, 4.0 μM, 300 nM, and 200 nM, respectively; both polyvalent conjugates showed higher potency than monovalent derivatives ( Fig. 3). There was no significant antiestrogenic activity or cytotoxicity observed from the test compounds at the concentrations studied (data not shown). EE2 derivatives 5 and 12 and ACD (3) represent control compounds for EACD: ACD was inactive, whereas 5 and 12 were 200-fold more potent than 7EACD (Fig. 3). Similarly, qE2 and CD derivative 2 represent controls for EqCD; 2 was inactive, whereas qE2 was a full estrogen (Fig. 3). All non-conjugated estrogen control compounds were substantially less potent than EE2 itself, for which EC50 was measured as 28 pM in Ishikawa cells. Katzenellenbogen has referred to potency for E2–dendrimer conjugates where observed potency is divided by the average number of E2 moieties in the conjugate.8 Applying this measure, the potency for: 5, 1EACD, and 7EACD were 27 nM, 4 μM, and 2 μM, respectively; and for qE2, 1EqCD, and 7EqCD were 27 nM, 4 μM, and 1.4 μM, respectively.
Figure 3.

Estrogenic activity determined measuring alkaline phosphatase activity induced in ER(+) Ishikawa cells. Left: concentration–response for cells treated with 1EACD (closed triangles), 1EqCD (open triangles), 7EACD (closed circles), 7EqCD (open circles): 100% estrogenic activity corresponds to E2 (1 nM). Data show mean and s.d. from triplicate experiments. Right: table of potency for all compounds for which concentration–response was measured in Ishikawa cells.
In the standard radioligand displacement assay for measuring relative binding affinity to recombinant human ERα, qE2 was observed to be a weak ERα ligand (IC50 = 0.4 ± 0.1 μM). In this assay, ER binding of EACD and EqCD derivatives was not observed within solubility limits, however, other compounds with submicromolar estrogenic potency in cell culture have shown low or negligible affinity in the radioligand displacement assay.38
Rapid signaling via non-classical estrogen-activated pathways has been recognized for some time in the CNS and cardiovascular systems.39,40 However, the quantification of non-classical estrogenic activity is not trivial, in part because different molecular identities and cellular localization have been proposed for these receptors.9 The debate on ligand-activated G-protein-coupled ER (GPR-ER, or GPR-30) has focused on the plasma membrane and endoplasmic reticulum,41 whereas ER itself has been proposed to be active in the plasma membrane, cytoplasm, or mitochondria. 42 Rapid assays of MAPK pathway activation and intracellular Ca levels have been used for extranuclear ER activation, however, several pathways may elicit such responses. The classical activity of ER as a nuclear receptor can be assayed by E2-responsive gene reporter assays such as the alkaline phosphatase assay presented above, however, it has been shown that of E2-responsive genes, a full 25% are induced by E2–dendrimer conjugates.43 Recently Tonetti and co-workers reported that T47D/PKCα cells (human breast cancer cells stably transfected with protein kinase C) were stimulated by E2-BSA, whereas the parent cell line (T47D/neo) was unresponsive.44 Therefore, the activity of 7EqCD was assayed in both T47D cell lines (Fig. 4). The classical ER-dependent potency of 7EqCD in T47D/neo cells was low: EC50 for 7EqCD was >100 nM in T47D/neo cells. However, the response of T47D/PKCα cells to 7EqCD was similar to that of E2-BSA; 7EqCD (10 nM) was a full estrogen, with the EC50 estimated as 2 nM. Taken together, the data shown in Figure 4 suggest that 7EqCD at 2–10 nM may be useful as an extranuclear ER selective ligand and warrants further study.
Figure 4.

Activation of ERE and induction of luciferase activity in T47D/neo and T47D/PKCα cell lines by 7EqCD compared to E2–BSA. In all experiments, relative luciferase activity was corrected for transfection efficiency and normalized to vehicle (0%) and E2 (1 nM; 100%). Data show mean and s.d. from triplicate measurements on two separate cell passages.
2.5. Membrane permeability and mechanism of action
Rhodamine was conjugated to 7EqCD (23R, 23R′) using two different coupling agents, and a control qCD (26R, 26R′) was conjugated in the same way to provide comparison. Both fluorescent E2-conjugates retained estrogenic activity at higher concentrations as shown by measured EC50 values in Ishikawa cells of 1.5 ± 0.47 μM and 2.6 ± 0.63 μM for 23R and 23R′, respectively. ER positive MCF-7 mammary cancer cells were incubated with either 23R, 23R′, or the control compound (26R) for 24 h at which time nuclei and conjugate were localized by confocal fluorescence microscopy (Fig. 5). Both 7EqCD conjugates were observed in the cytoplasm, but not the nucleus of MCF-7 cells, whereas no evidence for cell membrane permeation was obtained for the control compound.
Figure 5.

Localization of (A) 26R, (B) 23R, and (C) 23R′ in ER positive MCF-7 cells by confocal microscopy. Drugs (100 nM) were incubated with cells for 24 h. Clockwise from bottom-left: Ex/Em: 544/576 nm rhodamine-conjugates; Ex/Em: 345 nm/420 nm nuclear marker; differential interference contrast image; overlay of rhodamine and nuclear dyes showing cytoplasmic localization of rhodamine conjugates for 23R and 23R′. Data shown are representative of several images of different cells and planes.
The estradiol conjugates best described to date are E2–BSA, E2–peroxidase, and E2–dendrimers; at higher concentrations (≥10 nM for protein conjugates and ≥100 nM for dendrimer conjugates) all three demonstrate classical estrogenic actions reasoned to result from leaching of non-covalently bound estrogen (E2 or EE2) or degradation to release estrogens. There is no reason to expect that inclusion of EE2 would be an important factor for either 7EACD or 7EqCD, because EE2 has low reported affinity for β-CD (Kd = 8 mM),45,46 and a more polar CD annulus is known to reduce the affinity for hydrophobic aromatic guests further.18,21 The presence of EE2 in the 7EqCD product was tested in the same Ishikawa cell cultures used for assessment of classical estrogenic activity. For comparison, the presence of E2 was measured in incubations of E2–BSA in Ishikawa cell culture. After four days, the cell supernatant was collected and assayed for EE2 and E2 using LC–MS/MS quantitation, against standard curves, of estrogens derivatized with dansyl chloride (Fig. 6). In the 7EqCD incubation, EE2 was not observed above detection limits, but E2 was observed in incubations of E2–BSA in cell cultures as anticipated from literature reports.
Figure 6.

LC–MS/MS analysis of free estrogen in cell culture supernatant after four days incubation of E2–BSA or 7EqCD. Positive ion electrospray selected reaction monitoring (SRM) chromatograms of estrogens as dansyl derivatives: (A) EE2 (70 nM) incubated in Ishikawa cell culture (peak intensity 3.0 × 105); (B) 7EqCD (10 nM) incubated with Ishikawa cells (maximum y-axis intensity 1.4 × 103) – EE2 peak was not observed above detection limits (<50 pM); SRM conditions EE2–dansyl·[MH+] m/z 530→171 collision energy 53 eV; (C) E2 (70 nM) incubated in Ishikawa cell culture (peak intensity 5.5 × 105); (D) E2–BSA (1 μg/mL) incubated with Ishikawa cells (peak intensity 1.1 × 104) showing approximately 1 nM based upon standard curves; SRM conditions E2–dansyl [MH+] m/z 506→171 collision energy 57 eV.
