

Abstract
Hexamethoxy-calix[6]arene has been fully functionalized with p-phosphonic acid groups on the upper rim in 57% yield over three steps, and has been authenticated in the solid state by X-ray diffraction as either a nitrate salt or one of two calcium complexes. The latter differ by the ratio of calcium ions per calixarene, either 3:1 or 4:1. In both structures the coordination sphere of the calcium ions is made up of oxygen atoms from the phosphonic acid groups and from water of crystallization, as part of extended polymeric layers in the extended 3D packing. Hirshfeld surface analysis shows extensive O…H and O…Ca interactions for the phosphonic acid moieties in both calcium structures. MALDI-TOF MS of the hexaphosphonic acid shows nano-arrays consisting of up to a maximum of 28 calixarene units.
Keywords: Calixarenes, self-assembly, phosphonic acids, supramolecular chemistry, mass-spectrometry, nanostructures
Introduction
Calixarenes are a class of macrocyclic compounds that have been utilized in a wide variety of roles, including sensors,1 gas absorption2 and the encapsulation of guest molecules, be it organic molecules such as fullerenes3 or ions such as the lanthanides.4 The main reason for the wide variety of uses for calixarenes is due in part to their ease of functionalization, and the attachment of acidic groups at the upper rim is an important strategy in self assembly studies, for example using sulfonic acid/sulfonate groups which affords a remarkable diverse range of structures.5 Attachment of alkyl chains to the lower rim of calixarenes allows the formation of bi-layers of differing thicknesses,6 which in combination with polar head groups imparts lipophilic character onto the calixarenes. Another reason for the wide use of calixarenes is their relative ease of preparation, with calix[4, 6 and 8]arene accessible in large quantities in high yields.7
Calixarenes bearing phosphonic acid/phosphonate groups in the para-position at the upper rim are relatively new, further highlighting the diversity of calixarenes in general, and are available in good yield, 62-68% overall for calix[4, 5, 6 and 8]arene involving robust five step procedures.8 These calixarenes are water soluble, and form bi-layers in the solid state, held together by strong hydrogen bonding between the diprotic phosphonic acid groups, and in solution and the gas phase they can form nano-rafts.8 O-alkylation at the lower rim while maintaining the integrity of the phosphonic acid/phosphate groups has also been reported, for methyl, n-butyl and n-octadecyl chains.9 The methyl substituted phosphonic acid calix[4]arene binds Cs+ ions in a 1,3-alternate conformation, through Cs…O and Cs…π interactions.
Calcium(II) ions play an important role in biological functions, with calcium channels thought to be involved in Alzheimer's disease,10 and certain cancers that increase the amount of calcium in the body through hypercalcemia.11 p-phosphonic acid calix[4]arene, devoid of O-alkyl groups, has been shown to bind Ca2+ ions in the solid state,12 and herein we show that phosphonic acid calix[6]arene bearing O-methyl groups at the lower rim binds Ca2+ ions, crystallizing in the classical “double partial cone” conformation for calix[6]arenes in general.
Hirshfeld surface analysis is a technique that is finding a plethora of uses in analysing intermolecular contacts and interactions of larger molecules,13 including for example, both alkyl esters of phosphonated calixarenes, 6a and phosphonic acid analogues.12 The parameter dnorm is of particular interest in undertaking Hirshfeld surface analyses, as it can be easily discerned, using the red-white-blue color scheme, which contacts are shorter or longer than the sum of the van der Waals radii of the atoms, and thus identify close contacts in the structure.14 The fingerprint plots are also very useful, as they can be used to identify specific contacts, such as CH…π contacts, which have characteristic shapes and positions on the plots.14
Results and Discussion
The synthesis of p-phosphonic acid hexamethoxy-calix[6]arene, 5, was accomplished in 4 steps starting from calix[6]arene 1, Scheme 1. Alkylation of 1 using MeI and NaH in dimethylformamide gave methoxycalix[6]arene, 2, which was subsequently iodinated using CF3CO2Ag and I2 in chloroform affording 3 in 66% yield. The iodinated precursor, 3, was phosphorylated using P(OEt)3 and NiCl2 in benzonitrile to give the phosphonate ester 4 in 92% yield. De-esterification of 4 using Me3SiBr in acetronitrile gave the target phosphonic acid 5 in 95% yield.
Scheme 1.
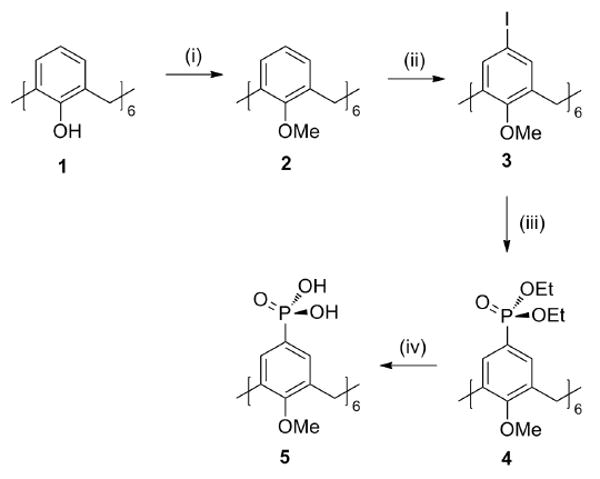
Synthetic procedure for p-phosphonic acid hexamethoxy-calix[6]arene. (i) MeI / NaH/ DMF (ii) CF3CO2Ag / I2/ CHCl3 (iii) P(OEt)3 / NiCl2 / PhCN (iv) BTMS / MeCN. Me = methyl, Et = ethyl, Ph = phenyl, BTMS = bromotrimethylsilane, DMF = dimethylformamide.
