

Abstract
When G-protein α subunits binds GTP and Mg2+, they transition from their inactive to their active conformation. This transition is accompanied by completion of the coordination shell of Mg2+ with electrons from six oxygens: two water molecules, the ß and γ phosphoryls of GTP, a helix-α1 Ser, and a switch I domain (SWI) Thr, and the repositioning of SWI and SWII domains. SWII binds and regulates effector enzymes and facilitates GTP hydrolysis by repositioning the γ-carbonyl of a Gln. Mutating the Ser generates regulatory GTPases that cannot lock Mg2+ into its place and are locked in their inactive state with dominant negative properties. Curiously, mutating the Thr appears to reduce GTP hydrolysis. The reason for this difference is not known because it is also not known why removal of the Thr should affect the overall GTPase cycle differently than removal of the Ser. Working with recombinant Gsα, we report that mutating its SWI-Thr to either Ala, Glu, Gln, or Asp results not only in diminished GTPase activity but also in spontaneous activation of the SWII domain. Upon close examination of existing α subunit crystals, we noted the oxygen of the backbone carbonyl of SWI-Thr and of the γ-carbonyl of SWII Gln to be roughly equidistant from the oxygen of the hydrolytic H2O. Our observations indicate that the Gln and Thr carbonyls play equihierarchical roles in the GTPase process and provide the mechanism that explains why mutating the Thr mimics mutating the Gln and not that of the Ser.
Keywords: ensymes/hydrolases, protein/metal ion interaction, catalytic water, heterotrimeric G proteins
The equilibrium dissociation constant of the interaction of Mg2+ with GTP is in the neighborhood of 60 μM at the physiologic pH of 7.4 (1).† This value changes more than a 1,000-fold to 10–15 nM when Mg interacts with GTP bound to an α subunit of a trimeric G protein (3). The increased affinity for Mg2+ is in good agreement with the apparent Km determined for Mg2+ supporting the intrinsic GTPase of a Giα subunit (4). The reason for the increased affinity of Mg2+ for the GTP complexed to the protein is that the protein contributes with two coordination bonds that replace weaker coordinating water oxygens. As a result, whereas Mg2+ is held by free GTP with only two coordination bonds, it is held in place by four coordination bonds in the context of protein-bound GTP (cf. figure 1 in ref. 5). Thus, upon promoting the nucleotide exchange reaction, receptors not only promote binding of GTP but also the simultaneous creation of a high affinity binding site for Mg2+. The combined binding of GTP and Mg2+ to G-protein α subunits causes the transition of α subunits from their inactive to their active conformation able to interact with effectors. In the case of the α subunit of Gs, these effectors are primarily the different adenylyl cyclase isoforms whose catalytic activity is enhanced by their interaction with the Gsα-Mg2+-GTP complex. In addition to leading to the switching into its effector-interacting conformation, occupancy of the α subunit by Mg2+-GTP also leads to activation of the subunit's second function, its intrinsic GTPase activity. The role, if any, that Mg2+ plays in this conformational transition has, to our knowledge, not been addressed.
G-protein α subunits, as well as regulatory GTPases that share the GTPase fold discovered in EF-Tu and ras (6, 7), all bind the Mg2+ of the GTP-Mg2+ complex in essentially the same fashion—i.e., with the participation of the OH groups of one Ser and one Thr: Ser17 and Thr35 in ras and Ser54 and Thr204 in Gsα. In the realm of structure activity studies, the effect of mutating the Ser has been extensively studied, owing in part to the finding that ras and ras-like GTPases with a Ser to Asp mutation have dominant negative properties (cf. ref. 8). The effect of mutating Thr35 in ras and the cognate Thr in other regulatory GTPases, including α subunits of heterotrimeric G proteins has, in contrast, received little attention. In small regulatory GTPases the only record we could find is an attempt to test the structural change of mutating Thr35 to Ser and Ala by analyzing crystals by X-ray diffraction and their structure in solution by NMR (9). No information could be collected from crystals because the Thr35 region, the so-called switch I domain (SWI) of ras, appears to be highly flexible and did not diffract constructively. In the same study, the flexibility of ras’s SWI domain was confirmed by NMR spectroscopy. In the heterotrimeric G-protein field, the sole data on record appear to be on the effect of mutating Thr182 to Ala in Gi2α (T182A-Gi2) by Nishina et al. (10). The mutant protein displayed increased rates of GDP release, and a decreased GTP hydrolysis rate, without changes in its interaction with Gβγ dimers or membrane-bound receptors. Whereas facilitated release of nucleotide might be an expected consequence of removing one of the coordination bonds that hold the Mg-nucleotide complexes in place, the mechanistic basis for a reduction in GTPase activity, which appears to be the opposite from that of mutating the Mg-coordinating Ser, was unexpected and not explored further.
