

Abstract
Objectives
Reduced caveolin-1 levels in scleroderma lung fibroblasts and the lungs of bleomycin-treated mice promote collagen overexpression and lung fibrosis. We now evaluate whether caveolin-1 is deficient in leucocytes from bleomycin-treated mice and scleroderma patients and examine the consequences of this deficiency and its reversal.
Methods
Mice or cells received the caveolin-1 scaffolding domain (CSD) peptide to reverse the pathological effects of reduced caveolin-1 expression. In bleomycin-treated mice, we examined caveolin-1 levels in leucocytes and the effect of CSD peptide on leucocyte accumulation in lung tissue. To validate our results in human disease and identify caveolin-1-regulated molecular mechanisms, we isolated monocytes and neutrophils from scleroderma patients and control subjects and evaluated caveolin-1, ERK, JNK, p38, CXCR4, and MMP-9 expression/activation. We also studied these parameters in monocytes treated with cytokines or CSD peptide.
Results
Leucocyte caveolin-1 is important in lung fibrosis. In bleomycin-treated mice, caveolin-1 expression is diminished in monocytes and CSD peptide inhibits leucocyte recruitment into the lungs. These observations are relevant to human disease. Scleroderma monocytes and neutrophils contain less caveolin-1 and more activated ERK, JNK, and p38 than their normal counterparts. CSD peptide treatment reverses ERK, JNK, and p38 hyperactivation. Scleroderma monocytes also overexpress CXCR4 and MMP-9. The overexpression of CXCR4 and MMP-9 is inhibited by the CSD peptide. Cytokine treatment of normal monocytes causes adoption of the scleroderma phenotype: low caveolin-1, high CXCR4 and MMP-9, and signaling molecule hyperactivation.
Conclusions
Caveolin-1 downregulation in leucocytes contributes to fibrotic lung disease, highlighting caveolin-1 as a promising therapeutic target in scleroderma.
Keywords: caveolin, monocytes, leukocytes, scleroderma, bleomycin
INTRODUCTION
Scleroderma is a complex autoimmune disease involving fibrosis of the skin, lungs, and other organs. Indeed, lung fibrosis is the primary cause of morbidity and mortality in scleroderma. Despite the fact that it is an autoimmune disease, little is known about the potential dysregulation of signaling molecules in cells of the immune system in scleroderma. Activated alveolar macrophages and polymorphonuclear cells (PMNs) are frequently observed in bronchoalveolar lavage fluid (BALF) from patients with scleroderma lung disease (1, 2). Indeed, the presence of these cells in BALF has been used to identify patients at high risk for progression of pulmonary fibrosis. Activated alveolar macrophages and PMNs are also present at high levels in the BALF and lung tissue in an animal model of scleroderma, mice treated with bleomycin. Thus, monocytes/macrophages and PMNs are particularly reasonable candidates to be immune cells in which signaling is altered during fibrotic lung disease.
Caveolin-1 serves as a central molecule in signaling cascades because it is a scaffolding protein that binds to a variety of kinases and thereby regulates their activity. The caveolin-1 scaffolding domain (CSD) peptide, when attached to the Antennapedia internalization sequence, is able to enter cells and mimic the activity of full-length caveolin-1 (3, 4). While many papers have reported the importance of caveolin-1 in inflammation, most of these papers have focused on caveolin-1 in endothelial cells where it is important in signaling cascades involving mediators such as I-CAM, LPS, and NO (3–9). Less is known about the functions of caveolin-1 in cells of the immune system. Caveolin-1 is present in various classes of leucocytes including monocytes/macrophages, PMNs, mast cells, and lymphocytes (10–14). Caveolin-1 inhibits the expression of proinflammatory cytokines in macrophages by regulating the activation of MAP kinase family members (15). In vivo experiments demonstrate roles for caveolin-1 in neutrophil activation, transendothelial migration, and resultant lung inflammation (16); and in inflammatory cell accumulation in the lungs of bleomycin-treated mice (17).
Caveolin-1 levels are significantly reduced in the fibrotic lung tissue of bleomycin-treated mice and of scleroderma and idiopathic pulmonary fibrosis (IPF) patients, and in fibroblasts derived from patient lung tissue (17–19). The current study demonstrates that the deficit in caveolin-1 in mice and humans with fibrotic lung disease is not limited to lung fibroblasts; peripheral blood cells are also deficient in caveolin-1.