7EACD bears comparison to the heterogeneous E2–dendrimer conjugate prepared by Katzenellenbogen and co-workers, since both bioconjugates are prepared from a similar EE2 synthon via an imine intermediate, with the same spacer unit linking macromolecular amine with EE2, and both are heterogeneous.8 In MCF-7 cells, the E2–dendrimer induced E2-responsive genes, but E2 itself was 3–4 orders of magnitude more potent than the conjugate (E2–dendrimer; EC50 ~ 10−9–10−8 M).8 These and other data were compatible with permeation of the E2–dendrimer conjugate into the cytoplasm, but not into the nucleus. Recently, the combination of E2–BSA (membrane selective estrogen) and E2–dendrimer (extranuclear selective estrogen) conjugates has been used to distinguish membrane and extranuclear inputs into E2-responsive gene induction.43
3. Conclusions
The classical mechanism of estrogen action is defined by: (i) binding of an estrogenic ligand to ER in the cytoplasm; (ii) ER dimerization and translocation to the nucleus; (iii) complexation with the ERE domain of DNA; (iv) recruitment of coregulator proteins; (v) transcription and expression of E2-responsive gene products. Non-classical mechanisms utilize extranuclear receptors that are activated by estrogenic ligands, often via rapid signaling cascades, however these may also induce ER-dependent, genomic activity.43 It is recognized that selective ligands are required to untangle the role of nuclear and extranuclear receptors and the cross-talk between classical and non-classical pathways. β-CD derivatives play an important role in drug delivery, and less frequently have been used as molecular scaffolds for biomimetics; 47–49 the expectation that these large, hydrophilic molecules seldom penetrate cellular membranes supports the exploration of E2–CD conjugates as cell compartment selective estrogens.
Several click chemistry approaches were attempted towards a β-CD persubstituted at the primary face with estradiol via a stable linker. Reactions using the amino-CD (ACD) scaffold proved insufficiently generalized; selected examples giving quantitative persubstitution, but others such as the ACD–estradiol conjugate, 7EACD, not providing an homogeneous persubstituted product. Conjugation via [1,3]-cycloaddition proved a suitable alternative method for preparation of a homogeneous, persubstituted E2–CD, namely 7EqCD. The four E2–CD conjugates studied in cell culture were not cytotoxic below solubility limits and all manifested estrogenic activity in cell culture. 7EqCD as an homogeneous E2–CD conjugate was preferred for further study. Labeling with rhodamine allowed localization of 7EqCD in the cytoplasm, but not the cell nucleus, suggesting that this E2–CD conjugate would provide a selective extranuclear estrogen comparable to E2–dendrimer conjugates. Furthermore, at concentrations giving negligible classical ER activity (1–10 nM), 7EqCD was shown to deliver activity in cell culture equivalent to E2, via a mechanism attributed in the literature to extranuclear ER. Thus, 7EqCD bears comparison to E2– dendrimer conjugates as a biological probe to explore extranuclear ER mechanisms; 7EqCD is the first homogeneous probe of this type.
4. Experimental
4.1. Synthesis of compounds
4.1.1. Materials and general methods
All chemicals and solvents were obtained from Sigma–Aldrich and Fisher unless otherwise stated and purified prior to use according to known procedures when necessary: 17β-ethynylestradiol (Steraloids, Newport, RI), NHS–rhodamine (PIERCE, Rockford, IL), and all other reagents were used as received. DMF and Et3N were distilled from CaH2. β-Cyclodextrin and all CD derivatives were dried under vacuum in a drying pistol over P2O5 with refluxing acetone prior to use. Moisture sensitive reactions were performed under nitrogen using oven dried glassware. Analytical thin layer chromatography (TLC) was carried out on 60 Å silica gel-flexible baked plates (Whatman), and compounds were visualized by UV or by staining with I2, KMnO4, or phosphomolybdic acid solutions. Flash column chromatography was performed using silica gel (32–63 μm, Selecto Scientific).
4.1.2. Instrumentation
NMR spectra were recorded on Bruker Ultrashield (400 MHz) or Advance (300 MHz) spectrometers using CDCl3, DMSO-d6, or CD3CN at 300 K. Variable temperature 1H NMR at 323 K was performed on 7EqCD in order to improve chemical splitting resolution. Chemical shifts of 1H NMR spectra were reported using TMS (0 ppm) as a reference, and 13C NMR spectra were referenced to internal deuterated NMR solvents. All 13C NMR spectra of CD derivatives were obtained from overnight runs at optimized condition (D1; 3–5 s, TD; 32 K, SI; 265 K). Analytical HPLC (C8 column, 5 μm, 4.6 × 150 mm, Agilent Technologies) was carried out with Perkin–Elmer (Fremont, CA) equipment, and semi-preparative reversed- phase C8 column (5 μm, 10 × 250 mm, VYDAC) was used for purification on the same equipment. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra were obtained on a Voyager (Applied Biosystems Co. Ltd) using DHB as a matrix. Mass spectra for monomer precursors were recorded on an Agilent 1100 series LC/MSD ion trap instrument, using APCI as ionization method.
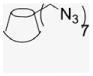
4.1.3. Per-6-azido-6-deoxy-β-cyclodextrin (2)50
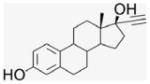
Per-6-iodo-β-cyclodextrin (1; 4.5 g, 2.36 mmol) obtained by literature procedures was dissolved in DMF (51 mL), and sodium azide (1.32 g, 20.3 mmol) was added. The suspension was stirred under N2 atmosphere at 70 °C for 20 h. The resulting yellow solution was concentrated under reduced pressure to half volume, and was poured into 90% EtOH (280 mL) to precipitate the final product, a fine white powder that was filtered and was washed with H2O and dried in a P2O5 drying pistol for one day (2.8 g, 90% yield). 1H NMR (300 MHz, DMSO-d6) δppm 5.91 (br d, J = 6.7 Hz, 7H), 5.76 (s, 7H), 4.92 (s, 7H), 3. 80–3.70 (m, 14H), 3.61–3.56 (m, 14H), 3.40–3.33 (m, 14H). 13C NMR (75 MHz, DMSO-d6) δppm 102.05, 83.20, 72.60, 72.00, 70.33, 51.33. MALDI-TOF: calcd C42H63O28N21 [M]+ 1310.08, found [M+Na]+ 1333.91.
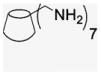
4.1.4. Per-6-amino-6-deoxy-β-cyclodextrin (3)
Per-6-azido-β-cyclodextrin 2 (1.53 g, 1.17 mmol) was dissolved in DMF (30.8 mL), and Ph3P (4.85 g, 18.5 mmol) was added. The evolution of N2 was observed by the formation of bubbles in the reaction vessel. After 1 h, the evolution of N2 ceased, 35% aqueous NH3 solution (6.0 mL) was added dropwise into the solution. After the addition of aqueous NH3 was completed, the reaction mixture turned to a white suspension that was stirred at room temperature for 18 h. The resulting mixture was condensed under vacuum at 60 °C to one fourth of volume, and then EtOH (77 mL) was added to precipitate the product. The white powder was washed with EtOH (77 mL, three times) and dried for one day (1 g, 82% yield). For NMR spectroscopy, the free amino cyclodextrin was converted to amine salt by addition of diluted HCl until the pH had reached 6. 1H NMR (300 MHz, DMSO-d6) δppm 5.21 (d, J = 2.4 Hz, 7H), 4.25 (ddd, 7H), 4.03 (t, J = 2.7 Hz, 7H), 3. 72 (dd, J = 2.4, 7.5 Hz, 7H), 3.63 (t, J = 6.9 Hz, 7H), 3.49 (dd, J = 2.1, 9.9 Hz, 7H), 3.33–3.28 (dd, J = 5.7 Hz, 7H), 13C NMR (75 MHz, DMSO-d6) δppm 100.94, 81.69, 71.67, 71.15, 67.29, 39.72. MALDI-TOF: calcd C42H77N7O28 [M]+ 1128.10, found [M+Na]+ 1151.96.