Given the central role that the OH groups of the Mg-coordinating Ser and Thr play in locking Mg-GTP into place with attendant “activation” of the protein, we sought to learn more about the role of Thr204 in Gsα by mutating it to Ala or Gln and studying the properties of the mutated α subunit as well as comparing the changes to previously observed properties of T182A-Gi2α. We report that mutation of the Mg-coordinating Thr to Gln, Glu, or Ala results in a similar loss of their intrinsic GTPase activity and, remarkably, in activation of the effector regulating function without requiring the cooperation of a G-protein coupled receptor. The activation by endogenous nucleotide was observed both in adenylyl cyclase assays and in tryptic digestion studies in which the mutant proteins display trypsin resistance in the absence of exogenous addition of nonhydrolyzable GTP analogue. The loss of the Mg-coordinating Thr has thus quite different consequences than the loss of the Mg-coordinating Ser. The role of the amide carbonyl group of the Mg-coordinating Thr in activation of the GTPase activity is discussed as the likely mechanism by which intrinsic GTPase is diminished in Thr to Ala or Gln mutants, highlighting a previously unappreciated role for this Thr.
Results
To study functional consequences resulting from mutating the Mg2+-coordinating Thr in Gsα (Thr204), we used purified membrane from S49 cyc- kin- (CK7) cells (deficient in Gsα activity) mixed with wild-type and mutant Gsα subunits synthesized in vitro by using reticulocyte lysates (11), in order to analyze the Gs-regulated adenylyl cyclase system. As shown in Fig. 1, under the conditions of the assay that used 10 μg CK7 cell membrane proteins, the presence of wild-type Gsα allows measurement of low levels of adenylyl cyclase in the absence of exogenous guanosine nucleotide, followed by a 35% and 17-fold Gsα-stimulated activity increased when GTP or GTγS are added at 100 μM. In contrast, Gsα with Thr204 (T204) mutated to Ala, Glu, or Gln led to much larger adenylyl cyclase activity that was increased neither by GTP nor by GTPγS and in the presence of GTP were between 63% and 74% of the activity seen with wild-type Gsα in the presence of GTPγS. GTP increased their activities between 20% and 35%, whereas GTPγS either did not affect activity (T204A-Gsα and T204Q-Gsα) or increased it to the same level as seen with GTP (T204E-Gsα). These findings indicated that the mutant proteins are constitutively active.
Fig. 1.

Reconstitution of Gs-regulated adenylyl cyclase activities by wt-Gsα, T204A-Gsα, T204E-Gsα, and T204Q-Gsα in S49 cyc- kin- (CK7) cell membranes shows high basal activities for the mutant Gsα subunits in the absence of added nucleotides. Membranes of CK7 cells (10 μL, 5–10 μG protein) were mixed with 10 μL in vitro synthesized Gsα subunits prepared as described in Materials and Methods and allowed to stand on ice for 10 min. Adenylyl cyclase assay reagents were then added to determine basal activities (no nucleotide addition, Open Bars) and activities in the presence of 100 μM GTP (Stippled Bars) or 100 μM GTPγS (Black Bars). Incubations were for 20 min at 32 °C. Conversion of α-[32P]ATP to [32P]cAMP was determined as described in Materials and Methods.
To explore the effect of mutating Gsα Thr204 on structural changes, we synthesized wild-type and mutant Gsα subunits in vitro in the presence of [35S]methionine, incubated the products in the presence of nucleotides and Mg, and followed this by partial tryptic digestion. The products were resolved by SDS-PAGE and visualized by autoradiography. Under these conditions, wild-type G-protein α subunits in their inactive conformation are fully digested but after activation with a nonhydrolyzable GTP analogue are partially resistant to tryptic digestion owing to the conformational change that occurs in their switch II domain (12). This conformational change diminishes access of trypsin to its triple basic amino acid sequence (RRK in Gsα and RKK in α subunits of the Gi/o and transducin family). As seen in Fig. 2A, activation of wild-type Gsα with GTPγS and Mg caused the protein to become resistant to partial tryptic digestion and accumulation of a fragment that migrated with an apparent molecular mass of ca. 37 kDa. This fragment is likely to be Gsα(1–231), cleaved between the two Arg of the switch II RRK motif (12). This fragment did not accumulate if no nucleotide was added, or if GTP or GDP were added, conditions known not to allow for accumulation of an activated form of Gsα (Fig. 1). The right panel of Fig 2A shows that, in contrast to wild-type Gsα, T204A-Gsα adopts the “activated” conformation even in the presence of GDP. We refer to this conformation as constitutively active. T204Q-Gsα and T204E-Gsα behaved essentially the same as T204A-Gsα (Fig. 2B). Thus, the appearance of the structural change in the T204 mutants of Gsα responsible for adenylyl cyclase activation was independent of the presence of the effector. The remainder of the experiments were performed with the T204A and T204Q mutants of Gsα.