RESULTS
CSD Peptide Inhibits Monocyte and Neutrophil Accumulation in the Lungs of Bleomycin-Treated Mice
We previously showed that the CSD peptide has a general inhibitory effect on the accumulation of inflammatory cells in the lungs of bleomycin-treated mice (17). To identify specific inflammatory cell types affected by CSD peptide, we have now quantified neutrophils and monocytes/macrophages. Bleomycin causes a massive increase in the number of these cells in lung tissue which is almost completely inhibited by CSD peptide (Fig. 1), both 3 d and 7 d after bleomycin treatment. In accord with the literature, more neutrophils are present 3 days after bleomycin treatment than 7 days after, whereas more monocytes/macrophages are present 7 days after bleomycin treatment.
Figure 1. CSD peptide inhibits the recruitment of monocytes/macrophages and neutrophils into the lungs of bleomycin-treated mice.

Sections from saline-treated mice receiving the CSD peptide (CSD) or control, scrambled (Scr) peptide were harvested 7 days after treatment. Sections from bleomycin-treated mice receiving the CSD peptide or scrambled peptide were harvested 3 or 7 days after treatment. Representative images are shown of sections stained with monoclonal antibodies Gr-1 (A) to detect neutrophils and Mac-3 (C) to detect monocytes/macrophages and counterstained with the nuclear stain DAPI. Bar = 10 μm. The number of cells per mm2 stained with Gr-1 (B) or with Mac-3 (D) was determined in images from 6 mice in each category, 5 fields per mouse.
Because CSD peptide reverses the effect of bleomycin on the accumulation of monocyte/macrophages and neutrophils in lung tissue, we tested the possibility that bleomycin inhibits the expression of caveolin-1 in monocytes and neutrophils, both in the peripheral circulation and in lung tissue. Seven days after bleomycin treatment, we observed a major decrease in caveolin-1 in circulating monocytes, but not in neutrophils (Fig. 2). We also observed modest, but statistically significant, decreases in caveolin-1 expression in monocyte/macrophages and neutrophils in the lung tissue of bleomycin-treated mice (Supplemental Fig. 1).
Figure 2. Bleomycin inhibits caveolin-1 expression in circulating monocytes.

Peripheral blood was collected from mice 7 days after treatment with bleomcyin or saline vehicle. Cells were collected by cytospin and stained with DAPI, with anti-caveolin-1, and with either Mac-3 or Gr-1. Representative images are shown of staining in Mac-3-positive (A) and Gr-1-positive (B) cells. Bar = 10 μm. Caveolin-1 staining intensity in randomly chosen Mac-3-positive cells (Cav/Mono) and Gr-1-positive cells (Cav/Neut) from saline- and bleomycin-treated mice is quantified (average ± SEM) in C. In each case, the data were obtained from 20 to 60 cells from 3 to 7 mice.
Signaling Molecule Profiles in Normal and Scleroderma Monocytes and PMNs
To validate the relevance to human disease of our results in the bleomycin model, we evaluated caveolin-1 levels in monocytes and PMNs isolated from the blood of scleroderma patients and control subjects. The clinical features of these patients are summarized in Supplemental Table 1. Among the most noteworthy observations were: Duration - 7.6 ± 6.8 years; Disease – Limited Cutaneous 6 patients, Diffuse Cutaneous 11 patients, UCTD 1 patient; Organ Involvement – Lungs, 18/18; GI, 18/18; Heart, 11/18; Kidneys, 1/18.
Caveolin-1 levels were 41 % as high in scleroderma monocytes as in normal monocytes (Fig. 3AD, p<0.001). We also confirmed this deficit in caveolin-1 expression in scleroderma monocytes by immunofluorescent microscopy (Fig. 3F). When the activation of MAP kinase family members regulated by caveolin-1 was examined, we found that activated ERK, p38, and JNK were present at much higher levels in scleroderma monocytes than in control monocytes (p<0.001), even though total ERK, p38, and JNK were present at similar levels (Fig. 3AD). Two additional molecules important in inflammation, Cox-2 and the chemokine receptor CXCR4, were also present at much higher levels in scleroderma monocytes than in control monocytes (p<0.001).
Figure 3. Signaling molecules differ between normal and scleroderma leucocytes.