Single crystal X-ray diffraction was used to authenticate calixarenes 4 and 5 as complexes 4a and 5a-c respectively. The crystalline complex 4a was prepared by slow evaporation of a saturated solution of 4 in toluene over 5 days to yield complex 4a as 4.H2O. Complex 5a was prepared by slow evaporation of a saturated solution of 5 in MeOH / 6M HNO3 / CsNO3 over 7 days to yield complex 5a as 5.4HNO3.6H2O. Slow evaporation of a solution of 5 with three or four equivalents of calcium acetate affords complex 5b as 56-.3Ca2+.28H2O and complex 5c as 58-.4Ca2+.26½H2O respectively. Only complexes 5b and 5c will be described in detail herein.
Complex 5b crystallizes in the space group P-1 (Z=2) with the asymmetric unit comprising two separate halves of two calixarene molecules located at two different inversion centers (0, 0, ½ and ½, ½, 0), 3 calcium atoms and 28 crystalline water molecules, Figure 1. According to our conventional atomic numbering scheme the aromatic rings 1-3 belong to calixarene A, and aromatic rings 4-6 belong to calixarene B. The coordination sphere of two of the calcium atoms (Ca1 and Ca3) consist of four crystalline water molecules and three (directly or symmetry connected) oxygen atoms from the p-phosphonic acid moieties of calixarene A, Ca-O distances 2.310(2) – 2.534(2) Å, and calixarene B, Ca-O distances 2.258(2) – 2.557(2) Å. The third calcium atom (Ca2) is seven-coordinated and involves two oxygen atoms from different calixarene molecules and five water molecules into its coordination sphere, Ca-O distances 2.326(2) – 2.559(2) Å. This combination of interactions between calixarene molecules and calcium atoms forms a continuous polymeric mono-layer along the [111] direction in the overall crystal packing, Figure 2.
Figure 1.
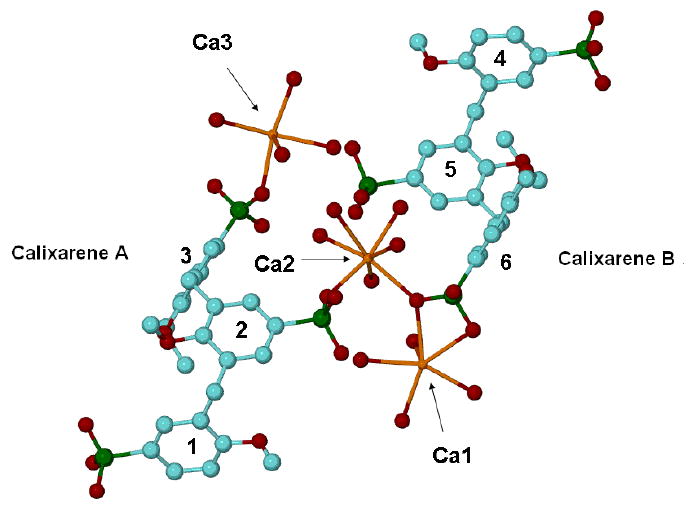
Ball and stick representation of the asymmetric unit for complex 5b showing the co-ordination spheres of the calcium ions. Hydrogen atoms and non-calcium co-coordinated water molecules have been omitted for clarity. For all crystallographic figures the following colour scheme is used; blue for carbon atoms, green for phosphorus atoms, red for oxygen atoms, gold for calcium atoms and silver for hydrogen atoms.
Figure 2.
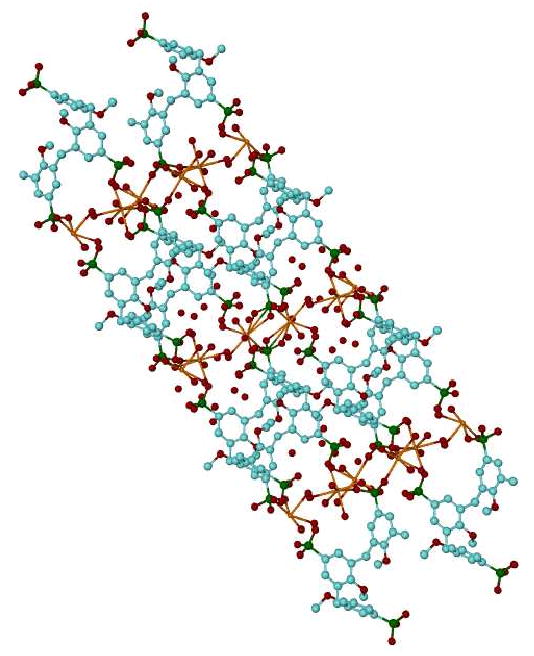
Projection of the overall packing for complex 5b along the [111] direction showing the continuous polymeric mono-layer. Hydrogen atoms have been omitted for clarity.
All of the crystalline water molecules are involved in an H-bond network with O…O distances in the range of 2.522(3) – 3.209 Å. The 3D packing of the molecules in the crystal has a sandwich structure with layers of the calixarene molecules alternated with layers of the Ca/water environment extended along the a-axis in crystal, Figure 2. The conformation of the calixarene molecules, A and B, are remarkably similar and can be described as a container macrocycle, Figure 3. For calixarene A a side wall is formed by the parallel-oriented aromatic rings 1 and 2 in the form of parallelepiped and with ring 3 at the top and bottom of the container. The dihedral angles between the middle-cross plane (0) of calixarene A and the aromatic rings are 86.3(1) (planes 0-1), 87.6(1) (0-2), 28.2(1) (0-3) and 75.0(1)° (1-2). The corresponding values calculated for calixarene B are 87.3(1) (0-4), 88.8(1) (0-5), 28.7(1)(0-6) and 77.1(1)° (4-5). In addition to the H-bonds, there are no specific interactions between the calixarene molecules.
Figure 3.