Fig. 2.

Incubation with trypsin reveals that Gsα with Thr204 mutated to Ala or Gln displays partial resistance to tryptic digestion in the absence of added guanine nucleotide or in the presence of GDP or GTP, as is seen in the wild-type Gsα only in the presence of nonhydrolyzable GTP analogues, in this instance GTPγS. Wild-type and Thr mutants of Gsα were synthesized in vitro in the presence of [35S]methionine and incubated for 15 min at 25 °C in 20 μL of 20 mM Hepes-NaOH, pH 7.5 and 5 mM MgCl2 with 10 μL/mL trypsin in the absence or presence of 200 μM GDP or GTP, followed by addition of 2 μL of 100 μg/mL trypsin and a second incubation for 15 min at 25 °C. The reactions were stopped by addition of 20 μL of Laemmli's sample buffer with 1% SDS. The samples were subjected to SDS-PAGE through a 4–12% polyacrylamide gel and visualized by autoradiography.
Which are the functional changes responsible for the constitutive activity of T204 mutants? To answer this question, we prepared highly purified recombinant G-protein α subunits and studied their intrinsic GTPase activities and the rates of association and dissociation of GTPγS and analyzed the GTP/GDP ratio of the guanine nucleotide after exposing the purified proteins to GTP. His-tagged G-protein α subunits were expressed in Escherichia coli (13) and purified as follows: (i) chromatography over a nitrilotriacetate-Ni column, eluting with buffer containing EDTA–i.e., removing the Ni from the column and thereby avoiding the deleterious effect that imidazole has on the stability of Gsα activity. This was followed by (ii) size-exclusion chromatography over a Superdex 200 GL column (Fig. 3 Inset). The Gα proteins obtained in this manner were more than 95% pure. The ca. 22–23 kDa contaminant remaining in the preparations was determined by mass spectrometry to be a fragment of Gsα formed after the Ni-affinity purification step. Analysis of the intrinsic GTPase activity of wild-type and mutant Gsα subunits showed the GTPase activities of the T204A and T204Q mutants of Gsα to be 1/20 and 1/10 of that of the wild-type Gsα (Table 1 and Fig. 3A).
Fig. 3.

Severely diminished GTP hydrolysis by Gsα-T204A and Gsα-T204Q (A) and T177A-*Tα (chimera 6) of Skiba et al. (14) (B). Recombinant wild-type and mutant Gsα or *Tα (5 pmol of purified protein GTPγS binding sites) were incubated with 500 nM [γ-32P]GTP (specific activity 40,000 cpm/pmol) at 32 °C for the indicated times and processed as described in Materials and Methods for determination of 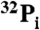 liberated. The data are representative of three different experiments with similar results. (Insets) Coomassie blue stained SDS-PAGE gels of the recombinant G-protein α subunits used for GTPase analysis.
liberated. The data are representative of three different experiments with similar results. (Insets) Coomassie blue stained SDS-PAGE gels of the recombinant G-protein α subunits used for GTPase analysis.
Table 1.
GTPase activity of mutant Gα subunits
| GTPase activity | ||
| pmol/ min /pmol Gα | % | |
| wt-Gsα | 1.34 ± 0.22 | 100 |
| T204A-Gsα | 0.07 ± 0.02 | 5 |
| T204Q-Gsα | 0.13 ± 0.01 | 10 |
| wt-*Tα | 0.091 ± 0.002 | 100 |
| T177A-*Tα | 0.007 ± 0.002 | 8 |
To establish whether the effect of mutating the Mg2+-coordinating Thr of Gsα was peculiar to Gsα, we sought to extend our observations to another G-protein α subunit in which the Mg2+-coordinating Thr of the GTPase domain was mutated to Ala or Gln. We chose *Tα, an α subunit engineered for greater stability (14) with better crystallization properties (15). *Tα has a transducin a (Tα) backbone in which 27 of its 354 amino acids were replaced with the corresponding amino acids of Gi1α. This engineered Tα has been referred to as chimera 6 (14) and *Tα (16). We use the *Tα name (17), as shown in Fig. 3B, when applied to E. coli expressed *Tα. The two-step purification protocol yielded a similarly pure Gα subunit as obtained with Gsα proteins and, as was the case with Gsα T204A and T204Q mutants, the intrinsic GTPase activity of the cognate T177A-*Tα has a severely diminished ability to hydrolyze GTP (Table 1 and Fig. 3B).