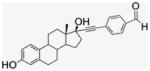
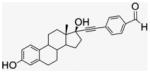
4.1.5. 17α-[(4-Formylphenyl)]ethynyl-estra-1,3,5-(10)-triene- 3,17β-diol, EE2–Ph–CHO (4)
Compound 4 was synthesized by literature procedures.25 After silica gel column chromatography (EtOAc/Hx, 1:2) and vacuum drying, a fine beige powder was obtained (945 mg, 70% yield). 1H NMR (300 MHz, DMSO-d6) δppm 10.00 (s, 1H), 8.99 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.1, 7.2 Hz, 2H), 6.51 (dd, J = 8.4, 2.4 Hz, 1H), 6.43 (d, J = 2.4 Hz, 1H), 5.59 (s, 1H), 2.70–1.23 (m, 14H), 0.82 (s, 3H). 13C NMR (75 MHz, CDCl3) δppm 192.5, 154.9, 137.2, 135.2, 131.9, 130.2, 129.7, 128.9, 126.1, 114.9, 112.8, 99.2, 83.6, 78.8, 49.5, 47.4, 43.4, 33.0, 29.2, 27.0, 26.2, 22.6, 12.9. APCI-MS: calcd for C27H28O3 400.52, found [M−H]− 399.70.
4.1.6. 17α-[(4-Hydroxymethylphenyl)]ethynyl-estra-1,3,5-(10)-triene-3,17β-diol (5)
EE2–Ph–CHO 4 (470 mg, 1.17 mmol) was dissolved in MeOH (4 mL) and DMF (1.3 mL). Under N2 atmosphere NaBH4 (110 mg, 2.91 mmol) was quickly added. The dark brown reaction mixture was stirred at room temp for 3 h. After removal of solvent, the residue was purified by silica gel column chromatography (EtOAc/Hx, 2:1) giving a white solid (423 mg, 90% yield). 1H NMR (400 MHz, DMSO-d6) δppm 9.00 (s, 1H), 7.37 (d, J = 8 Hz, 2H), 7.30 (s, J = 7.6 Hz 2H), 7.07 (d, J = 8.4 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 6.44 (s, 1H), 5.44 (s, 1H), 5.25 (s, 1H), 4.50 (s, 2H), 2.71 (s, 2H), 2.32 (d, J = 11.6 Hz, 1H), 2.24–1.69 (m, 9H), 1.37–1.19 (m, 5H), 0.86 (m, 1H), 0.81 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δppm 155.0, 142.7, 137.2, 131.0, 130.3, 126.5, 126.1, 121.1, 115.0, 112.7, 94.5, 84.1, 78.6, 62.5, 49.4, 47.2, 43.4, 39.0, 33.0, 29.3, 27.1, 26.3, 22.7, 13.0. APCI-MS: calcd for C27H30O3 402.53, found [M−H]− 401.50.
4.1.7. Per-6-(estradiol-17-ethynylbenzylamino)-6-deoxy-β-cyclodextrin (6)
Per-6-amino-β-cyclodextrin (50 mg, 0.044 mmol) and EE2–Ph–CHO 4 (176 mg, 3.08 mmol) were dissolved in DMF (1 mL), and the reaction mixture was refluxed at room temperature or 65 °C for 4–10 d under N2 atmosphere. The resulting brown imine suspension was reduced in situ by freshly prepared 0.1 M NaBH4 solution (2.1 mL) at 0 °C and allowed to stir for 2 h. The warmed reaction mixture was poured into acetone (25 mL) to precipitate the desired product. The beige solid was repeatedly washed and filtered until no corresponding alcohol 5 from EE2–Ph–CHO reduction was detected on TLC. The resulted precipitates were collected and dried yielding a beige powder (51 mg, 30% yield). MALDI-TOF: m/z calcd for C231H273N7O42 3818.9, C204H245N7O40 3434.7, C177H217N7O38 3050.5; found [M+Na]+ 3843.46, 3458.81, 3073.22.
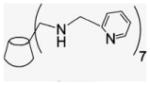
4.1.8. Per-6-(pyridine-2-ylmethylamino)-6-deoxy-β-cyclodextrin (7)
2-Pyridinecarboxaldehyde (33.5 μL, 0.35 mmol) was syringed into a suspension of 3 (50 mg, 0.044 mmol) in DMF (100 μL). The yellow reaction solution was stirred at room temperature for 5 hr. The resulting imine was reduced in situ by freshly prepared 0.1 M NaBH4 solution (2.58 mL) at 0 °C and allowed to stir for 2 h. The warmed reaction mixture was poured into Et2O (50 mL) to precipitate product. After washing and filtration, the white precipitate was collected (60 mg, 77% yield). For NMR assay, the HBr salt was prepared after acidification with 5% HBr to bring pH to 4.0. After water was evaporated, the beige powder was taken for NMR spectroscopy. 13C NMR (75 MHz, HBr salt in D2O) δppm 148.1, 138.5, 124.3, 123.6, 100.9, 81.8, 71.7, 71.2, 67.3, 50.8, 48.4. MALDI-TOF: calcd for C84H112N14O28 1765.89, found [M+Na]+ mass 1788.31.
4.1.9. Per-6-(4-(benzyloxy)benzylamino)-6-deoxy-β-cyclodextrin (8)
4-Benzyloxybenzaldehyde (312 mg, 1.47 mmol) was added to a suspension of 3 (40 mg, 0.035 mmol) in DMF (580 μL) and MeOH (1.17 mL). The reaction solution was stirred at room temperature for seven days. The resulting imine was reduced in situ with freshly prepared 0.1 M NaBH4 solution (2.1 mL) stirring at 0 °C for 2 h. The warmed reaction mixture was poured into acetone (25 mL) to precipitate the product. After repetitive washing followed by filtration, a white precipitate was collected and dried (71 mg, 81% yield). MALDI-TOF: calcd for C140H161N7O35 2501.84, found [M+Na]+ 2523.94.
4.1.10. 4-(4-Iodophenyl) butanal (9)
Compound 9 was prepared by adaptation of literature procedures. 51,52 To a mixture of 1,4-diiodobenzene (5 g, 0.015 mol), palladium(II) acetate (68 mg, 0.304 mmol), sodium hydrogen carbonate (3.2 g, 0.038 mol), and tetra-N-butyl ammonium chloride (4.2 g, 0.015 mol) purged with nitrogen gas was added DMF until dissolution. 3-Butene-1-ol (2 mL, 0.023 mol) was added via syringe, and the mixture stirred for 2 d at 40 °C. During the reaction, the mixture turned from orange to dark brown in color. After cooling acetone (20 mL) was added, and the resulting precipitates were removed by filtration. This procedure was repeated until no more precipitate was produced, and the filtrates then concentrated. Purification by silica gel chromatography (EtOAc/Hx, 1:7) afforded a light-yellow oil (1.08 g, yield 26%; 80:20 to branched aldehyde). 1H NMR (300 MHz, CDCl3) δppm 9.72 (s, 1H), 7.57 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 8.1 Hz, 2H), 2.56 (t, J = 7.8, 7.2 Hz, 2H), 2.41 (td, J = 7.2, 1.2 Hz, 2H), 1.89 (p, J = 7.2, 7.5 Hz, 2H). 13C NMR (75 MHz, CDCl3) δppm 202.0, 141.0, 137.5, 130.6, 101.78, 81.24, 72.43, 69.74, 68.93, 59.61, 21.23.