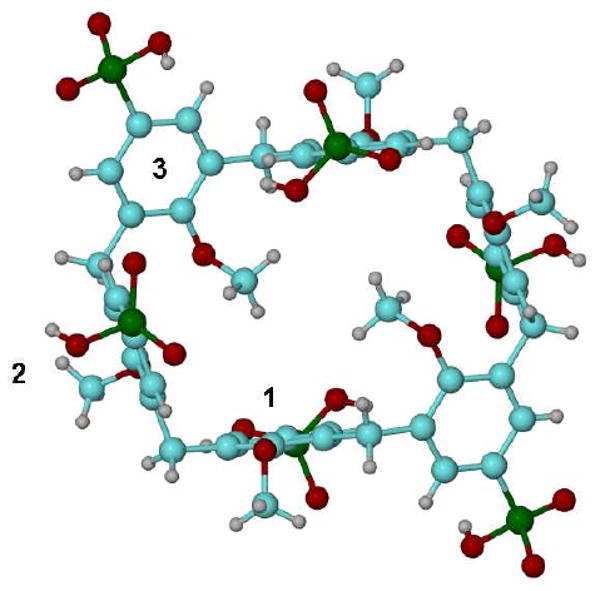
View of the container for calixarene A with aromatic rings 1-3 numbered.
Complex 5c crystallizes in the space group P-1 (Z=1) with the asymmetric unit comprising one calixarene molecule situated at an inversion centre (½, ½, ½), two calcium atoms, 13 fully occupied water molecules and one half occupied water molecule, Figure 4. The container conformation of the calixarene molecule is similar to that described above for the calixarene molecules in complex 5b. The dihedral angles between middle-cross plane (0) of the calixarene and aromatic rings are 89.1(1) (planes 0-1), 86.9(1) (0-2), 20.4(2) (0-3) and 71.0(1)° (1-2). One of the p-phosphonic acid moieties and its symmetry equivalent are completely deprotonated, making the total charge of the calixarene equal to -8.
Figure 4.
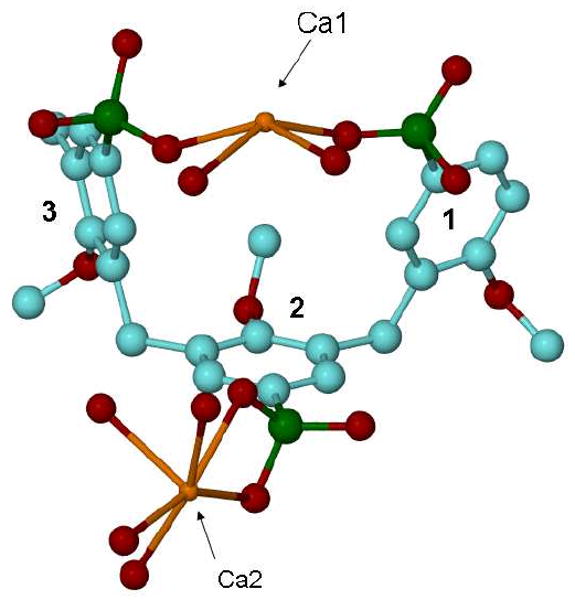
Ball and stick representation of the asymmetric unit for complex 5c. Hydrogen atoms and non-calcium co-coordinated water molecules have been omitted for clarity.
The coordination spheres of the calcium are different, with Ca1 eight coordinate through five water molecules and three oxygen atoms from the p-phosphonic acid moieties of the calixarene, with two water molecules bridging Ca1-Ca1 atoms to form a polymeric chain. Ca2 is seven coordinate with four crystalline water molecules and three oxygen atoms belonging to p-phosphonic acid groups, with two water molecules bridging Ca2-Ca1 atoms to complete the polymeric chain. This is associated with the calixarene molecules being linked in an extended polymeric layer, parallel to the c-axis with Ca-O distances in the range of 2.293(3) - 2.817(4) Å, Figure 5. The 3D-packing of the molecules is effectively a sandwich structure with layers of the calixarene molecules alternating with layers of Ca/water moieties, extended parallel to the a-b plane. There are water molecules and hydroxyls of phosphonic groups involved in a H-bond network with O…O distances, 2.476(4) - 3.271(5) Å.
Figure 5.
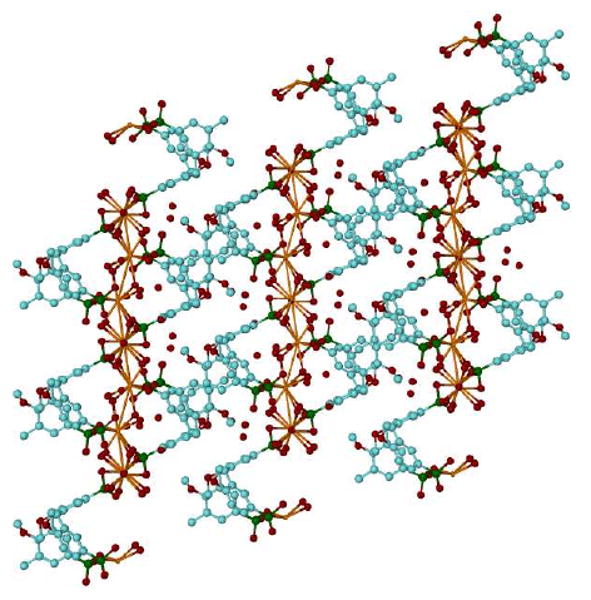
Projection of the overall packing for complex 5c as viewed down the b axis showing the extended polymeric layer.
Hirshfeld surface analysis was performed on complexes 5b and 5c in order to gain insight into the nature of the intermolecular interactions within the crystal structures. Both molecules of the phosphonated calix[6]arene in the asymmetric unit of 5b show similar C…H contacts, which make up 7.3 and 7.2% of the Hirshfeld surface for calixarene A and calixarene B respectively. These contacts represent CH…π acceptor contacts and are caused by the O-methyl group of a neighbouring calixarene pointing towards a phenyl ring of calixarene A or B.
A large portion of the interactions that comprise the Hirshfeld surface of the calixarenes in 5b are due to O…H interactions which are quite similar for both molecules, Figure 6. Of the six phosphonate groups present at the upper rim of the calixarene, four are involved in O…H interactions. Where there are deprotonated phosphonate ligands present in the structure, they interact solely with water molecules. There are points in the expanded structure where phosphonate groups are in close proximity to each other, in which case they are bridged by a water molecule. Other interactions also involve contacts between water molecules and the phenolic oxygen of the calix[6]arenes, and O…H interactions make up 29.5 and 29.8% of the surface for calixarene A and B respectively, and can be viewed as a spike in the bottom left hand corner of the fingerprint plot, Figure 7(A).