The expression/activation of the indicated proteins in freshly isolated leucocytes from healthy donors (Norm, Normal) and scleroderma patients (SSc, Scleroderma) was determined by Western blotting (50 μg total protein per lane for monocytes, 100 μg for PMNs) and quantified by densitometry. (A) Representative Western blot performed using monocytes enriched by adherence. (B) Representative Western blot from one of three independent experiments performed using monocytes enriched by immunodepletion rather than adherence. (C) Representative Western blot performed using PMNs. (D) Densitometric quantification (average ± SEM) of the data from seven independent experiments similar to A using seven different subjects in each category. The level of each protein in normal monocytes was set to 100 arbitrary units. (E) Densitometric quantification (average ± SEM) of the data from seven independent experiments similar to C using seven different subjects in each category. The level of each protein in normal PMNs was set to 100 arbitrary units. The expression of caveolin-1 in isolated monocytes (F) and PMNs (G) was evaluated by immunofluorescent microscopy. Bars = 5 μm. Note that in both Normal and Scleroderma monocytes, caveolin-1 is present primarily in a punctate pattern forming a ring around the perimeter of the cells and to a lesser extent over the rest of the cell. However, the ring is much brighter and more continuous in Normal monocytes than in Scleroderma monocytes. Note that in Normal PMNs, caveolin-1 is present in a punctate pattern at high levels over the nucleus and at a moderate level at the cell surface. In contrast, Scleroderma PMNs show no staining at the cell surface and only occasional punctate staining over the nucleus. (*** p < 0.001, ** p < 0.01, * p < 0.05)
To rule out the possibility that the differences between normal and scleroderma monocytes in caveolin-1, pERK, pJNK, pp38, and CXCR4 expression were induced by the adherence step used in their isolation, monocytes were also enriched by negative selection (to remove T cells, B cells, NK cells, erythrocytes, and granulocytes). This approach yielded a cell population that we found by both flow cytometry and immunocytochemistry of cells collected by cytospin to contain about 95 % Mac-1-positive monocytes, an improvement over enrichment by adherence which we found to yield about 75 % Mac-1-positive monocytes. Results obtained with negative selection (Fig. 3B) were indistinguishable from results obtained with adherence. These observations indicate that the properties we have attributed to SSc monocytes are indeed the properties of these cells and not of contaminating cells in the preparation.
To determine whether the decrease in caveolin-1 expression observed in scleroderma monocytes is also observed in other types of immune cells, we examined caveolin-1 expression in PMNs, T cells, and B cells. Scleroderma PMNs, like scleroderma monocytes, were also altered in caveolin-1, ERK, JNK, p38, and Cox-2 expression or activation. Caveolin-1 levels were 12 % as high in scleroderma PMNs as in normal PMNs (Fig. 3 CE p<0.001). This deficit in caveolin-1 expression was confirmed by immunofluorescent microscopy (Fig. 3G). Activated ERK, p38, and JNK were present at much higher levels in scleroderma PMNs than in control cells (p<0.001), even though total ERK, p38, and JNK were present at similar levels (Fig. 3 CE). Cox-2 levels were also upregulated (p<0.001). There was also a small but statistically significant decrease in caveolin-1 level in scleroderma T cells (Supplemental Fig. 2). In contrast, there was no difference in caveolin-1 expression between normal and scleroderma B cells (Supplemental Fig. 2).
Activated Normal Monocytes are Similar to Scleroderma Monocytes
To determine whether cytokines present in the blood and tissues of scleroderma patients activate monocytes by decreasing caveolin-1 expression, normal monocytes were treated with the classic proinflammatory cytokine TNFα or the pro-fibrotic cytokine TGF-β. Each treatment caused an almost complete loss of caveolin-1 (about 90 %) (Fig.4). Each treatment also increased the activation of ERK and JNK and CXCR4 expression. Only TNFα increased p38 activation; neither TGF-β nor TNFα increased Cox-2 expression (Fig. 4). Thus, much of the phenotype of scleroderma monocytes (low caveolin-1, high pERK, pJNK, pp38 and CXCR4) is mimicked by treating normal monocytes with TGF-β and/or TNFα.
Figure 4. Cytokine treatment of monocytes alters caveolin-1 and CXCR4 expression and ERK, JNK, and p38 activation.