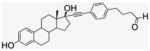
4.1.11. 17α-[(4-Butanalphenyl)]ethynyl-estra-1,3,5 (10)-triene-3,17β-diol (10)
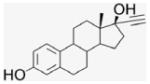
Compound 10 was prepared by literature procedures25 as a light-yellow powder (900 mg, 60% yield). 1H NMR (400 MHz, CD3CN) δppm 9.75 (s, 1H), 7.45 (d, J = 8 Hz, 2H), 7.25 (d, J = 8 Hz, 2H), 7.20 (d, J = 8 Hz, 2H), 6.91 (s, 1H), 6.69 (d, J = 8 Hz, 1H), 6.62 (s, 1H), 3.64 (s, 1H), 2.85 (m, 2H), 2.69 (t, J = 8 Hz, 2H), 2.48–2.46 (m, 4H), 1.96–1.86 (m, 6H), 1.49–1.34 (m, 4H), 0.98 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δppm 203.2, 154.9, 141.8, 137.1, 131.2, 130.3, 128.6, 126.1, 120.5, 114.9, 112.7, 94.5, 84.0, 78.6, 49.4, 47.3, 43.4, 42.3, 40.4, 39.0, 34.2, 32.9, 29.2, 27.0, 26.3, 23.2, 12.9. APCI-MS: calcd C30H32O3 442.25, found [M−H]− 442.10.
4.1.12. Per-6-(4-iodobenzylamino)-6-deoxy-β-cyclodextrin (11)
ACD 3 (50 mg, 0.044 mmol) and 4-iodobenzaldehyde (85.1 mg, 0.352 mmol) were dissolved in DMF (0.1 mL), and the reaction mixture was stirred at 60–70 °C for 18 h under N2 atmosphere. The resulted brown imine solution was reduced in situ by freshly prepared 0.1 M NaBH4 solution (2.1 mL) at 0 °C and allowed to stir for 2 h. The warmed reaction mixture was poured into acetone (25 mL) to precipitate the product as beige powder that was repeatedly washed and filtered until no corresponding alcohol from 4-iodobenzaldehyde was detected by TLC. The precipitates were collected, and overnight drying gave a light-yellow powder that by MALDI-TOF analysis gave signals corresponding to a mixture of 1–7-fold substitution.
4.1.13. 17α-(4-Aminophenyl)ethynyl-estra-1,3,5 (10)-triene-3,17β-diol (12)53–55
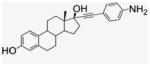
A solution of Pd(OAc)2 (80.7 mg, 0.169 mmol) and P(Ph)3 (90.4 g, 0.338 mmol) in TEA (20 mL) was stirred under N2 for 10 min. CuI (65.7 mg, 0.338 mmol) and 4-iodoaniline (748 mg, 3.376 mmol) were added, and after 5 min, 17β-ethynylestradiol (1 g, 3.376 mmol) was transferred. The dark brown mixture was stirred at room temperature for 5 h. TEA was evaporated, and the residue was purified by silica gel column chromatography (CH2Cl2/MeOH, 20:1). After drying under vacuum, yellow powder was obtained (1.10 g, 84% yield). 1H NMR (400 MHz, CD3CN) δppm 8.98 (s, 1H), 7.06 (s, 2H), 6.50 (d, J = 7.2 Hz, 2H), 6.43 (s, 1H), 5.39 (s, 2H), 5.25 (s, 1H), 3.33 (s, 1H), 2.70 (s, 2H), 2.31 (d, J = 9.6 Hz, 1H), 2.15–1.68 (m, 9H), 1.31 (m, 4H), 0.79 (s, 3H). 13C NMR (100 MHz, CD3CN) δppm 154.9, 148.8, 137.1, 132.3, 130.3, 126.1, 114.9, 113.6, 112.7, 109.1, 91.4, 85.2, 78.6, 49.2, 47.1, 43.4, 32.9, 29.2, 27.0, 26.3, 22.6, 12.9. APCI-MS: calcd For C26H29NO2 387.2, found [M−H]− 386.6.
4.1.14. (2-Phthalimidoethyl) isothiouroniumhydrobromide (13)
Compound 13 was synthesized according to literature procedures. 54,55 as a white solid (5.1, 79% yield). 1H NMR (400 MHz, DMSO-d6) δppm 9.14 (s, 4H), 7.91 (m, 4H), 3.39 (t, J = 5.2 Hz, 6.4 Hz, 2H), 3.55 (t, J = 5.2 Hz, 6.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δppm 169.2, 167.6, 134.7, 131.4, 123.2, 36.0, 29.3. APCI-MS: calcd for C11H12BrN3O2S 328.98, found [M−HBr]+ 249.80.
4.1.15. Per-6-(phthalimidoethylthio)-6-deoxy-β-cyclodextrin (14)
Compound 14 was synthesized according to literature procedures27,56 as a white solid (112 mg, 17% yield). 1H NMR (400 MHz, DMSO-d6) δppm 7.71 (s, 4H), 5.84 (d, J = 23 Hz, 2 Hz, 2H), 4.86 (s, 1H), 3. 94 (br s, 1H), 3.81 (m, 2H), 3.59 (m, 1H), 3.42–3.33 (m, overlaps with DOH), 3.04 (d, J = 11.6 Hz, 1H), 2.91–2.78 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δppm 167.4, 134.2, 131.3, 122.9, 102.1, 84.2, 72.5, 72.2, 71.2, 36.9, 33.0, 30.8. MALDI-TOF: calcd for C112H119N7O42S7 2459.66, found [M+Na]+ 2482.62.
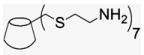
4.1.16. Per-6-(2-aminoethylthio)-6-deoxy-β-cyclodextrin (15)
Compound 15 was prepared by literature procedures27 as a white powder (50 mg, 81% yield). 1H NMR (400 MHz, DMSO-d6) δppm 5.45 (d, J = 24, 4 Hz, 2H), 4.51 (br s, 1H), 3.40 (br s, 1H), 3. 19 (t, J = 8.4 Hz, 8.8 Hz, 1H), 3.02–2.48 (m, overlaps with DOH), 2.09 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δppm 102.1, 84.5, 72.6, 72.2, 71.4, 38.6, 32.8, 29.7. MALDI-TOF: calcd for C56H105N7O28S7 1548.94, found [M]+ 1548.90.
4.1.17. Per-6-(estradiol-17-(4-((2-(propylthio)ethylamino)-methyl)phenyl)ethynyl)-6-deoxy-β-cyclodextrin (16)
Compounds 15 (40 mg, 0.0258 mmol) and 4 (310 mg, 0.774 mmol) were dissolved in DMF, and stirred at 60–70 °C for 10 days. 2 mL of aqueous NaOH (1 N in MeOH) and stirred for a further 2.5 h. After cooling and concentration, the residue was precipitated in acetone (25 mL) giving a beige powder that was repeatedly washed and filtered until no trace of 4 was detected by TLC. The precipitates were dried to give a light-yellow solid product containing 16 as the major product in a heterogeneous mixture. MALDI-TOF: calcd for C245H301N7O42S7 4240.49, found [M+H2O+H]+ 4259.84.