In addition, as shown in Fig. 4, the ability to retain a nonhydrolyzable GTP, GTPγS, was found to be decreased. Thus, whereas the rate of association of [35S]GTPγS was unaltered (Fig. 4A), its dissociation was markedly increased in T04A-Gsα and T204Q-Gsα compared to wild-type Gsα (Fig. 4B). In agreement with these findings, exposure of the recombinant Gsα proteins to GTP in the presence of Mg2+ led to accumulation of proteins with a higher proportion of resident guanine nucleotide being in the GTP form in the mutant Gsα when compared to wild-type Gsα. The GDP/GTP ratio on wild-type Gsα was 11.3 , whereas on T204A-Gsα and T204Q-Gsα this ratio was reduced to 0.91 and 0.54, respectively (Fig. 5); i.e., whereas only 8% of the nucleotide bound was GTP on the wild-type Gsα, in the mutants it was 52% and 65% (ratio of open bar to hatched bar in Fig. 5). Total occupancy of wild-type, T204A, and T204Q Gsα proteins by GDP + GTP (hatched bar in Fig. 5) was 9.5%, 43%, and 51%, respectively, expressed in terms of GTPγS binding capacity (black bar in Fig. 5).
Fig. 4.

Increased rates of dissociation of GTPγS from T204A-Gsα and T204Q-Gsα. (A) Association of GTPγS to wild-type Gsα, T204AGsα, and T204Q-Gsα. Five pmol of GTPγS binding sites were incubated at 32 °C with 1,000 nM [35S]GTPγS (specific activity 16,000 cpm/pmol). At the indicated times, aliquots were removed and the bound [35S]GTPγS was determined as is described in Materials and Methods. (B) Dissociation of GTPγS from wild-type and mutant Gsα. Wild-type and mutants Gsα were first incubated as in A with 1,000 nM [35S]GTPγS for 15 min at 32 °C. At time 15 min (time zero on the graph) unlabeled GTPγS was added to a final concentration of 1 mM, and 100-μL samples were collected at the indicated times in order to analyze the [35S]GTPγS remaining in a protein-bound state.
Fig. 5.

Guanine nucleotide bound at equilibrium after exposure to GTP shows a higher proportion of the resident guanine nucleotide in the GTP as opposed to GDP from mutant Gsα subunits. Wild-type Gsα, T204A-Gsα, and T204Q-Gsα (5 pmol GTPγS binding sites) were incubated at 32 °C with 1,000 nM [35S]GTPγS (specific activity 17,000 cpm/pmol), 1,000 nM [α-32P]GTP (specific activity 16,000 cpm/pmol), or 1,000 nM [γ-32P]GTP (specific activity 15,000 cpm/pmol). After 15 min, triplicate aliquots were removed and bound nucleotides analyzed by liquid scintillation counting as described in Materials and Methods. Radioactive nucleotide recovered from Gsα exposed to [γ-32P]GTP (Open Bars) is taken as the GTP bound; that recovered from Gsαs exposed to [α-32P]GTP (Hatch Bars) is the sum of GTP plus GDP. The difference between GTPγS (Black Bars) binding sites and [α-32P]GTP is assumed to be nucleotide-free Gsα.
The intrinsic GTPase activity of G-protein α subunits varies inversely with the ionic diameter of the divalent cation used to stabilize the activated conformation of the protein. This relationship is lost in the T204A mutant, in agreement with the fact that one of the constraining bonds, which completes the cation's coordination sphere, has been removed (Fig. 6).
Fig. 6.

The GTPase activity of Gsα varies inversely with the atomic radius of the divalent cation. The hydrolysis of GTP by 20 pmol each of wild-type Gsα (Black Bars) and T204A-Gsα (Open Bars and Inset) was assessed as described in Materials and Methods in the absence and presence of 10 mM of the indicated divalent cations. The data are representative of three different experiments with similar results.
Discussion
The major finding in the present studies has been that mutating the Mg-coordinating Thr204 in Gsα to either Gln or Ala results in a major decrease in the catalytic rate of the protein's GTPase activity without affecting its ability to be activated by GTP. Indeed, as shown in Fig. 1 for the T204A, T204Q, and T204D forms of Gsα, activation by GTP is facilitated when compared to wild-type Gsα. Our finding that the Thr to Ala mutant has decreased GTPase activity recapitulates the effect mutating the cognate Thr182 to Ala had on the GTPase activity of Gi2α (10). We conclude that preventing the establishment of the coordination bond with Mg2+ carries as a consequence the loss of the structural change needed to activate the G protein's GTP hydrolyzing capacity. Given that we recapitulated this result with *Tα, the Tα-Gi1 chimera, it appears that the tethering of the Thr to Mg2+ is a general requirement for activation of the GTPase activity. The “facilitation” of activation by GTP seen in the mutants compared to wild-type Gsα is likely because of the fact that exit of GDP from the wild-type protein is rate limiting and slower than the intrinsic GTPase activity, so that the GTP form cannot accumulate unless the release of GDP is facilitated, which is what G-protein coupled receptors do. As shown in Fig. 4, GTPγS dissociation is faster in Gsα subunits with Thr204 mutated to Ala or Gln, and GDP release is likely to be faster as well.