Figure 6.
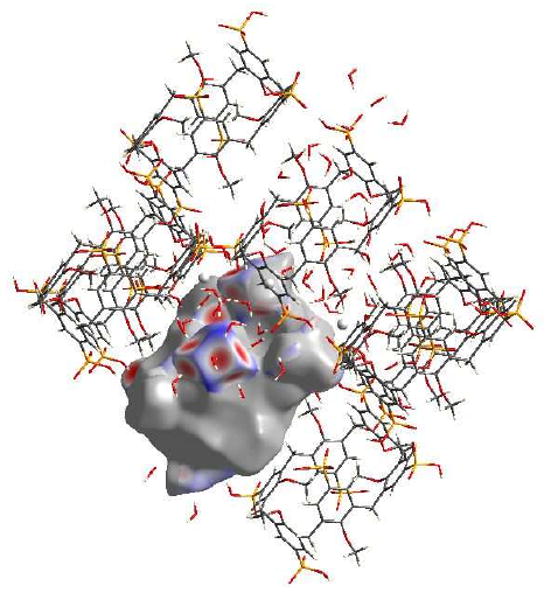
View of the O…H interactions for calixarene B in complex 5b, with red areas highlighting short contacts.
Figure 7.
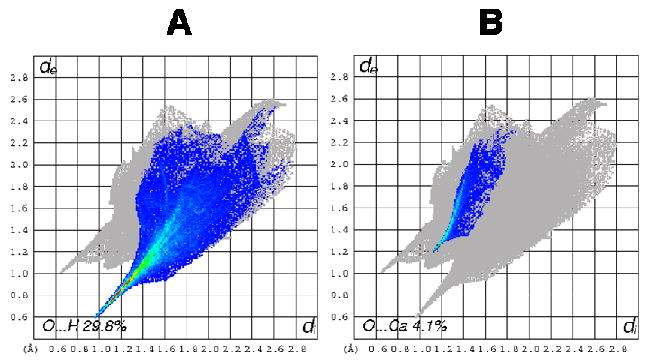
Fingerprint plots of calixarene B in complex 5b: (A) O…H interactions and (B) O…Ca interactions.
Interestingly, the two phosphonate groups that do not show strong O…H interactions, display strong O…Ca interactions instead, Figure 8. These O…Ca interactions appear on the fingerprint plot as a bright streak between the O…H and H…O spikes, and they make up 4.1 and 4.0% of the surface respectively for calixarenes 1 and 2, Figure 7(B). Also for the phosphonic acid groups that have O…Ca interactions, every oxygen atom in that group, be it from a formal P…O double bond, protonated or deprotonated hydroxyl group, interacts with a unique calcium atom.
Figure 8.
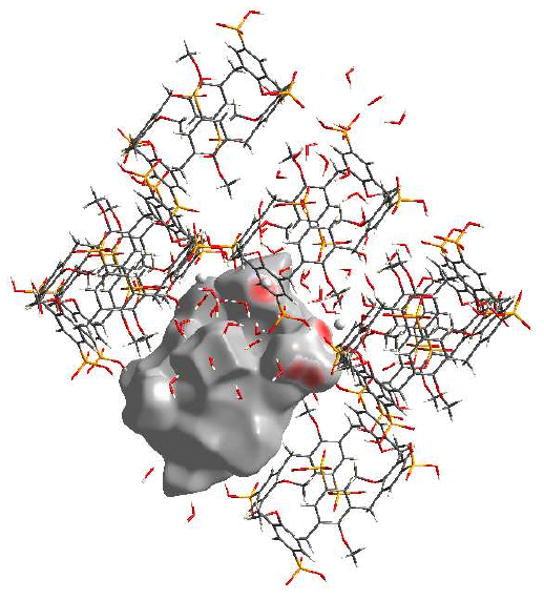
View of the O…Ca interactions for calixarene B in complex 5b, with red areas highlighting short contacts.
Of the four phosphonate groups that show O…H interactions, only two out of the three oxygen atoms participate in these types of contacts. The remaining protonated oxygen atoms take part in H…O interactions, which make up 11.7 and 10.8% of the Hirshfeld surfaces of calixarenes A and B respectively. On calixarene A, these contacts are due to the protonated hydroxyl interacting with either an oygen atom from a water molecule, or a deprotonated hydroxyl group from a neighbouring calixarene. However, on calixarene B these interactions are restricted to the interaction of a protonated hydroxyl and an oxygen atom from a close water molecule.
Structure 5c is different from 5b in that two of the six phosphonate ligands are completely deprotonated and that there is only one calixarene in the asymmetric unit. C…H interactions comprise 7.4% of the Hirshfeld surface for the calix[6]arene and are notable for a lack of short contacts. The shortest contact is where methylene protons from a neighbouring calixarene points into a phenyl ring of the calixarene. O…H contacts make up 29.3% of the surface of the calix[6]arene and are represented by the large spike in the bottom left hand corner of the plot, Figure 9(A). As in 5b, four of the six phosphonate groups display O…H interactions, with the two completely deprotonated phosphonate ligands displaying only O…H interactions, Figure 10. Contacts between deprotonated hydroxyl groups and water molecules, or deprotonated hydroxyl groups and protonated phosphonate groups comprise the majority of these interactions, however there are contacts between phenolic oxygen atoms and bound water molecules which also contribute.
Figure 9.
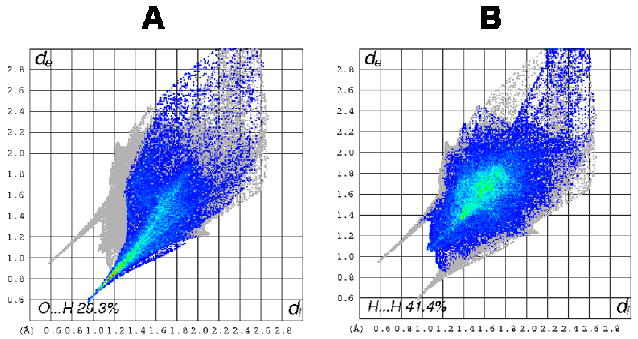
Fingerprint plots of the calixarene molecule in complex 5c: (A) O…H interactions and (B) H…H interactions.