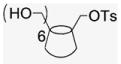
4.1.18. Mono-6-(p-tolylsulfonyl)-6-deoxy-β-cyclodextrin (17)
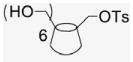
Compound 17 was prepared by adaptation of literature procedures57 as a white powder was obtained (1 g, 10% yield). 1H NMR (300 MHz, DMSO-d6) δppm 7.76 (d, J = 7.6 Hz, 2H), 7.44 (d, J = 7.2 Hz, 2H), 5.71 (br s, 14H), 4.84 (s, 4H), 4.77 (s, 3H), 4.33 (d, J = 10.4 Hz, 3H), 4.20 (m, 3H), 3.66–3.51 (m, overlaps with HOD), 3.38–3.22 (m, overlaps with HOD), 2.09 (s, 3H), 13C NMR (100 MHz, DMSO-d6) δppm 144.9, 132.7, 129.9, 127.6, 102.0 (m), 81.5 (m), 73.1–72.8, 60.0, 21.3. MALDI-TOF: calcd C49H76O37S [M]+ 1289.19, found [M+Na]+ 1313.43.
4.1.19. Mono-6-azido-6-deoxy-β-cyclodextrin (18)57
Compound 17 (350 mg, 0.271 mmol) and NaN3 (189 mg, 2.90 mmol) were suspended in H2O (3.53 mL), which was heated and to a clear solution with stirring at 80 °C for 15 h. The reaction mixture was cooled, poured into acetone (60 mL) to precipitate the product as a white powder that was dried in a drying pistol at 56 °C under vacuum. A pure white solid was obtained (492.8 mg, 90% yield). 1H NMR (400 MHz, DMSO-d6) δppm 5.73 (br s, 14 H), 4.84 (m, 7 H), 3.75–3.55 (m, 28 H), 3.41–3.28 (m, overlaps with HOD), 13C NMR (100 MHz, DMSO-d6) δppm 102.0 (m), 83.0, 81.6 (m), 73.0 (m), 72.4, 72.2, 72.1, 70.2, 60.2 (m), 51.1. MALDI-TOF: calcd C42H69N3O34 [M]+ 1160.00, found [M+Na]+ 1183.74.
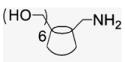
4.1.20. Mono-6-amino-6-deoxy-β-cyclodextrin hydrochloride (19)57
Compound 18 (300 mg, 0.259 mmol) and PPh3 (116 mg, 0.442 mmol) were dissolved in DMF (5.6 mL) to which 30% NH4OH (2.7 mL) was added by syringe. The reaction mixture was stirred at room temperature for 18 h, and poured into acetone (50 mL) yielding after successive washes and drying a white powder (200.0 mg, 68% yield). 1H NMR (400 MHz, DMSO-d6) δppm 5.15 (br s, 7 H), 4.06–4.00 (m, 21 H), 3.73–3.63 (m, 10 H). 13C NMR (100 MHz, DMSO-d6) δppm 101.8, 101.5, 82.8, 81.0, 80.8, 73.1, 73.0, 72.1, 71.8, 71.7, 60.2, 41.2. MALDI-TOF: calcd C42H71NO34 [M]+ 1134.01, found [M+Na]+ 1157.57.
4.1.21. Mono-6-(17-estradiolphenylmethylamino)-6-deoxy-β-cyclodextrin (20)
Compounds 19 (50 mg, 0.044 mmol) and 4 (370.6 mg, 0.926 mmol) were dissolved in DMF (1 mL), and the reaction mixture was refluxed at 65 °C for 3 d under N2. The resulting brown imine suspension was reduced in situ by the addition of a freshly prepared 0.1 M NaBH4 solution (3 mL) at 0 °C with stirring for 2 h. The warmed reaction mixture was precipitated into acetone (25 mL) and the precipitate repeatedly washed and filtered until no presence of 5 was detected by TLC. After drying a white powder was obtained (30 mg, 45% yield). MALDI-TOF: calcd for C69H99NO36 1518.53, found [M+Na]+ 1541.97.
4.1.22. Mono-6-(estradiol-17-ethynylbenzylamino)-6-deoxy-β-cyclodextrin (21)
Compounds 17 (100 mg, 0.078 mmol) and 12 (630 mg, 1.63 mmol) were dissolved in DMF (0.5 mL), and the reaction mixture was refluxed at 60–70 °C for 7 d under N2. The solvent was evaporated under vacuum, and the dark brown residue was precipitated in acetone (50 mL). The beige powder was repeatedly washed and filtered until no trace of compound 12 was detected by TLC. The precipitates were collected, and overnight drying gave a light-yellow solid (51 mg, 43%). MALDI-TOF: calcd for C69H99NO36 1518.53, found [M+Na]+ 1541.60.
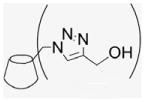
4.1.23. Mono-6-(estradiol-17-(1H-1,2,3-triazol-4-yl))-6-deoxy-β-cyclodextrin (22)
Compound 17 (100 mg, 0.078 mmol), 17β-ethynylestradiol (23.1 mg, 0.078 mmol), and sodium azide (6.6 mg, 0.10 mmol) were dissolved in DMF and H2O (3:1, 0.9 mL). A solution of CuSO4–5H2O (400 mg, 1.60 mmol) and sodium ascorbate (360 mg, 1.82 mmol) was prepared separately in the same DMF/H2O solvent system (2 mL) and added via syringe. The brown suspension was stirred at 60–70 °C for four days. The solvent was evaporated, and the residue was precipitated in H2O (50 mL). This washing process was repeated three times for complete removal of residual copper ions. After centrifugation at 4 °C (4000 rpm, 20 min), a fine beige powder was obtained (60 mg, 53%). MALDI-TOF: calcd for C62H93N3O36 1456.42, found [M+Na]+ 1479.44.
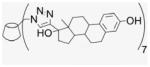
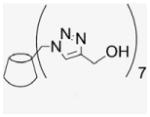
4.1.24. Per-6-(estradiol-17-(1H-1,2,3-triazol-4-yl))-6-deoxy-β-cyclodextrin (23)
Compound 2 (100 mg, 0.076 mmol) and EE2 (157.6 mg, 0.532 mmol) were dissolved in 50% of MeOH and DMF (1.9 mL). CuI (72.4 mg, 0.38 mmol) was added, and DIEA (66.2 μL) added by syringe to the reaction mixture. The green suspension was stirred at room temperature for 1 d until there was no EE2 detected by TLC. The solvent was evaporated under vacuum, and the residue was dissolved with 6 mL of aqueous NH4OH (30%). The deep blue solution was precipitated into cold acetone (50 mL). This washing process was repeated three times for removal of residual copper. After addition of H2O and centrifugation at 4 °C three times (4000 rpm, 20 min), a fine beige powder was obtained (150 mg, 58%). 1H NMR (300 MHz, DMSO-d6, 323 K) δppm 8.61 (br s, 1H), 7.78 (s, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.46 (s, 1H), 5.61 (s, 3H), 5.12 (s, 1H), 4.66 (s, 2H), 4.60 (s, 2H), 4.14 (m, 1 H), 3.74 (t, J = 8.7 Hz, 2H), 3.27–3.15 (m, overlaps with DOH), 2.653 (s, 3H), 2.20–1.26 (18H), 0.903 (s, 3H), 0.69 (m, 1H). 13C NMR (75 MHz, DMSO-d6, 300 K) δppm 154.8, 153.6, 137.1, 130.4, 125.9, 124.4, 114.9, 112.6, 101.5, 82.0, 81.0, 72.4, 71.8, 69.5, 49.5, 47.6, 46.8, 43.1, 37.1, 32.6, 30.7, 29.2, 27.2, 26.1, 23.4, 14.3. MALDI-TOF: m/z calcd for C182H231N21O42 3383.7, found [M+H2O]+ 3402.90. HRMS: m/z calcd for C182H233N21O42 [MH2]2+ 1692.3366; found 1692.3302.