Which critical interaction(s) of Thr needed for GTPase activation are disrupted in the mutant Gsα subunits? The mechanism by which GTP is hydrolyzed by regulatory GTPases has been extensively studied. The current consensus appears to be that it is of the dissociative type (18). That is, the β-γ phosphate bond is weakened by a nucleophilic attack of H2O from the distal side, opposite to the diester bond. This nucleophilic attack subtracts charge from the β-γ ester bond so that its oxygen remains with the β phosphoryl and the γ phosphoryl leaves the reaction as a phosphate carrying the oxygen of the attacking water. It has been shown previously that the hydrolytic attack is facilitated by a Gln—227 in Gsα, 204 in Gi1α, and 200 in Tα—located at the beginning of the so-called switch II domain (SWII), which positions the attacking water, and by an Arg—201 in Gsα, 178 in Gi1α, and 174 in Tα—which further withdraws electron density from the β-γ ester bond. As is visible in crystals of both transducin α (e.g., 1GOT, 1TAG) and Gi1α (e.g., 1GP2, 1BOF) in their GDP and GDP-Mg2+ liganded, GTPase-inactive conformations, the catalytic Arg points “away” from the β-γ phosphodiester bond as does the Mg2+-coordinating Thr. Close inspection of the difference between the crystals with GDP and crystals with GTPγS or GMP-P(NH)P reveals that the attacking water is also positioned in front of the γ phosphoryl by the backbone carbonyl of a Thr on SWI. Thus, the coordinating Mg bond serves the function of pulling the backbone carbonyl of the Thr into position to, in turn, stabilize the attacking H2O allowing for better catalysis of the diester bond between the β and γ phosphates of the GTP. Thus, the process by which the Mg-GTP complex is locked into place causes not only the activation of the protein's effector regulating function but also through stabilization of a H2O molecule at ca. 3 Å from the γ phosphoryl that is to be removed from GTP. The importance of the Thr carbonyl oxygen in positioning the hydrolytic H2O for the hydrolytic reaction to proceed is further strengthened by the fact that in the crystal of the Mg2+-GDP-Al4F- complex the carbonyl oxygen of the Thr has moved to just 2.86 Å (1FQK, Fig. 7B), significantly closer than it is found when the nonhydrolyzable GTPγS is bound to the protein (3.40 Å—1TND, Fig. 7A).
Fig. 7.

The crystal structures of transducin α show that in the MgGTP activated conformation (1TND), the amide carbonyl group of the Mg-coordinating Thr177 and the γ-carbonyl group of Gln200 are equidistant from the catalytic H20 located in “front” of the γ-phosphory l of GTP. (A) In the GTPγS-Mg liganded form of the protein, SWI, SWII (to which Gln200 belongs), and Arg174 are oriented in such a way that the catalytic H2O that will capture the γPO3 split off the GTP and leave the active site as part of the newly formed Pi. The Mg-coordinating Ser43 and Thr177 are shown in gray, with the O of the OH groups shown as pink balls, and Mg is shown with four of the six coordinating oxygens: one from the brown γP, one from the brown βP, and one each from the OH groups of Ser and Thr. The coordination shell is completed by the electrons from two additional water oxygens [not shown here, but seen in figure 1 of Zurita and Birnbaumer (17)]. Mg-O distances are shown as green lines with corresponding lengths in Å. Distances between the catalytic water oxygen (w) and the Thr177 amide carbonyl O, the γ carbonyl O of Gln200, and the γ-phosphoryl O are also shown as green lines with corresponding lengths in Å. The distance between the β-γ ester bond (white) and Arg174, referred to as the “Arg finger” by Wittinghofer and colleagues (24) when GTPase activating G proteins (GAPs) are available to interact with ras-like regulatory GTPases, is also shown as a green line with the corresponding length in Å. (B) In the Mg-GDP-Al4F liganded conformation of *Tα stabilized by RGS4 (1FQK)—which is presumed to be equivalent to the transition form of the GTPase—one sees that the backbone carbonyl of Thr177 as well as the γ carbonyl of the Gln have both moved closer to the catalytic H2O oxygen. (C) In the GDP-Mg liganded conformation of Tα (1TAG) none of the critical amino acids are close enough to establish meaningful contacts; especially Thr177 cannot be pulled in to confer an effector regulating conformation to SWII. Crystal structures of Gi1α in the MgGTP conformation [1GIA with GTPγS or 1CIP with GMP-P(NH)P] and GDP conformation (1BOF, 1GP2) and of Gsα in the MgGTPγS conformation (e.g., 1AZT, 1AZS) are in agreement with structural features highlighted in this figure.
The same, roughly equal distances of the Thr amide carbonyl and Gln γ carbonyl oxygens to the hydrolytic water (3.03 and 2.93 Å, respectively) are also seen in the transition state crystal of rasGAP bound to H-ras in complex with Mg2+-GDP-Al4F- (1WQ1). This suggests that the observations made here, assigning an equihierarchical role for the two oxygens in GTP hydrolysis in heterotrimeric GTPase α subunits, is likely to be a general feature of all regulatory GTPases.