Figure 10.
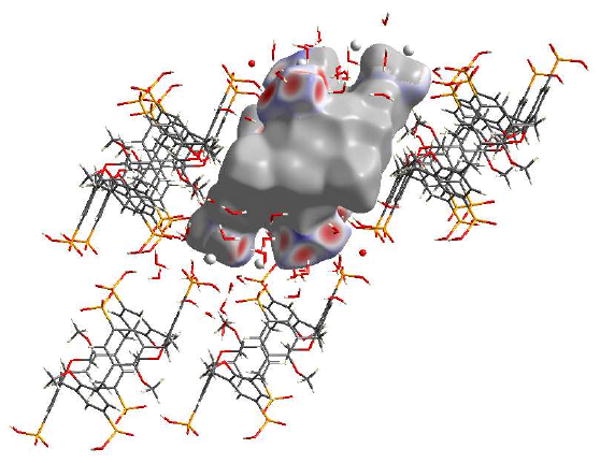
View of the O…H interactions for the calixarene molecule in complex 5c, with red areas highlighting short contacts.
For the six phosphonate groups, four of them participate in O…H bonding; however the other two are involved in O…Ca interactions, with similar contacts to the O…Ca interactions seen in 5b, Figure 11. These O…Ca interactions make up 5.7% of the Hirshfeld surface. Noteworthy is that in the other four phosphonate groups, there are four short contacts, each corresponding to a calcium atom which bridges two of these phosphonate groups. This feature is not observed in structure 5b.
Figure 11.
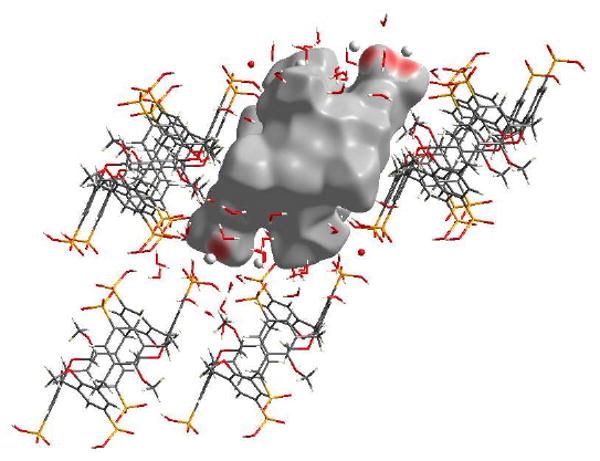
View of the O…Ca interactions for the calixarene molecule in complex 5c, with red areas highlighting short contacts.
The majority of the surface is made up of H…H contacts, which account for 41.4% of the Hirshfeld surface, Figure 9(B). An interesting feature of the fingerprint plot is the “double spike”, seen towards the bottom left of the plot where de ≈ di. This is due to the interaction of a hydrogen from the O-methyl group of the calixarene with a meta hydrogen from a neighbouring calixarene. The atoms are not a uniform distance away from the Hirshfeld surface generated, and are manifested in the “double spike” in the plot.
Previously we have reported that a series of p-phosphonic acid calix[n]arenes (n = 4, 5, 6 and 8) form stable nano-arrays (nano-particles) or nano-rafts (n = 4) in the gas phase using MALDI-TOF-MS with around 20 calixarene units (18-30kDa) per nano-array.8 This has been extended to alkylated p-phosphonic acid calix[4]arenes with successive peaks out to 15, 33 and 16 calixarene units for the methyl, butyl and octadecyl derivatives respectively.9 MALDI-TOF-MS of methylated p-phosphonic acid calix[6]arene, Figure 12, shows nano-arrays up to 28 calixarene units in size. Presumably this association arises also from the multiple hydrogen bonding between calixarenes through the phosphonic acid moieties as in the well characterized structures of the p-phosphonic acid calix[4]arene, and its calcium complexes,12 and also interactions between the aromatic rings (π….π and π….H-O). Interestingly all these structure form bilayers with the calixarene in the ‘up-down’ arrangement, and it is therefore likely that the parent O-methylated calix[4]arene in the present study is also assembled into bilayers, and for the calcium complexes reported herein, that it also assembles into bilayers, and these are persistent to a degree in the gas phase. This is also consistent with the closely related p-sulfonated calix[6]arene also assembling into bilayers with the calixarenes in the ‘up-down’ arrangement.19
Figure 12.
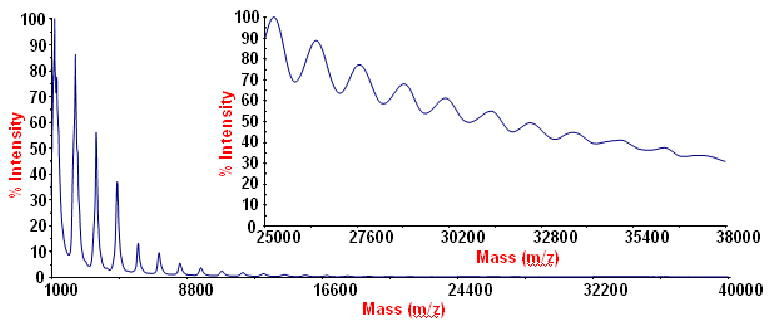
MALDI-TOF-MS showing nano-arrays for p-phosphonic acid hexamethoxy-calix[6]arene.