4.1.25. Mono-6-(17-estradiol-17-(1H-1,2,3-triazol-4-yl))-6-deoxy-β-cyclodextrin (24)
Mono-6-azido-β-cyclodextrin 2 (100 mg, 0.086 mmol) and 17β-ethynylestradiol (25.5 mg, 0.086 mmol) were dissolved in 50% of MeOH and DMF (2.15 mL). CuI (82.0 mg, 0.43 mmol) was added, and the reaction mixture formed brown suspension. DIEA (75.0 μL) was added via syringe into the reaction mixture. The green suspension was stirred at room temperature for one day until there was no EE2 detected on TLC. The solvent was evaporated under vacuum, and the residue was dissolved with 6 mL of aqueous NH4OH (30%). The deep blue colored solution was precipitated into cold Ac2O (50 mL). This washing process was repeated three times for the complete removal of copper ions. After centrifugation at 4 °C (4000 rpm, 20 min), fine beige powder was obtained, 1EqCD (80 mg, 64% yield). MALDI-TOF: calcd for C62H93N3O36 1456.42, found [M+Na]+ 1479.20.
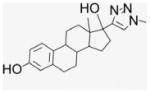
4.1.26. 17α-(17-1,2,3-Triazolmethyl)ethynyl-estra-1,3,5 (10)-triene-3,17β-diol (25)
This procedure was modified from the literature.31 Dimethylsulfate (90 mL, 0.676 mmol) was added to the solution of 17β-ethynylestradiol (100 mg, 0.338 mmol) and sodium azide (46 mg, 0.676 mmol) in t-BuOH/H2O (0.61/0.2 mL) at 0 °C. CuSO4 (4.22 mg, 5 mol%) in 200 μL of water and sodium ascorbate (6.7 mg, 10 mol%) in 200 μL of water were added stepwisely. This reaction suspension was gradually warmed up to room temperature and stirred for 1 d. It was worked up by addition of water and extracted with EtOAc. The combined organic layers were washed with brine, and the solvent was removed. The resulting mixture was evaporated under pressure, and the residue was purified by silica gel column chromatography (EtOAc/Hx, 1:5). After drying under vacuum, white powder was obtained (71.7 mg, 60% yield). 1H NMR (400 MHz, DMSO-d6) δppm 8.94 (s, 1H), 7.79 (s, 1H), 6.95 (d, J = 8 Hz, 1H), 6.46 (d, J = 8.4 Hz, 1H), 6.40 (s, 1H), 5.04 (s, 1H), 4.01 (s, 3H), 2.68 (m, 2H), 2.50 (m, 1H), 2.10 (d, J = 12 Hz, 1H), 1.88–1.70 (m, 5H), 1.42–1.26 (m, 6H), 0.95 (s, 3H), 0.65 (t, J = 12 Hz, 11.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δppm 154.8, 154.2, 137.1, 130.4, 126.0, 123.5, 114.8, 112.6, 81.1, 47.5, 46.6, 43.1, 37.1, 36.0, 32.6, 29.2, 27.2, 26.1, 23.5, 14.3.
4.1.27. Per-6-(4-hydroxymethyl-1,2,3-triazolmethyl)-6-deoxy-β-cyclodextrin (26)
Azide 2 (50 mg, 0.038 mmol) and CuI (36.0 mg, 0.19 mmol) were dissolved in 50% of MeOH and DMF (1.0 mL) to which propagyl alcohol (15.8 μL, 0.27 mmol) and DIEA (33.0 μL, 0.19 mmol) were added by syringe, and the mixture then stirred at room temperature for 1 d. The resulting green suspension was clarified using 2.0 mL of aqueous 30%NH4OH, and Chelex 100 sodium form was added to the deep blue solution to complex copper ions. The solution was allowed to stir vigorously overnight, and the yellow supernatant obtained was concentrated to give a beige powder (11 mg, 15% yield). 1H NMR (400 MHz, DMSO-d6) δppm 7.80 (s, 1H), 6.0 (br s, 4H), 5.1 (m, 6H), 4.6–4.3 (m, 11H), 4.0 (m, 3H), 3.7 (t, J = 8.8, 8.4 Hz, 4H), 2.9 (br s, 11H), 2.1 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δppm 147.6, 124.1, 101.5, 82.5, 72.5, 71.8, 69.7, 54.6, 49.3, MALDI-TOF: m/z calcd for C63H91N21O35 1701.6, found [M+Mg]+ 1726.0.
4.1.28. General rhodamine conjugation procedure
Standard synthetic procedures were used. The appropriate qCD derivative (50 mg), the appropriate rhodamine coupling reagent (1 equiv of NHS–rhodamine for 23R, 26R and rhodamine-19-perchlorate for 23R′, 26R′) and DMAP (1 equiv) were dissolved in DMF (1 mL) under N2. The reaction mixture was cooled to 0 °C and EDC–HCl salt (1 equiv) was quickly added and the solution allowed to warm, before stirring overnight at room temperature in the dark. The resulting dark red suspension was precipitated in acetone (15 mL) giving a pink powder after filtration, which was washed twice with acetone followed by centrifugation at 4 °C for 1 h (10,000 rpm). The collected solids were lyophilized overnight to give a pink solid and kept at −20 °C in the dark.
4.1.29. Rhodamine coupled 7EqCD (23R)
Yield: 35 mg, 61%. MALDI-TOF: calcd for C210H259N23O46 3841.43, found [M+Na]+ 3864.41.
4.1.30. Rhodamine coupled 7EqCD (23R′)
Yield: 36 mg, 63%. MALDI-TOF: calcd C209H259N23O43 3781.42, found [M+Na]+ 3804.41.
4.1.31. Rhodamine coupled per-6-(4-hydroxymethyl-1,2,3-triazolmethyl)-6-deoxy-β-cyclodextrin (26R)
Yield: 30 mg, 47%. MALDI-TOF: calcd for C91H119N23O38 2143.05, found [M+K]+ 2181.75.
4.1.32. Rhodamine coupled per-6-(4-hydroxymethyl-1,2,3-triazolmethyl)-6-deoxy-β-cyclodextrin (26R′)
Yield: 30 mg, 49%. MALDI-TOF: m/z calcd for C90H119N23O36 2099.04, found [M+K]+ 2138.15.
