

Abstract
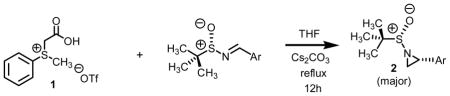
Reaction of sulfur ylide with a series of aryl substituted chiral non-racemic sulfinyl imines afforded the corresponding aziridines in high yield and good stereoselection. The sulfur ylides were generated by the thermally induced decarboxylation of carboxymethylsulfonium betaines. A drop in the diastereomeric ratio was observed when going from electron deficient to electron releasing aryl substituted imines. Sulfonium methylidene aziridinations involving the decarboxylation of carboxymethylsulfonium betaine functionality compliments existing technologies with the advantages of the reaction protocol, levels of conversion and scope.
Introduction
Nucleophilic alkylidene transfer agents have been shown to be extremely useful for the introduction of functionality into organic molecules.1 Once generated, they can and do react with a large variety of π-acceptors.2 A representative list of processes involving a π-acceptor would include formation of oxiranes from aldehydes and ketones, aziridines from imines, and cyclopropanes from electron deficient olefins. The direct and stereospecific assembly of small ring carbo- and heterocyclic building blocks represents a very attractive synthetic transformation, especially when considering the assembly of materials of biological and medicinal significance.3,4 Previous communications from our lab reported on a novel protocol for the preparation of nucleophilic alkylidene transfer agents. The protocol had as its key step the decarboxylation of carboxymethyl betaine functionality which generated the requisite sulfur ylides in situ (Scheme 1).5 Sulfur ylide intermediate ii is best described as a semi-stabilized ylide when compared to thioacetate scaffolds consisting of not alkyl aryl substitution but alkyl alkyl substitution on sulfur. While traditional points of stabilization would focus on the alkylidene carbon, attachments onto the sulfur moiety and even solvation of the intermediate itself have also been shown to improve to the half-lives of sulfur ylides.6 When ylide ii is used in excess quantities, clean conversion of aldehyde to oxirane functionality is observed. The ease by which the sulfur ylide is generated from sulfonium salt 1 is the key advantage of this method over other existing technologies. That is, the generation of the ylides by decarboxylation has distinct advantages over more traditional methods. First, the process does not require the use of strong and often pyrophoric bases such as butyl lithium or sodium hydride. Second, the method does not require expensive metal catalysts. Third this approach is compatible with ecologically benign solvent systems. And finally, fourth, through fine-tuning of the electronics of the betaine, a range of activity and lifetime of methylidene transfer agent can be “dialled in.”
Scheme 1.

While very satisfied with the chemistry, the methylidene transfer technology using sulfonium salt 1 has yet to reach its full potential when considering catalysis, additions to other π-acceptors and asymmetry. Efforts to address the latter two, additions to other π-acceptors and asymmetry, have been made and reported herein are our findings in the stereoselective assembly of heterocylic building blocks of aziridine functionality.
Results and Discussion
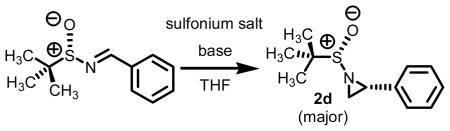
Stockman and co-workers have several reports on the Corey-Chaykovsky reaction of chiral sulfinyl imines (Scheme 2).7 Using methylidene transfer agents derived from trimethyl sulfonium iodide, a series of aliphatic, aromatic, and heterocyclic tert-butylsulfinyl aziridines were prepared.6d Reaction times ranging between three and ten hours using NaH as base, DMSO as solvent at 20°C lead to the highest levels of conversion and diastereoselectivity.
Scheme 2.

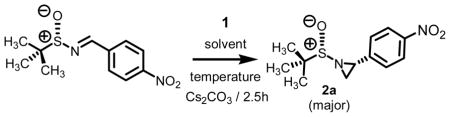
Intrigued by this report, we embarked upon an aziridination study using our technology involving the decarboxylation of carboxymethylsulfonium betaine functionality to generate a sulfur ylide in situ. One of our goals was to address the question if alternative reagents and solvent systems could be used without significantly compromising the levels of conversion while maintaining high levels of stereoselectivity. For comparative purposes, we chose to test a series of aryl substituted chiral non-racemic tert-butylsulfinyl imines against our sulfonium salt 1. As we found in our epoxidation studies,5 the incorporation of S-aryl functionality offers added stability of the sulfur ylide (vide infra) and thus we hypothesized that this would as well result in high levels of conversion when subjected to imine functionality. Except for this single modification, use of imine as opposed to aldehyde functionality, all other reaction conditions were kept the same as in our previously optimized work with aldehyde π-acceptors. A total of seven sulfinimine derivatives were surveyed. The data from the study is presented below in Table 1.
Table 1.
Sulfonium methylidene aziridinations using sulfonium salt 1.
 | |||
|---|---|---|---|
| entry | imine | yield (%)a | drb |
| 1, a | 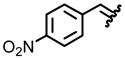 |
97 | 87:13 |
| 2, b | 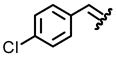 |
96 | 78:22 |
| 3, c | 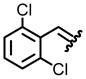 |
94 | 77:23 |
| 4, d | 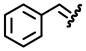 |
95 | 74:26 |
| 5, e | 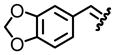 |
85 | 84:16 |
| 6, f | 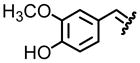 |
93 | 80:20 |
| 7, g | 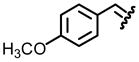 |
89 | 66:34 |
Isolated chromatographically pure material.
Determined by 1H NMR (crude reaction mixture).