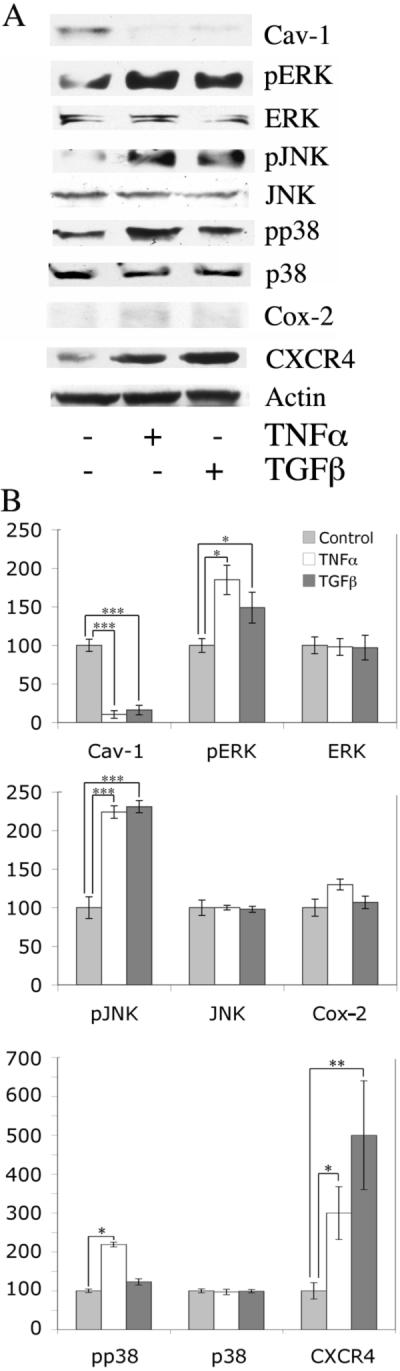
Normal PBMCs were plated in 6-well plates (2 × 107 cells/well), incubated 1 h, and washed with HBSS. Attached monocytes were further incubated for 3 h with RPMI 1640 containing 10 ng/ml of TNFα or TGF-β or no addition. The expression/activation of the indicated proteins was determined by Western blotting (50 μg total protein per lane) of cell extracts and quantified by densitometry. Actin serves as a loading control. (A) Representative blot. (B) Densitometric quantification (average ± SEM) of the data from three independent experiments similar to A using three different normal subjects. The level of each protein in untreated monocytes was set to 100 arbitrary units.
Mechanism of Signaling Molecule Activation in Scleroderma Monocytes
To validate that the hyperactivation of MAP kinase family members in scleroderma monocytes is due to decreased caveolin-1 expression/function, normal and scleroderma monocytes were treated with the CSD peptide to upregulate caveolin-1 function. CSD peptide inhibited ERK, JNK, and p38 activation (p<0.05, 0.001, 0.001 respectively), while total ERK, JNK, and p38 expression was unaffected (Fig. 5). CSD peptide treatment decreased CXCR4 expression in scleroderma monocytes, but not in normal monocytes where this molecule is expressed only at low levels. CSD peptide treatment did not affect Cox-2 expression in either normal or scleroderma monocytes, indicating that the upregulation of Cox-2 in scleroderma monocytes occurs via a caveolin-1 and MAPK-independent signaling mechanism.
Figure 5. CSD peptide inhibits ERK, JNK, and p38 activation in normal and scleroderma monocytes.
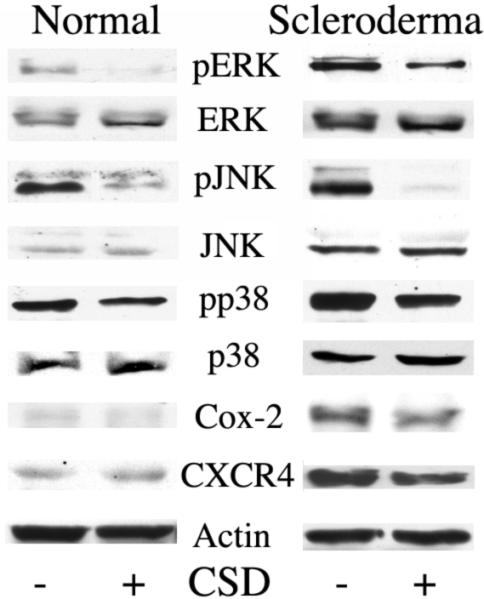
Normal and Scleroderma PBMCs were plated in 6-well plates (2 × 107 cells/well), incubated 1 h, and washed with HBSS. Attached monocytes were incubated for 3 h in RPMI 1640 containing 5 μM of the CSD peptide (+) or scrambled control peptide (−). The expression/activation of the indicated proteins was determined by Western blotting (50 μg total protein per lane). Actin serves as the loading control. Similar results were obtained in three independent experiments performed using cells isolated from different subjects.