Material and Methods
Radioisotopes α-[32P]ATP, [3H]cAMP, [35S]GTPγS, [γ-[32P]]GTP, and [35S]methionine were purchased from NEN-PerkinElmer.
Trypsin and GTP were from Sigma-Aldrich; and GTPγS was from Roche. All other chemicals and biochemicals were of the highest purity commercially available and used without further purification.
CK7 cells, derivatives of S49 thymoma cells that are cyc- and kin-, i.e., deficient in Gsα activity and in cAMP-dependent protein kinase activity, were obtained from the Cell Culture Facility at University of California at San Francisco.
Standard recombinant DNA techniques were used throughout (19). The mutants of Gsα were generated in the cDNA coding for the 394-aa splice variant of the human Gsα (20). Single codon mutations were made by using the Quikchange II XL Site-Directed Mutagenesis kit from Stratagene.
In Vitro Synthesis of Gsα Subunits.
Wild-type Gsα and the indicated T204 mutants of Gsα were synthesized by using the TNT Quick Coupled Transcription/Translation System kit from Promega following the manufacturer instructions by using 1 μg of plasmid pSP72 (Promega) carrying the cDNA template. After the in vitro translation, the samples (50 μL) were diluted into 4 mL of the desired buffer and concentrated to ∼50 μL by using a 30,000 NMWL Amicon Ultra filter from Millipore centrifuging at 3,500 × g for 35 min. 35S-labeled G-protein α subunits were synthesized as above by supplementing the in vitro translation step with 10 μCi of [35S]methionine (1,000 Ci/mmol).
Bacterial Expression and Purification Gα Proteins.
Two-liter overnight cultures of competent BL21-codon plus (DE3) RIPL cells (Stratagene), transformed with the vector pHis6 carrying the cDNAs of wild-type or mutants Gsα, were induced with 100 μg/mL IPTG for 14 h at 25 °C in order to induce expression of the desired protein. The cells were harvested by centrifugation at 4,000 rpm at 4 °C for 20 min in a Sorvall SH-3000 rotor. Cells in the pellet were resuspended in 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 5 mM MgCl2, 5 mM β-mercaptoethanol (lysis buffer) and lysed by a single decompression cycle in a French Press (Thermo) pressurized with N2 to 2,250 psi. The lysate was centrifuged at 43,000 rpm in a Beckman 45Ti rotor at 4 °C for 60 min. The supernatant was adjusted to 60 mM Tris-HCl, pH 8.0, 0.5 M NaCl, 0.5% Lubrol PX (buffer A), and 20 mM imidazole and applied to a 3-mL nickel-agarose column (Qiagen). The column was washed with 10 volumes of buffer A with 20 mM imidazole and eluted with buffer A plus 50 mM EDTA. The eluted fractions containing the recombinant protein of interest (located either by activity analysis or by SDS-PAGE followed by Coomassie blue staining) were pooled, concentrated to 0.5 mL by ultrafiltration (Amicon Ultracel-10K; Millipore) and applied to a 10 × 300 mm Superdex 200 GL column (GE Healthcare) equilibrated in buffer A. Fractions with wild-type and mutant Gsα subunits were concentrated by ultrafiltration and either stored at -70 °C for up to 1 yr or used immediately in the experiments reported below. Recombinant Gsα proteins prepared in this manner had a specific [35S]GTPγS binding activity between 0.8 and 0.92 pmol GTPγS per pmol protein using BSA as standard.
Tryptic Digestions.
Gα subunits, synthesized in vitro in the presence of [35S]methionine and subjected to two rounds of 100-fold dilution with 20 mM Hepes-NaOH, pH 8.0, 5 mM MgCl2 (dilution buffer) followed by concentration by ultrafiltration, were incubated for 15 min at room temperature in a final volume of 20 μL of dilution buffer with or without 200 μM GDP, GTP, or GTPγS, followed by tryptic digestion initiated by addition of 2 μL 100 μg/mL trypsin (Sigma-Aldrich). The reactions were terminated after 15 min at room temperature by addition of 20 μL of 2× Laemmli’s sample buffer (1× Laemmli;s buffer 62,5 Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, and 5% β-mercaptoethanol). The tryptic fragments were resolved by 4–12% SDS-PAGE. The gel slabs were dried under vacuum and subjected to autoradiography at room temperature for 15–30 min by using a Kodak BioMax MR film.
Adenylyl Cyclase Assays.