Each system, aldehyde to imine (experimental section and Supporting Information) and imine to aziridine, afforded the desired material in excellent overall yield. The yields for aziridine formation are of isolated and chromatographically pure material. All reactions reported here were clean as judged by GC and NMR analysis of the crude reaction mixture. The levels of diastereocontrol were obtained of the crude reaction mixture by 1H NMR.8 The diagnostic peaks for both the major and minor aziridine of each system are presented in the Supporting Information.
Table 1 reveals some interesting trends when looking at the levels of diastereocontrol. Electron deficient aryl imines (entries 1–3) resulted in levels of diastereocontrol ranging from 87:13 (entry 1) to 77:23 (entry 3). The levels of diastereocontrol with these two examples served as bookends when switching to electron neutral (entry 4) and the two examples representing disubstituted electron releasing aryl functionality (entries 5 and 6). Unique with the latter is that when going from a disubstituted aryl system consisting of electron releasing groups to a monosubstituted electron releasing group (entry 7), the level of diastereocontrol dropped to 66:34.
While significantly higher levels of diastereocontrol could be obtained with our technology, it was at the expense of aziridine conversion (dr = 90:10 at 50% conversion for 2d). Albeit lower levels of diastereocontrol were observed overall, we were encouraged by the fact that extremely high levels of conversion were obtained with a process which does not rely on DMSO as solvent and strong bases such as NaH or nBuLi.
The trend in diastereocontrol is supported by the model proposed by Aggarwal9 and Crudden10 which has as a key interaction in the formation of small ring heterocycles, the electrostatic interactions of the sulfur atom and hetereoatom. For aziridination reactions, while less is known about the transition state, there is evidence that the crucial electrostatic interactions (as is the case of oxirane formation) can be offset with unfavorable steric interactions when considering the formation of 1,2,3-trisubstituted aziridines.11
With our 1,2-disubstituted systems, a more electron rich system would predict that the sp2 hybridization of the carbon of the imine (or oxirane) is maintained. This downplays the electrostatic interactions of π-acceptor (nitrogen) and sulfonium salt (sulfur), which in turn, diminishes the steric effects of the tert-butyl moiety. As a result a switch in the transition state is proposed when analyzing the diastereoselectivities obtained (Figure 1). That is, a switch from a closed (cisoid) to an open (transoid) transition state may be operative, and would explain the trend in diastereoselectivities observed when electron deficient and releasing aryl imines are subjected to sulfonium methylidene aziridination processes using sulfonium salt 1. Figure 1 offers two models both result in formation of the same diastereomer. The illustration highlights the two limiting scenarios for a favorable electrostatic interaction (closed) and one which does not rely on the two highlighted developing charges (closed).
Figure 1.

Operative transition states in the aziridination reaction of sulfur ylides.
As for entries 5 and 6, which have two electron releasing groups, the substitution patterns at both the meta- and para- positions do offset one another when considering the overall electronics of the π-acceptor at the imine carbon.12 The highlight of this approach is the extent of conversion when looking at reaction of a series of electronically unique aldimines bearing as a directing group, tert-butyl sulfinyl functionality. Each system afforded the desired product in excellent yield. This was validated when we conducted a side-by-side comparison of this approach with reaction conditions in an earlier study (Table 2).
Table 2.
A Comparison of Conversion and Diastereocontrol.
 | |||||
|---|---|---|---|---|---|
| entry | sulfonium salt | base | temperature (°C) | yield (%)a | drb |
| 1 | 1 | Cs2CO3 | 78 | 95 | 74:26 |
| 2 | (CH3)3S⊕|⊖ | NaH | 78 | 86 | 76:24 |
| 3c | (CH3)3S⊕|⊖ | n-BuLi | −20 | 56 | 92:8 |
| 4c,d | (CH3)3S⊕|⊖ | NaH | 20 | 84 | 93:7 |
Isolated chromatographically pure material.
Determined by 1H NMR (crude reaction mixture).
Reference 7d.
Solvent is DMSO.
Differences in conversion were observed when comparing the two methods using different sulfonium salts (entry 1 (sulfonium salt 1) and entry 2 (trimethylsulfonium iodide)). Both ylides are effective methylidene transfer agents. Any differences in mass throughput to the desired product is most likely due to competing side reactions when going from an unstabilized to a semi-stabilized sulfur ylide.
Substantial differences were noted when looking at changes in reaction temperature (entry 3) and solvent (entry 4). An inverse relationship between temperature and diastereocontrol is not surprising. To address these differences using our method, we conducted a temperature survey using DMSO as solvent. Presented below in Table 3 are our results using DMSO as base as a function of reaction temperature.
Table 3.
Temperature Survey using DMSO.
 | ||||
|---|---|---|---|---|
| entry | temperature (°C) | solvent | ratioa (aziridine:imine) | dra |
| 1 | 21 | DMSO | 13:87 | 86:14 |
| 2 | 40 | DMSO | 42:58 | 83:17 |
| 3 | 60 | DMSO | 56:44 | 79:21 |
| 4 | 80 | DMSO | 92:8 | 70:30 |
| 5 | 80 | THF | >95:5 | 87:13 |
Determined by 1H NMR (crude reaction mixture).
For comparative purposes, entry 5 lists the extent of conversion using THF as solvent. For this study, we elected to use an electron deficient aryl imine so that a rapid screen could be conducted. The longer reaction times presented in Table 1 assures completion of reaction. Perhaps most striking with this survey is that in refluxing THF, the level of diastereoselectivity is high (entry 4 versus entry 5). Furthermore, the level of diastereocontrol at room temperature using DMSO (entry 1) is on par with that of refluxing THF (entry 5). As stated above, while levels of conversion dropped, significantly higher levels of diastereocontrol could be obtained with our technology in THF when operating closer to room temperature. The key however is the process. High levels of conversion can be obtained without the need to use DMSO as solvent and strong bases such as NaH or nBuLi.