CSD peptide regulates the inflammatory function of monocytes
To evaluate whether caveolin-1 can regulate inflammatory cell function, the effect of the CSD peptide on the release of MMP-9 by normal monocytes, normal monocytes treated with TGF-β, and scleroderma monocytes was determined. MMP-9 release was enhanced by about 50 % in both scleroderma monocytes and TGF-β-treated normal monocytes (Fig. 6). CSD peptide treatment decreased MMP-9 release to the same low level in normal monocytes, scleroderma monocytes, and TGF-β-treated normal monocytes (Fig. 6). These results demonstrate that caveolin-1 regulates the immune function of monocytes and that the deficient expression of caveolin-1 in scleroderma monocytes is directly responsible for their altered function.
Figure 6. CSD peptide inhibits MMP-9 release from TGF-β-activated Normal monocytes and Scleroderma monocytes.
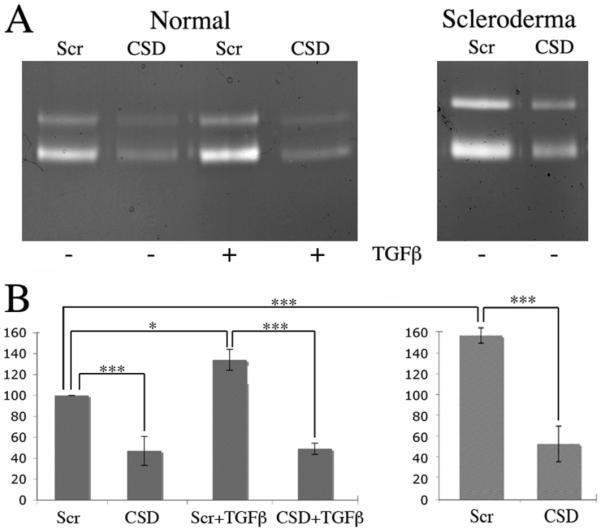
Normal and Scleroderma PBMCs were plated in 6-well plates (2 × 107 cells/well), incubated 1 h, and washed with HBSS. Attached monocytes were incubated in 6-well plates for 1 h at 37° C in RPMI 1640 supplemented with 10 ng/ml of TGF-β or no addition. The cells were then incubated an additional 3 h in fresh RPMI 1640 containing 5 μM of either CSD peptide or control, scrambled peptide. Scleroderma monocytes without TGF-β priming were also treated with CSD or control peptide for 3 h. The level of MMP-9 in equal aliquots of culture medium was determined by gelatin zymography and quantified by densitometry. (A) Representative zymogram. (B) Densitometric quantification (average ± SEM) of the data from four independent experiments similar to A using four different subjects in each category. The level of MMP-9 secreted by normal monocytes that were not treated with TGF-β and were treated with control peptide was set to 100 arbitrary units.
DISCUSSION
The reduced level of caveolin-1 present in fibroblasts from fibrotic lung tissue has been demonstrated to be directly responsible for the hyperactivation of several signaling molecules and the overexpression of collagen in vitro and to contribute to the progression of lung fibrosis in vivo (17, 18). The present study is the first demonstration that caveolin-1 levels are also reduced in leucocytes from humans and animals with fibrotic lung disease and that reduced leucocyte caveolin-1 expression regulates signaling and cell behaviour in a manner that promotes the progression of lung fibrosis.
CSD peptide has been observed to reverse the effects of caveolin-1 depletion in many systems by substituting for the full-length molecule (17, 19, 20). We demonstrate here that CSD peptide treatment inhibits the accumulation of monocytes/macrophages and neutrophils in the lung tissue of bleomycin-treated mice. Consistent with the idea that bleomycin treatment causes the loss of caveolin-1 from leucocytes and that CSD peptide treatment compensates for the loss of caveolin-1, we found lower caveolin-1 levels in lung monocytes/macrophages, lung neutrophils, and circulating monocytes from bleomycin-treated mice than from control mice. On the other hand, we did not detect reduced caveolin-1 levels in circulating neutrophils from bleomycin-treated mice. There are several reasonable explanations for the fact that CSD peptide reversed neutrophil accumulation in lung tissue in bleomycin-treated mice yet we did not detect low caveolin-1 levels in circulating neutrophils: 1) The highest level of monocyte/macrophage accumulation in lung tissue was seven days after bleomycin treatment whereas the highest level of neutrophil accumulation was at three days. We only quantified caveolin-1 at seven days. It is likely that at three days we would have detected diminished caveolin-1 in circulating neutrophils. 2) The population of neutrophils with reduced caveolin-1 may migrate extremely efficiently into the lungs whereas the population of neutrophils that remain in the circulation may not have reduced caveolin-1. 3) CSD peptide may inhibit neutrophil migration into injured lung tissue via an indirect mechanism in which it does not simply substitute for reduced caveolin-1 in circulating neutrophils. Given our results with leucocytes from scleroderma patients in which we did see reduced caveolin-1 levels in both circulating monocytes and PMNs, we favor the first explanation.