4.2. Bioassay procedures
4.2.1. Cell culture
The Ishikawa cell line was provided by Dr. R. B. Hochberg (Yale University, New Haven, CT) and the cells were grown in Dulbecco’s Modified Eagle medium (DMEM/F12) containing 1% sodium pyruvate, 1% non-essential amino acids (NEAA), 1% glutamax-1, 0.05% insulin, and 10% heat-inactivated FBS. All cultures were maintained at 37 °C with 5% CO2:95% air. For phenol red-free complete medium, estrogen-stripped FBS was used. Human breast cancer cell line T47D:A18 is a hormone-responsive clone: the stable transfectant cell lines T47D:A18/neo and T47D:A18/PKCα58 were maintained in RPMI 1640 (phenol red) supplemented with 10% fetal bovine serum-containing G418 (500 μg/mL). Before transient transfection, T47D:A18/neo and T47D:A18/PKCα cells were maintained for three days in phenol red-free, E2-depleted RPMI 1640 supplemented with G418. After stripping, cells were transiently transfected by electroporation as described previously.58
4.2.2. Measurement of ERE activation
T47D cells were transiently transfected by electroporation as described previously.58 Briefly, 8 × 106 cells were harvested and resuspended in 0.5 mL serum-free, phenol red-free RPMI 1640. ERE-tk-Luc plasmid containing the luciferase reporter gene controlled by the ERE (5 μg) and β-galactosidase (β-gal; 1 μg) expressing plasmid pCMVβ were added to the cell suspension and incubated and processed. On the following day, medium containing E2 (10−9 mol/L), 7EqCD or qE2 (10−9–10−7 mol/L), or vehicle (ethanol) was added. Luciferase activities were measured using Luciferase Reporter Gene Assay System from Applied Biosystems. β-gal signals were measured with Galacto-Light Plus assay systems (Applied Biosystems). After 18 h incubation, cells were washed with ice-cold PBS and lysed in the lysis buffer provided. The cell lysates were cleared by centrifugation and luciferase activity and β-gal signals were determined after adding corresponding substrates and read by a Monolight 2010 luminometer (Analytical Luminescence Laboratory). The results were normalized relative to pRL-TK activity, to account for transfection efficiency, by dividing the sum of the luciferase activity by the sum of the β-gal signals.
4.2.3. Measurement of induction of alkaline phosphatase
The procedure of Liu et al. was used as described previously.59 Test samples (10 μL at varying concentrations in DMSO) were added to determine EC50 values, and the cells in a total volume of 200 μL media/well were incubated at 37 °C for four days. For the determination of antiestrogenic activity, 2 × 10−8 M estradiol was added to the media. The enzyme activity was measured by reading the release of p-nitrophenyl phosphate at 405 nm every 15 s with a 10 s shake between readings for 16 readings using a Power Wave 200 microplate scanning spectrophotometer (Bio-Tek Instruments, Winooski, VT). For estrogenic determination, the percent induction as compared with the estradiol control was calculated. For antiestrogenic determination, the percent induction was calculated compared to the background induction. The sulforhodamine (SRB) assay was used as a measure of cytotoxicity in parallel with alkaline phosphatase (ALP) induction assays using ~1000 cells per well in a 96-well plate (5 × 106 cells/mL). Cells were treated with the same samples used in the ALP induction assay. The plates were read using the endpoint mode at 515 nm. Calculation of the percent cytotoxicity is essential to rule out cell death as a causal factor in reduced induction and apparent antiestrogenic activity.
4.2.4. ERα competitive binding assay
Competitive displacement of [3H] estradiol (E2) radioligand (Amersham Biosciences, Piscataway, NJ) from full-length recombinant human (h) ERα (PanVera/Invitrogen, Carlsbad, CA) was assayed as described.59 Briefly, the reaction mixture consisted of 5 μL of compound in DMSO, 5 μL of hERα (0.5 pmol) in ER binding buffer [consisting of 10 mM Tris–HCl (pH 7.5), 10% glycerol, 2 mM dithiothrietol, 1 mg/mL BSA], 5 μL of ‘Hot Mix’ [400 nM, prepared fresh using 95 Ci/mmol [3H] estradiol, diluted in 1:1 ethanol/ER binding buffer; obtained from NEN Life Science Products (Boston, MA)] and 85 μL of ER binding buffer. The incubation was carried out with test compounds at room temperature for 2 h. Radioactivity was counted using a Beckman LS 5801 liquid scintillation counter (Schamburg, IL). The IC50 was calculated from a concentration–response data obtained in triplicate experiments.
4.2.5. Fluorescence confocal microscopy
MCF-7 cells were grown (106 cells/mL) on each of eight wells on a sterile Nunc™ chambered cover glass and incubated for 48 h at 37 °C with 5% CO2 in phenol-free medium supplemented with 10% stripped fetal bovine serum medium. Cells were incubated with rhodamine conjugates (100 nM) for 24 h at 37 °C, 5% CO2. Cells were rinsed twice with PBS to remove the unincorporated dye and 0.2 μg/mL Hoechst stain was added to the cells to detect nuclear staining. Imaging was performed with a Zeiss LSM 510 laser-scanning confocal microscope with the detector gain adjusted to eliminate the background autofluorescence. The fluorescence signal from rhodamine conjugate was monitored with a 544 nm argon/krypton laser and a 576 nm band pass filter. Hoechst nuclear staining signal was monitored with a 345 nm UV laser and 420 nm band pass filter. A × 63 (1.2 numerical aperture) water immersion objective was used for all experiments. Images were analyzed using the analysis tool provided in the Zeiss biophysical software package.
Acknowledgments
This research was supported by the National Institutes of Health (CA 102590) and DOD contract W81XWH-07-1-0445. Dr. Zhiqiang Wang and Dr. Kuanqiang Gao are acknowledged for technical assistance.