The results for our decarboxylation approach show extremely high levels of conversions while operating a much warmer reaction conditions when compared to aziridination protocols found in the literature. It is interesting that a comparison of reaction of sulfonium salt 1 with trimethylsulfonium iodide resulted in nearly identical levels of diastereocontrol. To explain the above results we propose that at high temperatures, the decarboxylation rate is high providing relatively high levels of ylide that can be trapped by suitably modified π-acceptors. Attempts to react sulfonium salt 1 with unactivated imines resulted in recovery of the starting imine. As we noted in prior reports, as the electrophilicity of the carbonyl compound decreases, the forward rate of reaction slows down and fails to compete effectively with ylide fragmentation and other side reactions. These observations and the data presented herein documents the clear relationship between electronic control and diastereoselectivity. Research in our group continues in the areas of 1) understanding the electrostatic relationships of this process, 2) increasing the scope of substrate using modified sulfonium salts, and 3) the preparation and use of a chiral, non-racemic sulfide promoter. The results from these studies will be reported in due course.
Conclusion
Methylidene transfer to imine functionality activated and directed with a tert-butyl sulfinyl moiety using decarboxylation of arylcarboxymethylsulfonium betaines has been achieved. While extremely high levels of imine conversion were observed, the levels of diastereocontrol are at best considered to be moderate. The levels of conversion were significant especially when working with electron releasing aryl imines functionality. While higher levels of diastereocontrol could be achieved when operating at lower reaction temperatures, lower temperatures resulted in lower levels of conversion given that the protocol has as its key step the thermal decarboxylation of carboxymethyl betaine functionality. Notwithstanding the levels of diastereocontrol nor the thermal requirements when considering conversion, the ease by which the sulfur ylide is generated, the scope and levels of conversion obtained are, together, the key advantages of this method over other existing technologies.
Experimental Section
Representative procedure for the preparation of N-sulfinyl imines
To a 50 ml round-bottomed flask equipped with a stir bar and septum was added aldehyde (1.5 equiv), anhydrous copper sulfate (2.0 equiv), anhydrous dichloromethane (3.0 mL) and (S)-(−)-2-methyl-2-propane sulfinamide (1.0 equiv (2.0 mmol)). The reaction mixture was allowed to stir under an argon atmosphere for a period of 24h at which time the reaction was monitored by TLC analysis until complete. When complete as judged by TLC analysis, the reaction mixture was filtered. After concentration of the filtrate in vacuo, the crude reaction mixture was immediately purified by silica gel chromatography (radial chromatography) using a gradient eluent system of hexanes and EtOAc (9:1) to afford analytically pure N-sulfinyl imine.
[S(S), N(E)]-2-methyl-N-[(phenyl)methylene]-2-propanesulfinamide
From the combination of 0.242 g (2 mmol) (S)-(−)-2-methyl-2-propane sulfonamide, 0.318 g (3 mmol) benzaldehyde, and 0.640 g (4 mmol) CuSO4, 0.470 g (95%) of the title compound was isolated as an oil after silica gel chromatography. Analytical data: (c 1.0, CHCl3). [(R)-(−) lit [α]D20 −104 (c 1.0, CHCl3)].13 IR (cm−1) 2960, 2924, 1605, 1572, 1449, 1082; 1H NMR (300 MHz, CDCl3) δ 7.85 (d, J = 7.8, 2H), 7.52-7.45 (m, 3H), 1.23 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 162.8, 134.1, 132.5, 129.4, 129.0, 57.8, 22.7; TLC Rf 0.41 (EtOAc/hexane, 1/9); HRMS (ESI) calcd for C11H15NOS (M+H) 210.0953; Found 210.0946.
[S(S), N(E)]-2-methyl-N-[(4-nitrophenyl)methylene]-2-propanesulfinamide
From the combination of 0.242 g (2 mmol) (S)-(−)-2-methyl-2-propane sulfonamide, 0.453 g (3 mmol) 4-nitrobenzaldehyde, and 0.640 g (4 mmol) CuSO4, 0.401 g (96%) of the title compound was isolated as a yellow solid after silica gel chromatography. Analytical data: (c 1.0, CHCl3), mp 131–132°C; [(R)-(−) lit [α]D22−55 (c 1.0, CHCl3), mp 138–140°C].14 IR (cm−1) 3113, 2950, 1615, 1585, 1530, 1473, 1313, 1178, 1007; 1H NMR (300 MHz, CDCl3) δ 8.66 (s, 1H), 8.32 (d, J = 8.7, 2H), 8.01 (d, J = 7.8, 2H), 1.25 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 160.7, 149.9, 138.9, 130.1, 124.3, 58.5, 22.7; TLC Rf 0.38 (EtOAc/hexane, 1/9); HRMS (ESI) calcd for C11H14N2O3S (M+H) 255.0810; Found: 255.0808.
[S(S), N(E)]-2-methyl-N-[(4-chlorophenyl)methylene]-2-propanesulfinamide
From the combination of 0.242 g (2 mmol) (S)-(−)-2-methyl-2-propane sulfonamide, 0.423 g (3 mmol) 4-chlorobenzaldehyde, and 0.640 g (4 mmol) CuSO4, 0.447 g (92%) of the title compound was isolated as a white solid after silica gel chromatography. Analytical data: (c 1.0, CHCl3), mp 41–42°C, [lit mp ((R)-(−)) 41–42°C].15 IR (cm−1) 2982, 2955, 1608, 1589, 1466, 1213, 1083, 674; 1H NMR (300 MHz, CDCl3) δ 8.52 (s, 1H), 7.76 (d, J = 8.5, 2H), 7.43 (d, J = 8.2, 2H), 1.24 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 161.5, 138.7, 132.5, 130.6, 129.4, 58.0, 22.6; TLC Rf 0.40 (EtOAc/hexane, 1/9). HRMS (ESI) calcd for C11H14ClNOS (M+H) 244.0567; Found 244.0563.