In addition to a lower level of caveolin-1 expression in scleroderma PMNs, we also observed an altered caveolin-1 distribution. In normal PMNs caveolin-1 is present in a punctate pattern at high levels over the nucleus and at a moderate level at the cell surface. In contrast, scleroderma PMNs show no staining at the cell surface and only occasional punctate staining over the nucleus. Given that caveolin-1 cycles back and forth from cell-surface caveolae to the Golgi complex via recycling endosomes (21), we speculate that caveolin-1 trafficking is altered in scleroderma PMNs.
Experiments using monocytes and PMNs isolated from scleroderma patients and control subjects have validated the importance of caveolin-1 in the control of leucocyte behaviour in human disease and allowed us to study molecular mechanisms leading to the loss of caveolin-1 and resulting from the loss of caveolin-1. We observed in both scleroderma monocytes and PMNs that caveolin-1 expression was diminished and that, as observed in other cell types in which caveolin-1 expression is diminished, several members of the MAP kinase family of signaling molecules (ERK, JNK, p38) were hyperactivated. In addition, Cox-2 expression was increased in both cell types and expression of the pro-migratory chemokine receptor CXCR4 and of the inflammatory mediator MMP-9 was increased in scleroderma monocytes. While it may seem counterintuitive for increased MMP-9 activity to be associated with a fibrotic disease, this is a standard observation (22). One could imagine several reasons why upregulating MMP expression does not reverse fibrosis. It may be that fragments of collagen generated by MMPs signal cells to produce even more collagen. Another possibility is that MMPs secreted by monocytes do not have access to or are not effective in degrading the mature, highly-crosslinked collagen that makes up most of the collagen in fibrotic lesions.
The proinflammatory cytokine TNFα and the profibrotic cytokine TGF-β play major roles in inflammation and fibrosis in scleroderma (1) and have been shown to inhibit caveolin-1 expression in other cell types (18, 19, 23). Therefore, we examined the possibility that these treatments might inhibit caveolin-1 expression in monocytes. Indeed, when normal monocytes were treated with TNFα or TGF-β, they adopted several aspects of the phenotype of scleroderma monocytes (low caveolin-1, hyperactivation of ERK and JNK, high CXCR4). However, these treatments did not up-regulate Cox-2, only TNFα activated p38, and only TGF-β upregulated MMP-9 secretion.
To determine whether the low level of caveolin-1 in scleroderma monocytes is upstream from the other observed alterations in phenotype, these cells were treated with the CSD peptide. This treatment reversed the hyperactivation of ERK, JNK, and p38 and the upregulation of CXCR4 and MMP-9 expression, but did not significantly affect Cox-2 expression. In summary, our results suggest that the TGF-β- and TNFα-rich milieu in the blood and tissues of scleroderma patients leads to the reduction of caveolin-1 expression in monocytes. In turn, the low level of caveolin-1 in scleroderma monocytes promotes the migration of these cells into target tissues by increasing CXCR4 expression and promotes tissue damage by upregulating MMP-9 expression. In contrast, the upregulation of Cox-2 in scleroderma monocytes occurs via a caveolin-1-independent mechanism. While our observation that scleroderma monocytes and PMNs overexpress Cox-2 suggests that Cox-2 inhibitors might be a useful treatment for lung inflammation/fibrosis, this approach may fail because Cox-2 inhibitors are likely to promote the proliferation and production of collagen by lung fibroblasts (24, 25).