Activity was measured as described by Iyengar et al. (21) with modifications. Briefly, unless specified otherwise, the reactions were performed by incubating CK7 membranes (10 μG protein) resuspended in 20 mM Hepes-NaOH (pH. 8.0) with 50 mM NaCl, 1 mM DTT, and 1 mM EDTA for 20 min at 32 °C in a final volume of 50 μL containing 25 mM Tris-HCl, pH 8.0, 1 mM [3H]cAMP (10,000 cpm), 1 mM EDTA, 6 mM MgCl2, 0.1 mM [α-32P]ATP (5 × 106 cpm), and a nucleoside triphosphate-regenerating system consisting of 20 mM creatine phosphate, 0.2 mg/mL of creatine phosphokinase (282 units/mg), and 0.02 mg/mL of myokinase (1,500 units/mg). When present, GTP and GTPγS were 100 μM. The reactions were stopped by addition of 100 μL of 40 mM ATP, 10 mM cAMP, and 1% SDS, and the [32P]cAMP formed was isolated by the method of Salomon et al. (22) as modified by Bockaert et al. (23).
GTPase Assays.
GTP hydrolysis by wild-type and mutant G-protein α subunits was determined as described by Sunyer et al. (4) with modifications. Briefly, 1–5 pmol purified proteins were incubated in the presence of 500 nM [γ-32P]GTP (specific activity 40,000 cpm/pmol) in a final volume of 100 μL containing 20 mM Tris-bis-propane (pH 6.5, or as stated), 100 mM NaCl, 1 mM EDTA, 4 mM β-mercaptoethanol, and 10 mM MgCl2. The reactions were incubated at 32 °C and stopped at the desired times by addition 0.7 mL of an ice-cold 5% (wt/vol) suspension of charcoal (Sigma) in 20 mM phosphoric acid (pH 2.1). The mixtures were allowed to stand on ice for 15 min followed by centrifugation for 15 min at 10,000 × g at 4 °C. The 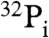 released from the 32P-labeled GTP was estimated in the supernatant by scintillation counting by using 2.8 mL of Ecolume liquid scintillation cocktail from MP Biomedical in a Tri-Carb 2800 TR liquid scintillation counter from PerkinElmer.
released from the 32P-labeled GTP was estimated in the supernatant by scintillation counting by using 2.8 mL of Ecolume liquid scintillation cocktail from MP Biomedical in a Tri-Carb 2800 TR liquid scintillation counter from PerkinElmer.
Guanine Nucleotide Binding.
Binding of guanine nucleotides to wild-type and mutant purified Gsα was determined as described in Graf et al. (5) with modifications. Briefly, purified Gsα proteins were incubated in the presence of 1,000 nM of [35S]GTPγS, 1,000 nM [α-32P]GTP, or 1,000 nM [γ-32P]GTP (specific activity 17,000 cpm/pmol), 20 mM Hepes-NaOH, pH. 8.0, 100 mM NaCl, 0.05% (wt/vol) BSA, 1 mM EDTA, 4 mM β-mercaptoethanol, and 2 mM MgCl2. Incubations were for 15 min, or as stated in the figures, at 32 °C and were stopped by addition of 1 mL ice-cold 20 mM Hepes-NaOH, pH. 8.0, 100 mM NaCl, 1 mM EDTA, 4 mM β-mercaptoethanol, and 2 mM MgCl2, followed by filtration through MF-Millipore membrane with 0.45-μm pore size. After filtration, the filters were washed two times with 7.5 mL of the same buffer, and the guanine nucleotide bound estimated by liquid scintillation counting as above.
Acknowledgments.
We thank Joseph M. Krahn from the Laboratory of Structural Biology for fruitful discussions and his suggestion to study the effect of divalent cation substitutions on the intrinsic GTPase activity of Gsα and its mutants. This work was supported by the Intramural Research Program of the National Institutes of Health (Z01-ES101643).
Footnotes
The authors declare no conflict of interest.
†Although the affinity of Mg2+ for GTPγS has apparently not been reported, that of Mg2+ for 5′AMPαS has been reported to be the same as that for 5′AMP (2). On this basis we assume that the affinity of Mg2+ for GTPγS is not significantly different from that of GTP—i.e., 60 μM.