[S(S), N(E)]-2-methyl-N-[(2,6-dichlorophenyl)methylene]-2-propanesulfinamide
From the combination of 0.242 g (2 mmol) of (S)-(−)-2-methyl-2-propane sulfonamide, 0.525 g (3 mmol) 2,6-dichlorobenzaldehyde, and 0.640 g (4 mmol) CuSO4, 0.371 g (67%) of the title compound was isolated as an oil after silica gel chromatography. Analytical data: (c 1.0, CHCl3). IR (cm−1) 2982, 2955, 1608, 1589, 1565, 1486, 1213, 1083, 674; 1H NMR (300 MHz, CDCl3) δ 8.90(s, 1H), 7.39-7.37 (m, 3H), 1.30 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 159.4, 135.8, 131.7, 130.4, 129.3, 58.0, 22.6; TLC Rf 0.39 (EtOAc/hexane, 1/9). HRMS (ESI) calcd for C11H13Cl2NOS (M+H) 278.0168; Found 278.0173.
[S(S), N(E)]-2-methyl-N-[(4-methoxyphenyl)methylene]-2-propanesulfinamide
From the combination of 0.242 g (2 mmol) (S)-(−)-2-methyl-2-propane sulfonamide, 0.408 g (3 mmol) 4-methoxybenzaldehyde, and 0.640 g (4 mmol) CuSO4, 0.458 g (96%) of the title compound was isolated as a white solid after silica gel chromatography. Analytical data: (c 1.0, CHCl3), mp 91–92°C, [(R)-(−)- lit [α]D23 −70 (c 1.0, CHCl3), mp 91–93°C]. IR (cm−1) 2982, 2959, 1588, 1567, 1508, 1456, 1305, 1256, 1078; 1H NMR (75 MHz, CDCl3) δ 8.50 (s, 1H), 7.79 (d, J = 8.5, 2H), 6.96 (d, J = 8.4, 2H), 3.86 (s, 3H), 1.19 (s, 9H); 13C NMR (300 MHz, CDCl3) δ 163.1, 161.8, 131.3, 127.3, 114.4, 57.6, 55.5, 22.6; TLC Rf 0.33 (EtOAc/hexane, 1/9); HRMS (ESI) calcd for C12H17NO2S (M+H) 240.1058. Found: 240.1051.
[S (S), N (E)]-2-methyl-N-[(3,4-methylenedioxyphenyl)methylene]-2-propanesulfinamide
From the combination of 0.242 g (2 mmol) (S)-(−)-2-methyl-2-propane sulfonamide, 0.450 g (3 mmol) 3,4-methylenedioxybenzaldehyde (piperonaldehyde), and 0.640 g (4 mmol) CuSO4, 0.445 g (88%) of the title compound was isolated as a white solid after silica gel chromatography. Analytical data: (c 1.0, CHCl3), mp 64–65°C; IR (cm−1) 2965, 2927, 2893, 1622, 1578, 1498, 1253, 1071; 1H NMR (300 MHz, CDCl3) δ 8.37 (s, 1H), 7.17 (d, J = 7.9, 2H), 6.76 (d, J = 7.9, 2H), 5.93 (s, 2H), 1.07 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 161.6, 151.4, 148.5, 129.0, 126.9, 108.3, 107.1, 101.8, 57.6, 22.5; TLC Rf 0.34 (EtOAc/hexane, 1/9); HRMS (ESI) calcd for C12H15NO3S (M+H) 254.0851. Found: 254.0847.
[S (S), N (E)]-2-methyl-N-[(4-hydroxy-3-methoxyphenyl)methylene]-2-propanesulfinamide
From the combination of 0.242 g (2 mmol) (S)-(−)-2-methyl-2-propane sulfonamide, 0.456 g (3 mmol) 4-hydroxy-3-methoxybenzaldehyde, and 0.640 g (4 mmol) CuSO4, 0.357 g (70%) of the title compound was isolated as a white solid after silica gel chromatography. Analytical data: (c 1.0, CHCl3), mp 109–110°C; IR (cm−1) 3203, 2957, 1573, 1509, 1455,1287, 1029, 865, 821; 1H NMR (75 MHz, CDCl3) δ 8.46 (s, 1H), 7.41-7.32 (m, 2H), 6.98 (d, J = 7.9, 1H), 6.21 (s, 1H), 3.94 (s, 3H), 1.25 (s, 9H); 13C NMR (300 MHz, CDCl3) δ 162.1, 150.0, 147.0, 127.1, 125.5, 114.6, 109.8, 57.7, 56.1, 22.6; TLC Rf 0.30 (EtOAc/hexane, 2/8); HRMS (ESI) calcd for C12H17NO3S (M+H) 256.1007. Found: 256.0998.
Representative procedure for the aziridination of N-sulfinyl imines
An oven-dried 25 mL round-bottomed flask was equipped with a stir bar, septum, and condenser. To this system was added (S)-N-sulfinyl imine (1.0 equiv (1.0 mmol)), cesium carbonate (4.0 equiv), THF (3.0 mL) and as a solution, sulfonium salt (3.0 equiv) in THF (2.0 mL). The solution of sulfonium salt was added in two equal portions at an interval of 6h via syringe. The system was externally heated to 80°C (sand bath). The reaction mixture was allowed to stir for a period of 12h to assure completion of reaction. After cooling to room temperature, the reaction mixture was concentrated in vacuo and immediately purified by chromatography using neutral alumina and a gradient eluent system of hexanes and EtOAc (9:1) to afford analytically pure aziridine.