In addition to helping create a milieu in injured lung tissue that promotes tissue damage and the activation of fibroblasts to secrete collagen and other ECM proteins, monocytes also play a direct role in fibrosis. While historically it was believed that the overexpression of collagen in fibrotic diseases occurred when resident fibroblasts became activated, it is now recognized that a subpopulation of activated, collagen-producing fibroblasts arise via the differentiation of monocytes into circulating, CD45+CXCR4+collagen+ cells known as fibrocytes that traffic into injured lung tissue where they further differentiate into fibroblasts (26–28). Given that: 1) scleroderma monocytes and fibroblasts are deficient in caveolin-1, 2) scleroderma monocytes overexpress CXCR4, 3) the overexpression of CXCR4 by scleroderma monocytes is reversed by the CSD peptide, and 4) TGFβ and TNFα inhibit caveolin-1 expression and promote CXCR4 expression in normal monocytes, it is an attractive idea that the low level of caveolin-1 in scleroderma monocytes promotes their expression of CXCR4, their differentiation into fibrocytes, the migration of fibrocytes into damaged lung tissue, and their further differentiation into fibroblasts.
Our previous studies demonstrated that manipulating caveolin-1 expression and function has profound effects on the behaviour of fibroblasts and epithelial cells in vitro and in vivo (17, 29). The current studies demonstrate that the CSD peptide can also reverse the effects of the deficient expression of caveolin-1 in monocytes and PMNs from bleomycin-treated mice and scleroderma patients. Thus, the beneficial effects of CSD peptide treatment in bleomycin-treated mice may result from a combination of its effects on both inflammation and fibrosis and may involve several cell types including fibroblasts, epithelial cells, monocytes, and PMNs. Because inflammation and fibrosis are intertwined in the bleomycin model, it would be impossible to deduce the relative contribution of each cell type to the effects of the CSD peptide without performing deletions of particular cell types (e.g. monocytes, PMNs, fibrocytes). In summary, the combined observations suggest that CSD peptide treatment may be beneficial for scleroderma and other human conditions characterized by interstitial lung disease because of its effects on several cell types.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. William Sessa (Yale University, New Haven, CT) and Dr. Pascal Bernatchez (University of British Columbia, Vancouver, Canada) for advice and discussion on the use of the CSD peptide.
This work was supported by the following grants: Scleroderma Foundation (to E. Tourkina), NIH NIAMS Grant K01 AR054143 (to E. Tourkina), NIH NHLBI Grant R01 HL73718 (to S. Hoffman), NIH NIAMS Grant P60 AR049459 (Multidisciplinary Clinical Research Center, to R. M. Silver), and NIH NCRR Construction Grant C06 RR015455.
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence on a worldwide basis to the BMJ Publishing Group Ltd to permit this article to be published in ARD and any other BMJPGL products and sublicences such use and exploit all subsidiary rights, as set out in our licence (http://ARD.bmjjournals.com/ifora/licence.pdf).
Locked and Research Funded Articles Acknowledgement This article has been accepted for publication in Annals of the Rheumatic Diseases (ARD). The definitive copyedited, typeset version, Tourkina E, Richard M, Oates J, Hofbauer, Bonner M, Gooz P, Visconti R, Zhang J, Znoyko S, Hatfield CM, Silver M, Hoffman. Caveolin-1 regulates leukocyte behaviour in fibrotic lung disease, Ann Rheum Dis 69 (6):1220-6, 2010 is available online at: http://ard.bmj.com/
REFERENCES
- 1.Atamas SP, White B. Cytokine regulation of pulmonary fibrosis in scleroderma. Cytokine Growth Factor Rev. 2003;14:537–50. doi: 10.1016/s1359-6101(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 2.Hussein MR, Hassan HI, Hofny ER, et al. Alterations of mononuclear inflammatory cells, CD4/CD8+ T cells, interleukin 1beta, and tumour necrosis factor alpha in the bronchoalveolar lavage fluid, peripheral blood, and skin of patients with systemic sclerosis. J Clin Pathol. 2005;58:178–84. doi: 10.1136/jcp.2004.019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bucci M, Gratton JP, Rudic RD, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–7. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 4.Bernatchez PN, Bauer PM, Yu J, et al. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc Natl Acad Sci U S A. 2005;102:761–6. doi: 10.1073/pnas.0407224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Millan J, Hewlett L, Glyn M, et al. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–23. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- 6.Garrean S, Gao XP, Brovkovych V, et al. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol. 2005;177:4853–60. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]