Conclusion
We have synthesized the novel compound p-phosphonic acid hexamethoxy-calix[6]arene in three steps starting from hexamethoxy-calix[6]arene with 57% overall yield. The hexaphosphonic acid shows nano-array formation in the gas phase with up to 28 calixarene units per array. This is analogous to the calix[4]arene phosphonic acids that are either non-alkylated or alkylated at the lower rim and form nano-arrays between 15 and 33 calixarene units in size. A pattern is now emerging in that the phosphonic acid upper rim groups of calixarenes is effective in assembling the molecules into assemblies, regardless of the nature of the O-alkyl group, or the absence of such a group, and this relates to each functional group being able to contribute to O-H groups in forming complex hydrogen bonding arrays. This is distinctly different from the ubiquitous sulfonated calixarenes, where each function group has only one O-H moiety, and hence less extensive hydrogen bonding capability. The hexaphosphonic acid forms two different calcium complexes with extended polymeric layers in the solid state with calcium ions and waters of sandwiched between layers of calixarenes. The different ratio of calcium to calixarene in these complexes, and for p-phosphonic acid calixarene itself,8 also highlights another pattern for phosphonic acid calixarenes, namely the variability of uptake/binding of metal ions arising from different degrees of deprotonation of the phosphonic acid moieties, and suggests a plethora of different metal ion complexes for phosphonated calixarenes in general. The new calcium complexes have potential uses as calcium delivery systems in the human body and in understanding cation-carrier interactions.
Experimental Section
Materials and Methods
All starting materials and solvents were obtained from commercial suppliers and used without further purification except otherwise noted. Acetonitrile was dried over 4 Å molecular sieves for 24 hours and NiCl2.6H2O was dried at 180 °C in vacuo for 8 hours before use. All moisture-sensitive reactions were performed under a positive pressure of nitrogen. Chromatographic purification was performed using 200-400 mesh silica gel. TLC analysis was performed on silica gel plates (absorbent thickness 250 μm) containing a fluorescent indicator. Melting points were determined using sealed and evacuated capillary tubes on an Electrothermal 9100 melting point apparatus and are uncorrected. IR spectra were recorded as KBr pellets on a Perkin Elmer Spectrum One spectrometer. 1H NMR (500 MHz), 13C NMR (126 MHz) and 31P NMR (202 MHZ) were recorded on a Bruker spectrometer and internally referenced to the solvent signal or phosphoric acid for 31P NMR. Elemental analysis was performed at The Campbell Microanalytical Laboratory, Otago, New Zealand and FAB-MS was performed on a HP5896 mass spectrometer.
MALDI-TOF-MS experiments were conducted on an Applied Biosystems Voyager DE-STR mass spectrometer. Data were collected in the negative ion, linear mode with delayed extraction. The accelerating voltage was set at 25 kV and delay time was 500 ns with the low mass gate set at 500 Da. Each spectrum was an average of 1000 shots. A standard mixture of 5 peptides of mass range from 1000 to 6000 Da was used for external calibration. p-Phosphonic acid hexamethoxy-calix[6]arene was dissolved in methanol to a final concentration of 1 mM and premixed with 2,5-DHB (150 mg/ml in 50% (v/v) methanol) at a ratio of 1:1. A 1-μL volume of the premixed solution was deposited onto a stainless steel 100-well MALDI target plate (Applied Biosystems) and allowed to air-dry at room temperature.
Calix[6]arene, 1,15 and hexamethoxycalix[6]arene, 2, were prepared as described previously in the literature.
5,11,17,23,29,35-Hexaiodo-37,38,39,40,41,42-hexamethoxycalix[6]arene (3)
A suspension of 1.84 g (8.32 mmol) of CF3CO2Ag and 0.50 g (0.69 mmol) of 2 was refluxed in 50 mL of chloroform for 4 h. After this time 4.23 g (16.65 mmol) of I2 was added and the purple solution refluxed for a further 2 h. After cooling to RT the mixture was filtered over Celite and the filtrate washed with a saturated solution of Na2S2O5 (2 × 10 mL) and then water (3 × 10 mL). The organic layer was dried over MgSO4, filtered and evaporated under reduced pressure to yield 0.68 g (66%) of 3 as a pale yellow solid. Trituration with dimethylformamide yielded 3 as a pure white solid suitable for micro analysis. m.p. >260 °C (dec.); 1H NMR (CDCl3, 25 °C, 500 MHz) δ7.24 (s, 12H, m-H), 3.83 (s, 12H, ArCH2Ar), 3.35 (s, 18H, OCH3); 13C NMR (CDCl3, 25 °C, 126 MHz) δ156.41, 138.00, 136.71, 87.97, 60.76, 29.79; HRMS (FAB) m/z calcd for (C48H42I6O6)+ 1475.7250, found 1475.7196; Anal. Calcd for C48H42I6O6: C 39.05, H, 2.87, found: C 39.13, H 3.07.
5,11,17,23,29,35-Hexa(diethoxyphosphoryl)-37,38,39,40,41,42-hexamethoxycalix[6]arene (4)
A solution of 1.50 g (1.02 mmol) of 3 and 0.20 g (1.53 mmol) of NiCl2 in 40 mL of benzonitrile (not completely soluble) was treated dropwise with 2.61 mL (15.25 mmol) of P(OEt)3 under nitrogen at 180 °C. The solution was stirred for 0.5 h and the volatiles removed under reduced pressure to leave an orange residue. The residue was dissolved in 100 mL of toluene, washed with 5% ammonia solution (5 × 30 mL), dried over MgSO4 and evaporated under reduced pressure to yield an orange oil. The oil was chromatographed over silica gel to yield 1.44 g (92%) of 4 as a white solid. Recrystallisation from toluene yielded X-ray quality single crystals, 4a, which were also submitted for microanalysis. Rf 0.35 (1:19 methanol-dichloromethane); m.p. 236-237 °C; IR (KBr) 2984 (m), 2907 (m), 1638 (w), 1593 (w), 1472 (m), 1272 (s), 1020 (s), 966 (s), 794 (m), 548 (m) cm-1; 1H NMR (CDCl3, 25 °C, 500 MHz) δ7.40 (d, 12H, ArH, JP-H = 13.5 Hz), 4.09-4.01 (m, 12H, POCHHCH3), 4.01-3.93 (m, 12H, POCHHCH3), 3.96 (s, 12H, ArCH2Ar), 3.30 (s, 18H, OCH3), 1.22 (t, 36H, POCH2CH3, J = 7.0 Hz); 13C NMR (CDCl3, 25 °C, 126 MHz) δ159.71 (d, 4JP-C = 3.8 Hz), 134.63 (d, 3JP-C = 16.0 Hz), 132.82 (d, 2JP-C = 10.8 Hz), 123.24 (d, 1JP-C = 189.8 Hz), 62.19 (d, 2JP-C = 5.5 Hz), 60.39, 30.02, 16.24 (d, 3JP-C = 6.4 Hz); 31P NMR (CDCl3, 25 °C, 202 MHz) δ19.26; HRMS (FAB) m/z calcd for (C72H102O24P6 + H)+ 1537.5265, found 1537.5202; Anal. Calcd for C72H102O24P6 + H2O: C 55.60, H 6.74, found: C 55.40, H 6.53.