1-[(1,1-dimethylethyl)sulfinyl]-2-phenylaziridine
From the combination of N-[t-butyl(S)-sulfinyl]-2-(phenyl)imine (0.209 g, 1.00 mmol), cesium carbonate (2.584 g, 8.00 mmol), and carboxymethylmethylphenylsulfonium trifluoromethanesulfonate (1.992 g, 6.00 mmol) in 2.0 mL of THF, measured was a 74:26 mixture of diastereomers as determined by 1H NMR of the crude reaction mixture. After purification by column chromatography using neutral alumina, 0.211 g (95% yield) of the title compound was obtained. Analytical data of mixture: IR (cm−1), 3067, 2982, 2868, 1605, 1497, 1073,; 1H NMR (300 MHz, CDCl3) δ 7.31-6.91 (m, 5H), 3.61 (dd, J = 4.1, J = 4.1, 1H), 3.11 (dd, J = 3.8, J = 3.8, 1H), 2.91 (d, J = 6.8, 1H), 2.44 (d, J = 7.0, 1H), 2.16 (d, J = 3.8, 1H), 2.01 (d, J = 3.8, 1H), 1.26 (s, 9H), 1.17 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 138.2, 137.5, 129.4, 129.1, 129.0, 128.3, 127.2, 126.9, 57.9, 57.4, 35.3, 32.4, 32.0, 29.2, 23.3; TLC Rf 0.39 (EtOAc/hexane, 1/4); HRMS (ESI) calcd for C12H17NOS (M+H): 224.1109. Found: 224.1108. Diagnostic analytical data of major isomer (1H NMR): δ 3.61 (dd, J = 4.1, J = 4.1, 1H), 2.44 (d, J = 7.0, 1H), 2.16 (d, J = 3.8, 1H), 1.17 (s, 9H). Diagnostic analytical data of minor isomer (1H NMR): δ 3.11 (dd, J = 3.8, J = 3.8, 1H), 2.91 (d, J = 6.8, 1H), 2.01 (d, J = 3.8, 1H), 1.26 (s, 9H).
1-[(1,1-dimethylethyl)sulfinyl]-2-(4-nitrophenyl)aziridine
From the combination of N[t-butyl-(S)-sulfinyl]-2-(4-nitrophenyl)imine (0.254 g, 1.00 mmol), cesium carbonate (2.584 g, 8.00 mmol), and carboxymethylmethylphenylsulfonium trifluoromethanesulfonate (1.992 g, 6.00 mmol) in 2.0 mL of THF, measured was an 87:13 mixture of diastereomers as determined by 1H NMR of the crude reaction mixture. After purification by column chromatography using neutral alumina, 0.260 g (97% yield) of the title compound was obtained. Analytical data of mixture: IR (cm−1) 2982, 2867, 1601, 1518, 1474, 1315, 1079; 1H NMR (300 MHz, CDCl3) δ 8.21 (d, J = 7.0, 2H), 7.44 (d, J = 7.0, 2H), 3.68 (dd, J = 3.8, J = 3.8, 1H), 3.17 (dd, J = 3.5, J = 3.5, 1H), 3.00 (d, J = 7.0, 1H), 2.50 (d, J = 7.0, 1H), 2.20 (d, J = 3.8, 1H), 2.01 (d, J = 3.8, 1H), 1.30 (s, 9H), 1.16 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 148.0, 145.8, 145.2, 128.0, 124.5, 124.2, 58.2, 57.6, 34.5, 33.0, 31.2, 29.7, 23.2, 23.1; TLC Rf 0.32 (EtOAc/hexane, 2/8); HRMS (ESI) calcd for C12H16N2O3S (M+H) 269.0960. Found: 269.0949. Diagnostic analytical data of major isomer (1H NMR): δ 3.68 (dd, J = 3.8, J = 3.8, 1H), 2.50 (d, J = 7.0, 1H), 2.20 (d, J = 3.8, 1H), 1.16 (s, 9H). Diagnostic analytical data of minor isomer (1H NMR): δ 3.17 (dd, J = 3.5, J = 3.5, 1H), 3.00 (d, J = 7.0, 1H), 2.01 (d, J = 3.8, 1H), 1.30 (s, 9H).
1-[(1,1-dimethylethyl)sulfinyl]-2-(4-chlorophenyl)aziridine
From the combination of N[t-butyl-(S)-sulfinyl]-2-(4-chlorophenyl)imine (0.243 g, 1.00 mmol), cesium carbonate (2.584 g, 8.00 mmol), and carboxymethylmethylphenylsulfonium trifluoromethanesulfonate (1.992 g, 6.00 mmol) in 2.0 mL of THF, measured was a 78:22 mixture of diastereomers as determined by 1H NMR of the crude reaction mixture. After purification by column chromatography using neutral alumina, 0.246 g (96% yield) of the title compound was obtained. Analytical data of mixture: IR (cm−1) 2981, 2903, 2871, 1493, 1455, 1080, 663; 1H NMR (300 MHz, CDCl3) δ 7.31-7.18 (m, 4H), 3.56 (dd, J = 3.8, J = 3.8, 1H), 3.05 (dd, J = 3.8, J = 3.8, 1H), 2.98 (d, J = 6.8, 1H), 2.45 (d, J = 6.8, 1H), 2.12 (d, J = 3.8, 1H), 1.95 (d, J = 3.8, 1H), 1.28 (s, 9H), 1.16 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 135.7, 128.9, 128.7, 128.1, 127.8, 57.5, 57.0, 34.2, 32.0, 30.9, 28.7, 22.8; TLC Rf 0.38 (EtOAc/hexane, 2/8); HRMS (ESI) calcd for C12H16NClOS (M+H) 258.0719. Found: 258.0717. Diagnostic analytical data of major isomer (1H NMR): δ 3.56 (dd, J = 3.8, J = 3.8, 1H), 2.45 (d, J = 6.8, 1H), 2.12 (d, J = 3.8, 1H), 1.16 (s, 9H). Diagnostic analytical data of minor isomer (1H NMR): δ 3.05 (dd, J = 3.8, J = 3.8, 1H), 2.98 (d, J = 6.8, 1H), 1.95 (d, J = 3.8, 1H), 1.28 (s, 9H).