References and notes
- 1.Khan AR, Forgo P, Stine KJ, D’Souza VT. Chem Rev. 1998;98:1977. doi: 10.1021/cr970012b. [DOI] [PubMed] [Google Scholar]
- 2.Dodziuk H. Cyclodextrins and Their Complexes: Chemistry, Analytical Methods, Applications. Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 3.Szejtli J. Chem Rev. 1998;98:1743. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- 4.Schaschke N, Assfalg-Machleidt I, Machleidt W, Lassleben T, Sommerhoff CP, Moroder L. Bioorg Med Chem Lett. 2000;10:677. doi: 10.1016/s0960-894x(00)00078-0. [DOI] [PubMed] [Google Scholar]
- 5.Duplex Yockot VM. Org Biomol Chem. 2003;1:1810. doi: 10.1039/b301670f. [DOI] [PubMed] [Google Scholar]
- 6.Schaschke N, Fiori S, Weyher E, Escrieut C, Fourmy D, Muller G, Moroder L. J Am Chem Soc. 1998;120:7030. [Google Scholar]
- 7.Attioui F, al-Omar A, Leray E, Parrot-Lopez H, Finance C, Bonaly R. Biol Cell. 1994;82:161. doi: 10.1016/S0248-4900(94)80018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Mol Endocrinol. 2006;20:491. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 9.Pedram A, Razandi M, Levin ER. Mol Endocrinol. 2006;20:1996. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- 10.Pietras RJ, Levin ER, Szego CM. Science. 2005;310:51. doi: 10.1126/science.310.5745.51. [DOI] [PubMed] [Google Scholar]
- 11.Losel RM, Falkenstein E, Feuring M, Schultz A, Tillmann HC, Rossol-Haseroth K, Wehling M. Physiol Rev. 2003;83:965. doi: 10.1152/physrev.00003.2003. [DOI] [PubMed] [Google Scholar]
- 12.Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Ronnekleiv OK, Kelly MJ. J Neurosci. 2003;23:9529. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. Endocrinology. 1999;140:5455. doi: 10.1210/endo.140.11.7247. [DOI] [PubMed] [Google Scholar]
- 14.Taguchi Y, Koslowski M, Bodenner DL. Nucl Recept. 2004;2:5. doi: 10.1186/1478-1336-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bulayeva NN, Gametchu B, Watson CS. Steroids. 2004;69:181. doi: 10.1016/j.steroids.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morales A, Diaz M, Ropero AB, Nadal A, Alonso R. Eur J Neurosci. 2003;18:2505. doi: 10.1046/j.1460-9568.2003.02997.x. [DOI] [PubMed] [Google Scholar]
- 17.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 18.Vizitiu D, Thatcher GRJ. J Org Chem. 1999;64:6235. [Google Scholar]
- 19.Borrajo AMP, Gorin BI, Dostaler SM, Riopelle RJ, Thatcher GRJ. Bioorg Med Chem Lett. 1997;7:1185. [Google Scholar]
- 20.Wang Z, Chang L, Klein WL, Thatcher GRJ, Venton DL. J Med Chem. 2004;47:3329. doi: 10.1021/jm034224e. [DOI] [PubMed] [Google Scholar]
- 21.Vizitiu D, Walkinshaw CS, Thatcher GRJ. J Org Chem. 1997;62:8760. [Google Scholar]
- 22.Gorin BD, Riopelle RJ, Thatcher GRJ. Tetrahedron Lett. 1996:4647. [Google Scholar]
- 23.Anstead GM, Carlson KE, Katzenellenbogen JA. Steroids. 1997;62:268. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 24.Wust F, Kniess T. J Lab Comp Radiopharm. 2003;46:699. [Google Scholar]
- 25.Kim SH, Katzenellenbogen JA. Angew Chem, Int Ed. 2006;45:7243. doi: 10.1002/anie.200601923. [DOI] [PubMed] [Google Scholar]
- 26.Wang K. PhD Thesis. Queen’s University; 2000. [Google Scholar]
- 27.Karginov VA, Yohannes A, Robinson TM, Fahmi NE, Alibek K, Hecht SM. Bioorg Med Chem. 2006;14:33. doi: 10.1016/j.bmc.2005.07.054. [DOI] [PubMed] [Google Scholar]
- 28.Kumar R, El-Sagheer A, Tumpane J, Lincoln P, Wilhelmsson LM, Brown T. J Am Chem Soc. 2007;129:6859. doi: 10.1021/ja070273v. [DOI] [PubMed] [Google Scholar]
- 29.Speers AE, Cravatt BF. Chem Biol. 2004;11:535. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J Am Chem Soc. 2003;125:3192. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 31.Sreedhar B, Reddy PS. Synth Commun. 2007;37:805. [Google Scholar]
- 32.Feldman AK, Colasson B, Fokin VV. Org Lett. 2004;6:3897. doi: 10.1021/ol048859z. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson BL, Bornaghi LF, Poulsen SA, Houston TA. Tetrahedron. 2006;62:8115. [Google Scholar]
- 34.Odlo K, Hoydahl EA, Hansen TV. Tetrahedron Lett. 2007;48:2097. [Google Scholar]
- 35.Rolf Breinbauer MKN. Chem Bio Chem. 2003;4:1147. doi: 10.1002/cbic.200300705. [DOI] [PubMed] [Google Scholar]
- 36.Busby MB, Lim P, Vigh G. Electrophoresis. 2003;24:351. doi: 10.1002/elps.200390045. [DOI] [PubMed] [Google Scholar]
- 37.Holinka CF, Hata H, Kuramoto H, Gurpide E. Cancer Res. 1986;46:2771. [PubMed] [Google Scholar]
- 38.Chang M, Peng KW, Kastrati I, Overk CR, Qin ZH, Yao P, Bolton JL, Thatcher GRJ. Endocrinology. 2007;148:4793. doi: 10.1210/en.2006-1568. [DOI] [PubMed] [Google Scholar]
- 39.Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly ES, Jr, Nethrapalli IS, Tinnikov AA. J Neurosci. 2002;22:8391. doi: 10.1523/JNEUROSCI.22-19-08391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Figtree GA, Lu Y, Webb CM, Collins P. Circulation. 1999;100:1095. doi: 10.1161/01.cir.100.10.1095. [DOI] [PubMed] [Google Scholar]
- 41.Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, Sklar LA, Arterburn JB, Prossnitz ER. ACS Chem Biol. 2007;2:536. doi: 10.1021/cb700072n. [DOI] [PubMed] [Google Scholar]
- 42.Levin ER. Mol Endocrinol. 2005;19:1951. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Mol Endocrinol. 2008;22:2116. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Zhao H, Asztalos S, Chisamore M, Sitabkhan Y, Tonetti DA. Mol Cancer Res. 2009;7:498. doi: 10.1158/1541-7786.MCR-08-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodriguez-Tenreiro C, Alvarez-Lorenzo C, Rodriguez-Perez A, Concheiro A, Torres-Labandeira JJ. Eur J Pharm Biopharm. 2007;66:55. doi: 10.1016/j.ejpb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 46.Shakalisava Y, Regan F. Electrophoresis. 2006;27:3048. doi: 10.1002/elps.200500842. [DOI] [PubMed] [Google Scholar]
- 47.Davis ME, Brewster ME. Nat Rev Drug Disc. 2004;3:1023. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- 48.Loftsson T, Duchene D. Int J Pharm. 2007;329:1. doi: 10.1016/j.ijpharm.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 49.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 50.Ashton PR, Koeniger R, Stoddart JF, Alker D, Harding VD. J Org Chem. 1996;61:903. [Google Scholar]
- 51.Larock RC, Leung WY, Stolz-Dunn S. Tetrahedron Lett. 1989;30:6629. [Google Scholar]
- 52.Gibson SE, Jones JO, Kalindjian SB, Knight JD, Mainol N, Rudd M, Steed JW, Tozer MJ, Wright PT. Tetrahedron. 2004;60:6945. [Google Scholar]
- 53.Arterburn JB, Rao KV, Perry MC. Tetrahedron Lett. 2000;41:839. [Google Scholar]
- 54.Gabano E, Cassino C, Bonetti S, Prandi C, Colangelo D, Ghiglia A, Osella D. Org Biomol Chem. 2005;3:3531. doi: 10.1039/b507716h. [DOI] [PubMed] [Google Scholar]
- 55.Osella D, Nervi C, Milone L, Galeotti F, Vessieres A, Jaouen G. J Organomet Chem. 2000;593–594:232. [Google Scholar]
- 56.Karginov VA, Nestorovich EM, Yohannes A, Robinson TM, Fahmi NE, Schmidtmann F, Hecht SM, Bezrukov SM. Antimicrob Agents Chemother. 2006;50:3740. doi: 10.1128/AAC.00693-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petter RC, Salek JS, Sikorski CT, Kumaravel G, Lin FT. J Am Chem Soc. 1990;112:3860. [Google Scholar]
- 58.Tonetti DA, Chisamore MJ, Grdina W, Schurz H, Jordan VC. Br J Cancer. 2000;83:782. doi: 10.1054/bjoc.2000.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J, Burdette JE, Xu H, Gu C, van Breemen RB, Bhat KP, Booth N, Constantinou AI, Pezzuto JM, Fong HH, Farnsworth NR, Bolton JL. J Agric Food Chem. 2001;49:2472. doi: 10.1021/jf0014157. [DOI] [PubMed] [Google Scholar]