5,11,17,23,29,35-Hexa(dihydroxyphosphoryl)-37,38,39,40,41,42-hexamethoxycalix[6]arene (5)
2.89 mL (21.87 mmol) of Bromotrimethylsilane was added to 1.40 g (0.91 mmol) of 4 in 50 mL of dry acetonitrile and the solution refluxed for 16 hours. The volatiles were removed under reduced pressure and the resulting residue was triturated with 30 mL of acetonitrile and 1 mL of water. The precipitate formed was filtered off, washed with acetonitrile (3 × 10 mL) and recrystallized from methanol to yield 1.04 g (95%) of 5 as a white solid. Recrystallisation from methanol / 6M HNO3 / CsNO3 yielded X-ray quality crystals, 5a, which were also submitted for microanalysis. m.p. >280 °C (dec); IR (KBr) 3180 (br), 2937 (m), 2834 (m), 2300 (br), 1641 (w), 1595 (w), 1472 (m), 1272 (m), 1118 (s), 1003 (s), 965 (s), 516 (m) cm-1; 1H NMR (DMSO-d6, 25 °C, 500 MHz) δ7.28 (d, 12H, ArH, JP-H = 13.2 Hz), 6.60 (br s, POH, shifts downfield with increasing [H]+), 3.96 (s, 12H, ArCH2Ar), 3.13 (s, 18H, OCH3); 13C NMR (DMSO-d6, 25 °C, 126 MHz) δ158.12, 133.93 (d, 3JP-C = 15.5 Hz), 131.38, 128.47 (d, 1JP-C = 186.0 Hz), 59.92, 29.92; 31P NMR (DMSO-d6, 25 °C, 202 MHz) δ15.17; HRMS (FAB) m/z calcd for (C48H54O24P6)+ 1200.1431, found 1200.1491; Anal. Calcd for C48H54O24P6 + 0.55HNO3 + 0.1H2O: C 46.60, H 4.46, found: C 46.61, H 4.44.
X-ray Crystallography
Suitable crystals of 5a and 5b for diffraction studies were prepared by dissolving 4.2 mmol of 5 and 11.4 (for 5a) or 17.0 (for 5b) mmol of Ca(acetate)2.H2O in 2 mL water close to reflux, followed by filtration, cooling to room temperature, followed by slow evaporation over several days. Uniformity of the samples was checked by cell determinations of several crystals for each sample. The X-ray diffracted intensities were measured from single crystals on one of two machines. The first machine was an Oxford Diffraction Gemini-R Ultra CCD diffractometer at about 100 K using monochromatized Cu-Kα (λ = 1.54178 Å.). The second machine was a Bruker ASX SMART CCD diffractometer at about 100 K or 153 K using monochromatized Mo-Kα (λ = 0.71073 Å). Data were corrected for Lorentz and polarization effects and absorption correction applied using multiple symmetry equivalent reflections. The structures were solved by direct method and refined on F2 using the SHELX-97 crystallographic package17 and X-seed interface.18 A full matrix least-squares refinement procedure was used, minimizing w(Fo2-Fc2), with w = [σ2(Fo2)+(AP)2+BP]-1, where P= (Fo2+2Fc2)/3. Agreement factors (R = Σ‖Fo|-|Fc‖/Σ|Fo|, wR2 = {Σ[w(Fo2-Fc2)2]/Σ[w(Fo2)2]}1/2 and GOF = {Σ[w(Fo2-Fc2)2]/(n-p)}1/2 are cited, where n is the number of reflections and p the total number of parameters refined). All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms partly were localized from difference Fourier map, partly calculated from geometrical consideration and their atomic parameters were constrained to the bonded atoms during the refinement. CCDC deposition numbers 766115-766116, 766252-766253.
Crystal/refinement details for complex 4a: C72H104O25P6, M = 1555.37, F(000) = 6608 e, monoclinic, C2/c (No. 15), Z = 8, T = 153(2) K, a = 40.464(5), b = 17.430(2), c = 27.483(4) Å, β = 123.113(1) °, V = 16235(4) Å3, Dc = 1.273 gcm-3, μMo = 0.205 mm-1, sinθ/λmax = 0.5946, N(unique) = 13879 (merged from 49484, Rint = 0.0311, Rsig = 0.0339), No (I > 2σ(I)) = 9507, R = 0.1489, wR2 = 0.3559 (A,B = 0.15, 190.0), GOF = 1.041, |Δρmax| = 2.1(1) e Å-3.
Crystal/refinement details for complex 5a: C48H70N4O42P6, M = 1560.90, F(000) = 812 e, triclinic, P-1 (No. 2), Z = 1, T = 100(2) K, a = 12.182(2), b = 12.332(2), c = 13.108(2) Å, α = 113.447(3), β = 94.212(3), γ = 108.695(3) °, V = 1664.9(5) Å3, Dc = 1.557 gcm-3, μMo = 0.270 mm-1, sinθ/λmax = 0.5946, N(unique) = 5816 (merged from 10181, Rint = 0.0251, Rsig = 0.0465), No (I > 2σ(I)) = 4259, R = 0.0697, wR2 = 0.1849 (A,B = 0.10, 3.70), GOF = 1.054, |Δρmax| = 1.1(1) e Å-3.