References
- 1.Alberty R. Standard Gibbs free energy, enthalpy, and entropy changes as a function of pH and pMg for several reactions involving adenosine phosphates. J Biol Chem. 1969;244:3290–3302. [PubMed] [Google Scholar]
- 2.Sigel R, Bin S, Sigel H. Acid-base and metal ion-binding properties of adenosine 5[prime ]-[[alpha ]-thio]-monophosphate (AMPS2-) J Inorg Biochem. 1995;59:293–295. [Google Scholar]
- 3.Raw A, Coleman D, Gilman A, Sprang S. Structural and biochemical characterization of the GTPγS, GDP.Pi, and GDP-bound forms of a GTPase-deficient Gly42 -->Val mutant of Gi1α. Biochemistry. 1997;36:15660–15669. doi: 10.1021/bi971912p. [DOI] [PubMed] [Google Scholar]
- 4.Sunyer T, Codina J, Birnbaumer L. GTP hydrolysis by pure Ni, the inhibitory regulatory component of adenylyl cyclases. J Biol Chem. 1984;259:15447–15451. [PubMed] [Google Scholar]
- 5.Graf R, Mattera R, Codina J, Estes M, Birnbaumer L. A truncated recombinant alpha subunit of Gi3 with a reduced affinity for beta gamma dimers and altered guanosine 5′-3-O-(thio)triphosphate binding. J Biol Chem. 1992;267:24307–24314. [PubMed] [Google Scholar]
- 6.Jurnak F. Structure of the GDP domain of EF-Tu and location of the amino acids homologous to ras oncogene proteins. Science. 1985;230:32–36. doi: 10.1126/science.3898365. [DOI] [PubMed] [Google Scholar]
- 7.Milburn M, et al. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 8.Feig L. Handbook of Experimental Pharmacology. In: Dickey B, Birnbaumer L, editors. GTPases in Biology I. Vol 108. Heidelberg, Germany: Springer; 1993. pp. 289–298. [Google Scholar]
- 9.Spoerner M, Herrmann C, Vetter I, Kalbitzer H, Wittinghofer A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc Natl Acad Sci USA. 2001;98:4944–4949. doi: 10.1073/pnas.081441398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishina H, et al. Significance of Thr182 in the nucleotide-exchange and GTP-hydrolysis reactions of the alpha subunit of GTP-binding protein Gi2. J Biochem. 1995;118:1083–1089. doi: 10.1093/jb/118.5.1083. [DOI] [PubMed] [Google Scholar]
- 11.Olate J, Mattera R, Codina J, Birnbaumer L. Reticulocyte lysates synthesize an active alpha subunit of the stimulatory G protein Gs. J Biol Chem. 1988;263:10394–10400. [PubMed] [Google Scholar]
- 12.Hurley J, Simon M, Teplow D, Robishaw J, Gilman A. Homologies between signal transducing G proteins and ras gene products. Science. 1984;226:860–862. doi: 10.1126/science.6436980. [DOI] [PubMed] [Google Scholar]
- 13.Linder M, Ewald D, Miller R, Gilman A. Purification and characterization of Goα and three types of Giα after expression in Escherichia coli. J Biol Chem. 1990;265:8243–8251. [PubMed] [Google Scholar]
- 14.Skiba N, Bae H, Hamm H. Mapping of effector binding sites of transducin alpha-subunit using Gtα/Gi1α chimeras. J Biol Chem. 1996;271:413–424. doi: 10.1074/jbc.271.1.413. [DOI] [PubMed] [Google Scholar]
- 15.Slep K, et al. Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature. 2001;409:1071–1077. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- 16.Pereira R, Cerione R. A switch 3 point mutation in the alpha subunit of transducin yields a unique dominant-negative inhibitor. J Biol Chem. 2005;280:35696–35703. doi: 10.1074/jbc.M504935200. [DOI] [PubMed] [Google Scholar]
- 17.Zurita A, Birnbaumer L. The same mutation in Gsalpha and transducin alpha reveals behavioral differences between these highly homologous G protein alpha-subunits. Proc Natl Acad Sci USA. 2008;105:2363–2368. doi: 10.1073/pnas.0712261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maegley K, Admiral S, Heschlag D. Ras-catalyzed hydrolysis of GTP: A new perspective from model studies. Proc Natl Acad Sci USA. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maniatis T, Fritsch E, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- 20.Mattera R, et al. Identification by molecular cloning of two forms of the alpha subunit of the human liver stimulatory (Gs) regulatory component of adenylyl cyclase. FEBS Lett. 1986;206:36–42. doi: 10.1016/0014-5793(86)81336-9. [DOI] [PubMed] [Google Scholar]
- 21.Iyengar R, Abramowitz J, Bordelon-Riser M, Blume A, Birnbaumer L. Regulation of hormone-receptor coupling to adenylyl cyclases: Effects of GTP and GDP. J Biol Chem. 1980;255:10312–10321. [PubMed] [Google Scholar]
- 22.Salomon Y, Londos C, Rodbell M. A highly sensitive adenylate cyclase assay. Anal Biochem. 1974;58:541–548. doi: 10.1016/0003-2697(74)90222-x. [DOI] [PubMed] [Google Scholar]
- 23.Bockaert J, Hunzicker-Dunn M, Birnbaumer L. Hormone-stimulated desensitization of hormone-dependent adenylyl cyclase. Dual Action of luteinizing hormone on pig graafian follicle membranes. J Biol Chem. 1976;251:2653–2663. [PubMed] [Google Scholar]
- 24.Ahmadian R, Stege P, Scheffzek K, Wittinghofer A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat Struct Biol. 1997;4:686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]