1-[(1,1-dimethylethyl)sulfinyl]-2-(2,6-dichlorophenyl)aziridine
From the combination of N-[t-butyl-(S)-sulfinyl]-2-(2,6-dichlorophenyl)imine (0.277 g, 1.00 mmol), cesium carbonate (2.584 g, 8.00 mmol), and carboxymethylmethylphenylsulfonium trifluoromethanesulfonate (1.992 g, 6.00 mmol) in 2.0 mL of THF, measured was a 77:23 mixture of diastereomers as determined by 1H NMR of the crude reaction mixture. After purification by column chromatography using neutral alumina, 0.274 g (94% yield) of the title compound was obtained. Analytical data of mixture: IR (cm−1) 2983, 2921, 2864, 1560, 1455, 1205, 1082, 837, 729; 1H NMR (300 MHz, CDCl3) δ 7.29-7.07 (m, 3H), 4.18 (s, 1H), 3.14-3.07 (m, 2H), 2.35 (d, J = 7.1, 1H), 2.25 (d, J = 3.8, 1H), 1.25 (s, 9H), 1.18 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 137.3, 136.7, 133.4, 130.5, 130.0, 129.9, 129.5, 129.0, 57.8, 57.4, 33.5, 30.6, 30.2, 27.9, 23.2, 23.0; TLC Rf 0.37 (EtOAc/hexane, 2/8); HRMS (ESI) calcd for C12H15Cl2NOS (M+H) 292.0327. Found: 292.0330. Diagnostic analytical data of major isomer (1H NMR): δ 2.35 (d, J = 7.1, 1H), 1.18 (s, 9H). Diagnostic analytical data of minor isomer (1H NMR): δ 2.25 (d, J = 3.8, 1H), 1.25 (s, 9H).
1-[(1,1-dimethylethyl)sulfinyl]-2-(4-methoxyphenyl)aziridine
From the combination of N-[t-butyl-(S)-sulfinyl]-2-(4-methoxyphenyl)imine (0.239 g, 1.00 mmol), cesium carbonate (2.584 g, 8.00 mmol), and carboxymethylmethylphenylsulfonium trifluoromethanesulfonate (1.992 g, 6.00 mmol) in 2.0 mL of THF, measured was a 66:24 mixture of diastereomers as determined by 1H NMR of the crude reaction mixture. After purification by column chromatography using neutral alumina, 0.225 g (89% yield) of the title compound was obtained. Analytical data of mixture: IR (cm−1) 3053,2907, 1608, 1504, 1445, 1262, 1037; 1H NMR (300 MHz, CDCl3) δ 7.19 (d, J = 8.5, 2H), 6.86 (d, J = 8.5, 2H), 3.86 (s, 3H), 3.79 (s, 3H), 3.55 (dd, J = 3.8, J = 3.8, 1H), 3.05 (dd, J = 3.8, J = 3.8, 1H), 2.94 (d, J = 6.8, 1H), 2.41 (d, J = 6.8, 1H), 2.13 (d, J = 4.0, 1H), 1.96 (d, J = 3.8, 1H), 1.24 (s, 9H), 1.16 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 159.8, 129.4, 129.3, 128.3, 128.0, 114.5, 114.4, 57.9, 57.4, 55.85, 55.82, 35.0, 32.2, 31.7, 30.2, 23.39, 23.36; TLC Rf 0.34 (EtOAc/hexane, 1/9); HRMS (ESI) calcd for C13H19NO2S (M+H) 254.1215. Found: 254.1215. Diagnostic analytical data of major isomer (1H NMR): δ 3.79 (s, 3H), 3.55 (dd, J = 3.8, J = 3.8, 1H), 2.41 (d, J = 6.8, 1H), 2.13 (d, J = 4.0, 1H), 1.16 (s, 9H). Diagnostic analytical data of minor isomer (1H NMR): δ 3.86 (s, 3H), 3.05 (dd, J = 3.8, J = 3.8, 1H), 2.94 (d, J = 6.8, 1H), 1.96 (d, J = 3.8, 1H), 1.24 (s, 9H).