Crystal/refinement details for complex 5b: C48H104Ca3O52P6, M = 1819.37, F(000) = 1916 e, triclinic, P-1 (No. 2), Z = 2, T = 100(2) K, a = 14.0340(4), b = 16.2130(5), c = 19.4062(6) Å, α = 110.878(3), β = 95.287(2), γ = 99.200(2) °, V = 4019.0(2) Å3, Dc = 1.503 gcm-3, sinθ/λmax = 0.5981, N(unique) = 14088 (merged from 42363, Rint = 0.0499, Rsig = 0.0770), No (I > 2σ(I)) = 8081, R = 0.0362, wR2 = 0.0704 (A,B = 0.024, 0), GOF = 1.004, |Δρmax| = 0.55(6) e Å-3.
Crystal/refinement details for complex 5c: C48H99Ca4O50.5P6, M = 1830.41, F(000) = 961 e, triclinic, P-1 (No. 2), Z = 1, T = 100(2) K, a = 11.4113(7), b = 12.9903(9), c = 15.4684(11) Å, α = 112.047(7), β = 95.814(5), γ = 108.467(6) °, V = 1951.6(2) Å3, Dc = 1.557 gcm-3, sinθ/λmax = 0.5984, N(unique) = 6828 (merged from 22732, Rint = 0.0977, Rsig = 0.1485), No (I > 2σ(I)) = 3372, R = 0.0495, wR2 = 0.0942 (A,B = 0.025, 0), GOF = 1.008, |Δρmax| = 1.30(8) e Å-3.
Supplementary Material
Acknowledgments
We thank the ARC, NSF and NIH (grant P41RR000954) for financial support of this work and the University of Western Australia for SIRF, GRST and PRT awards to TEC.
Footnotes
Supplementary Supporting Information
Crystallographic information file (cif). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Diamond D, McKervey MA. Chem Soc Rev. 1996;25:15–24. [Google Scholar]
- 2.(a) Atwood JL, Barbour LJ, Jerga A. Science. 2002;296:2367–2369. doi: 10.1126/science.1072252. [DOI] [PubMed] [Google Scholar]; (b) Atwood JL, Dalgarno SJ, Thallapally PK. J Am Chem Soc. 2006;128:15060–15061. doi: 10.1021/ja065499x. [DOI] [PubMed] [Google Scholar]
- 3.Atwood JL, Koutsantonis GA, Raston CL. Nature. 1994;368:229–231. [Google Scholar]
- 4.Kotek J, Lukes I, Plutnar J, Rohovec J, Zak Z. Inorg Chim Acta. 2002;335:9. [Google Scholar]
- 5.(a) Atwood JL, Bott SG, Hamada F, Means C, Orr GW, Robinson KD, Zhang H. Inorg Chem. 1992;31:603–607. [Google Scholar]; (b) Atwood JL, Barbour LJ, Orr GW. Science. 1999;285:1049–1052. doi: 10.1126/science.285.5430.1049. [DOI] [PubMed] [Google Scholar]; (c) Hardie MJ, Raston CL. J Chem Soc, Dalton Trans. 2000:2483–2492. [Google Scholar]
- 6.(a) Clark TE, Makha M, McKinnon JJ, Raston CL, Sobolev AN, Spackman MA. Cryst Eng Comm. 2007;9:566–569. [Google Scholar]; (b) Coleman AW, Kuduva SS, Shahgaldian P, Zaworotko MJ. Chem Commun. 2005:1968–1970. doi: 10.1039/b412925n. [DOI] [PubMed] [Google Scholar]
- 7.Arduini A, Casnati A. Macrocycle Synthesis. 1st. Oxford University Press; Oxford: 1996. [Google Scholar]
- 8.(a) Clark TE, Makha M, Sobolev AN, Su D, Rohrs H, Gross ML, Atwood JL, Raston CL. New J Chem. 2008;32:1478–1483. [Google Scholar]; (b) Clark TE, Makha M, Sobolev AN, Rohrs H, Atwood JL, Raston CL. Chem Eur J. 2008;14:3931–3938. doi: 10.1002/chem.200701472. [DOI] [PubMed] [Google Scholar]
- 9.Clark TE, Makha M, Sobolev AN, Su D, Rohrs H, Gross ML, Raston CL. Cryst Growth Des. 2009;9:3575–3580. doi: 10.1021/cg900315h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng B, Mattson MP. J Neurosci. 1992;12:1558–1666. doi: 10.1523/JNEUROSCI.12-04-01558.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thackery E. The Gale Encyclopedia of Cancer. 1st. The Gale Group; 2002. [Google Scholar]
- 12.Martin AD, Raston CL, Sobolev AN, Spackman MA. Cryst Growth Des. 2009;9:3759–3764. [Google Scholar]
- 13.(a) Clark TE, Makha M, Sobolev AN, Raston CL. Cryst Growth Des. 2008;8:890–896. doi: 10.1021/cg900315h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jayatilaka D, Spackman MA. Cryst Eng Comm. 2009;11:19–32. [Google Scholar]
- 14.Jayatilaka D, McKinnon JJ, Spackman MA. Chem Commun. 2007:3814–3816. doi: 10.1039/b704980c. [DOI] [PubMed] [Google Scholar]
- 15.Parker D, editor. Macrocycle Synthesis: A Practical Approach. Oxford University Press; Oxford: 1996. [Google Scholar]
- 16.Gutsche CD, Lin L. Tetrahedron. 1986;42:1633. [Google Scholar]
- 17.Sheldrick GM. Acta Cryst. 2008;A64:3. [Google Scholar]
- 18.Barbour LJ. J Supramol Chem. 2001;1:189. [Google Scholar]
- 19.Dalgarno SJ, Hardie MJ, Makha M, Raston CL. Chem Eur J. 2003;9:2384. doi: 10.1002/chem.200304963. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.