1-[(1,1-dimethylethyl)sulfinyl]-2-(3,4-methylenedioxyphenyl)aziridine
From the combination of N-[t-butyl-(S)-sulfinyl]-2-(4-nitrophenyl)imine (0.253 g, 1.00 mmol), cesium carbonate (2.584 g, 8.00 mmol), and carboxymethylmethylphenylsulfonium trifluoromethanesulfonate (1.992 g, 6.00 mmol) in 2.0 mL of THF, measured was a 84:16 mixture of diastereomers as determined by 1H NMR of the crude reaction mixture. After purification by column chromatography using neutral alumina, 0.225 g (85% yield) of the title compound was obtained. Analytical data of mixture: IR (cm−1) 2962, 2934, 1605, 1562, 1439, 1062; 1H NMR (300 MHz, CDCl3) δ 6.85-6.66 (m, 3H), 5.91 (dd, J = 1.6, 2H), 3.49 (dd, J = 3.8, J = 3.8, 1H), 3.01 (dd, J = 3.8, J = 3.8, 1H), 2.90 (d, J = 6.8, 1H), 2.36 (d, J = 6.8, 1H), 2.06 (d, J = 3.8, 1H), 1.90 (d, J = 3.8, 1H), 1.24 (s, 9H), 1.14 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 148.5,148.4, 147.8, 147.7, 132.1 131.4, 121.1, 120.7, 108.8, 108.7, 106.9, 106.8, 101.6, 57.9, 57.4, 35.2, 32.3, 32.0, 29.0, 23.3; TLC Rf 0.34 (EtOAc/hexane, 2/8); HRMS (ESI) calcd for C13H17NO3S (M+H) 268.1007. Found: 268.1006. Diagnostic analytical data of major isomer (1H NMR): δ 3.49 (dd, J = 3.8, J = 3.8, 1H), 2.36 (d, J = 6.8, 1H), 2.06 (d, J = 3.8, 1H), 1.14 (s, 9H). Diagnostic analytical data of minor isomer (1H NMR): δ 3.01 (dd, J = 3.8, J = 3.8, 1H), 2.90 (d, J = 6.8, 1H), 1.90 (d, J = 3.8, 1H), 1.24 (s, 9H).
1-[(1,1-dimethylethyl)sulfinyl]-2-(4-hydroxy-3-methoxyphenyl)aziridine
From the combination of N-[t-butyl-(S)-sulfinyl]-2-(4-hydroxy-3-methoxyphenyl)imine (0.127 g, 0.50 mmol), cesium carbonate (1.292 g, 4.00 mmol), and carboxymethylmethylphenylsulfonium trifluoromethanesulfonate (0.996 g, 3.00 mmol) in 2.0 mL of THF, measured was a 80:20 mixture of diastereomers as determined by 1H NMR of the crude reaction mixture. After purification by column chromatography using neutral alumina, 0.125 g (93% yield) of the title compound was obtained. Analytical data of mixture: IR (cm−1) 3201, 2953, 2853, 1574, 1462, 1267, 1236, 1078; 1H NMR (300 MHz, CDCl3) δ 6.97-6.77 (m, 3H), 3.70 (s, 3H), 3.89 (s, 3H), 3.57 (dd, J = 4.0, J = 4.0, 1H), 3.09 (dd, J = 3.8, J = 3.8, 1H), 2.98 (d, J = 6.8, 1H), 2.45 (d, J = 6.8, 1H), 2.15 (d, J = 3.8, 1H), 2.01 (d, J = 3.8, 1H), 1.30 (s, 9H), 1.20 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 148.2, 148.0, 147.7, 129.1, 128.4, 118.3, 117.7, 110.1, 109.3, 108.2, 108.1, 56.4, 55.9, 54.8, 33.7, 30.7, 30.4, 27.5, 21.8, 21.5; TLC Rf 0.29 (EtOAc/hexane, 2/8); HRMS calcd for C13H19NO3S (M+H) 270.1170. Found: 270.1170. Diagnostic analytical data of major isomer (1H NMR): δ 3.89 (s, 3H), 3.57 (dd, J = 4.0, J = 4.0, 1H), 2.45 (d, J = 6.8, 1H), 2.15 (d, J = 3.8, 1H), 1.20 (s, 9H). Diagnostic analytical data of minor isomer (1H NMR): δ 3.90 (s, 3H), 3.09 (dd, J = 3.8, J = 3.8, 1H), 2.98 (d, J = 6.8, 1H), 2.01 (d, J = 3.8, 1H), 1.30 (s, 9H).
Control Reaction: Reaction of N-Sulfinyl Imine with Trimethylsulfonium Iodide
To a suspension of NaH (3 equiv of pentane washed 60% in mineral oil) in THF (2 mL), was added trimethylsulfonium iodide (3 equiv). After 10 min at room temperature, the cloudy reaction mixture turned clear at which point in time a solution of N-sulfinyl imine (1 equiv) in THF (2 mL) was added dropwise to the mixture. The reaction mixture was externally warmed to 80°C. After stirring at this temperature for a period of 12h, the reaction mixture was allowed to cool to room temperature. Analysis of the crude reaction mixture revealed at dr of 76:24. The crude reaction mixture was isolated and purified as described above to afford the desired material in 86% yield.
Control Reaction: Reaction of N-Sulfinyl Aziridine with Thioanisole
To a 25 ml of round-bottomed flask thioanisole 0.25 mmol (0.031 g, 1 equiv), THF (5 ml), 1-[(1,1-dimethylethyl)sulfinyl]-2-(4-methoxyphenyl)aziridine 0.25 mmol (0.063 g, 1 equiv) and triethylamine 1 mmol (0.10 g, 4 equiv) was added. The reaction mixture was externally warmed to reflux (80°C) while under an argon atmosphere and allowed to react for a period of 10h. When cooled to room temperature, the reaction mixture was analyzed by 1H NMR which revealed no change in the initial aziridine to thioanisole ratio indicating no reversion of the aziridine to betaine intermediate under these reaction conditions.
Control Reaction: Reaction of N-Sulfinyl Aziridine with Thioanisole in the Presence of N-Sulfinyl Imine
To a 25 ml of round-bottomed flask thioanisole 0.25 mmol (0.031 g, 1 equiv), THF (5 ml), 1-[(1,1-dimethylethyl)sulfinyl]-2-(4-methoxyphenyl)aziridine 0.25 mmol (0.063 g, 1 equiv), [S(S), N(E)]-2-methyl-N-[(4-nitrophenyl)methylene]-2-propanesulfinamide 0.25 mmol (0.063 g, 1 equiv), and triethylamine 1 mmol (0.10 g, 4 equiv) was added. The reaction mixture was externally warmed to reflux (80°C) while under an argon atmosphere and allowed to react for a period of 10h. When cooled to room temperature, the reaction mixture was analyzed by 1H NMR which revealed no change in the initial ratio of aziridine to thioanisole to imine indicating no reversion of the aziridine to betaine intermediate under these reaction conditions. If reversion was to occur, trapping the ylide with the more reactive imine was anticipated and having no evidence of this derivative in solution, reversion under these reactions conditions is not proposed.
Acknowledgments
DCF would like to thank NSF (CHE 0514004) and the Camille and Henry Dreyfus Foundation (TH-06-008) for partial funding of this research. SRA, CJB, and AML would like to acknowledge financial support through NSF (OISE 0405210), the Alabama Space Grant Scholars Program and the University of South Alabama (UCUR and the University Honor Program).
Footnotes
Supporting Information Available: General experimental considerations, summaries and copies of NMR spectra of all compounds prepared.
References
- 1.(a) Yudin A, editor. Aziridines and Epoxides in Organic Synthesis. Wiley; New York: 2006. [Google Scholar]; (b) Trost BM, Melvin LS., Jr . Sulfur Ylides Emerging Synthetic Intermediates. Academic Press; New York: 1975. p. 31. [Google Scholar]; (c) Clark JS, editor. Nitrogen, Oxygen and Sulfur Ylides Chemistry: A Practical Approach. Oxford University Press; Oxford: 2002. [Google Scholar]
- 2.McGarrigle EM, Myers EL, Illa O, Shaw MA, Riches SL, Aggarwal VK. Chem Rev. 2007;107:5841. doi: 10.1021/cr068402y. [DOI] [PubMed] [Google Scholar]
- 3.(a) Dalko PI, Moisan L. Angew Chem, Int Ed. 2004;43:5138. doi: 10.1002/anie.200400650. [DOI] [PubMed] [Google Scholar]; (b) Dai LX, Hou XL, Zhou YG. Pure and Applied Chemistry. 1999;71:369. [Google Scholar]
- 4.(a) Blot V, Brier JF, Davoust M, Miniere S, Reboul V, Metzner P. Phosphorus, Sulfur, and Silicon and the Related Elements. 2005;180:1171. [Google Scholar]; (b) Aggarwal VK. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. II. Springer-Verlag; Heidelberg: 1999. pp. 679–693. [Google Scholar]; (c) Li AH, Dai LX, Aggarwal VK. Chem Rev. 1997;97:2341. doi: 10.1021/cr960411r. [DOI] [PubMed] [Google Scholar]; (d) Stockman RA. Ann Rep Prog Chem, Sec B. 2004;100:149. [Google Scholar]
- 5.(a) Forbes DC, Standen MC, Lewis DL. Org Lett. 2003;5:2283. doi: 10.1021/ol034612a. [DOI] [PubMed] [Google Scholar]; (b) Forbes DC, Amin SR, Bean CJ, Standen MC. J Org Chem. 2006;71:8287. doi: 10.1021/jo061370u. [DOI] [PubMed] [Google Scholar]
- 6.(a) Cheng JP, Liu B, Zhang XM. J Org Chem. 1998;63:7574. doi: 10.1021/jo981129i. [DOI] [PubMed] [Google Scholar]; (b) Johnson AW, Amel RT. Can J Chem. 1968;46:461. [Google Scholar]
- 7.(a) Chigboh K, Nadin A, Stockman RA. Synlett. 2007;18:2879. [Google Scholar]; (b) Morton D, Stockman RA. Tetrahedron. 2006;62:8869. [Google Scholar]; (c) Morton D, Pearson D, Field RA, Stockman RA. Org Lett. 2004;6:2377. doi: 10.1021/ol049252l. [DOI] [PubMed] [Google Scholar]; (d) Morton D, Pearson D, Field RA, Stockman RA. Synlett. 2003;13 1985. [Google Scholar]
- 8.The analytical data of the major diastereomer for aziridine 2d is consistent with that of previous reports (reference 7).
- 9.Aggarwal VK, Harvey JN, Richardson J. J Am Chem Soc. 2002;124:5747. doi: 10.1021/ja025633n. [DOI] [PubMed] [Google Scholar]
- 10.Edwards DR, Du J, Crudden CM. Org Lett. 2007;9:2397. doi: 10.1021/ol070875j. [DOI] [PubMed] [Google Scholar]
- 11.Robiette R. J Org Chem. 2006;71:2726. doi: 10.1021/jo052559t. [DOI] [PubMed] [Google Scholar]
- 12.Using the dissociation constants for benzoic acid, the σmeta value for OCH3 is +0.12 whereas the σpara value for OCH3 is −0.27 (−0.37 for OH). For a more extended list, see: Ritchie CD, Sager WF. Prog Phys Org Chem. 1964;2:323. and Hansch C, Leo A, Unger S, Kim KH, Nikaitani D, Liem EJ. J Med Chem. 1973;16:1207. doi: 10.1021/jm00269a003.
- 13.Ardej-Jakuhisiak M, Kawecki R, Swietlinska A. Tetrahedron Asymmetry. 2007;18:2507. [Google Scholar]
- 14.Morton D. Ph. D. Thesis. University of East Anglia; Norwich: 2005. [Google Scholar]
- 15.Plobeck N, Powell D. Tetrahedron Asymmetry. 2002;13:303. [Google Scholar]