- 7.Tiruppathi C, Shimizu J, Miyawaki-Shimizu K, et al. Role of NF-kappaB-dependent caveolin-1 expression in the mechanism of increased endothelial permeability induced by lipopolysaccharide. J Biol Chem. 2008;283:4210–8. doi: 10.1074/jbc.M703153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maniatis NA, Shinin V, Schraufnagel DE, et al. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1−/− mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L865–73. doi: 10.1152/ajplung.00079.2007. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu G, Vogel SM, Schwartz DE, et al. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ Res. 2008;102:e120–31. doi: 10.1161/CIRCRESAHA.107.167486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiss AL, Turi A, Mullner N, et al. Caveolin isoforms in resident and elicited rat peritoneal macrophages. Eur J Cell Biol. 2000;79:343–9. doi: 10.1078/S0171-9335(04)70038-2. [DOI] [PubMed] [Google Scholar]
- 11.Yan SR, Fumagalli L, Berton G. Activation of SRC family kinases in human neutrophils. Evidence that p58C-FGR and p53/56LYN redistributed to a Triton X-100-insoluble cytoskeletal fraction, also enriched in the caveolar protein caveolin, display an enhanced kinase activity. FEBS Lett. 1996;380:198–203. doi: 10.1016/0014-5793(96)00029-4. [DOI] [PubMed] [Google Scholar]
- 12.Shin JS, Gao Z, Abraham SN. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 2000;289:785–8. doi: 10.1126/science.289.5480.785. [DOI] [PubMed] [Google Scholar]
- 13.Harris J, Werling D, Koss M, et al. Expression of caveolin by bovine lymphocytes and antigen-presenting cells. Immunology. 2002;105:190–5. doi: 10.1046/j.1365-2567.2002.01362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fra AM, Williamson E, Simons K, et al. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc Natl Acad Sci U S A. 1995;92:8655–9. doi: 10.1073/pnas.92.19.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang XM, Kim HP, Song R, et al. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. Am J Respir Cell Mol Biol. 2006;34:434–42. doi: 10.1165/rcmb.2005-0376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu G, Ye RD, Dinauer MC, et al. Neutrophil caveolin-1 expression contributes to mechanism of lung inflammation and injury. Am J Physiol Lung Cell Mol Physiol. 2008;294:L178–86. doi: 10.1152/ajplung.00263.2007. [DOI] [PubMed] [Google Scholar]
- 17.Tourkina E, Richard M, Gooz P, et al. Antifibrotic properties of caveolin-1 scaffolding domain in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2008;294:L843–61. doi: 10.1152/ajplung.00295.2007. [DOI] [PubMed] [Google Scholar]
- 18.Wang XM, Zhang Y, Kim HP, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203:2895–906. doi: 10.1084/jem.20061536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galdo FD, Sotgia F, de Almeida CJ, et al. Decreased expression of caveolin 1 in patients with systemic sclerosis: Crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 2008;58:2854–65. doi: 10.1002/art.23791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chidlow JH, Jr, Greer JJ, Anthoni C, et al. Endothelial caveolin-1 regulates pathologic angiogenesis in a mouse model of colitis. Gastroenterology. 2009;136:575–84. doi: 10.1053/j.gastro.2008.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Head BP, Insel PA. Do caveolins regulate cells by actions outside of caveolae? Trends Cell Biol. 2007;17:51–7. doi: 10.1016/j.tcb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28:12–24. doi: 10.1165/rcmb.2002-0166TR. [DOI] [PubMed] [Google Scholar]
- 23.Fakhrzadeh L, Laskin JD, Laskin DL. Regulation of caveolin-1 expression, nitric oxide production and tissue injury by tumor necrosis factor-alpha following ozone inhalation. Toxicol Appl Pharmacol. 2008;227:380–9. doi: 10.1016/j.taap.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keerthisingam CB, Jenkins RG, Harrison NK, et al. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol. 2001;158:1411–22. doi: 10.1016/s0002-9440(10)64092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lovgren AK, Jania LA, Hartney JM, et al. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2006;291:L144–56. doi: 10.1152/ajplung.00492.2005. [DOI] [PubMed] [Google Scholar]
- 26.Moore BB, Kolodsick JE, Thannickal VJ, et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–84. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips RJ, Burdick MD, Hong K, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–46. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. 2006;8:145–50. doi: 10.1007/s11926-006-0055-x. [DOI] [PubMed] [Google Scholar]
- 29.Tourkina E, Gooz P, Pannu J, et al. Opposing effects of protein kinase Calpha and protein kinase Cepsilon on collagen expression by human lung fibroblasts are mediated via MEK/ERK and caveolin-1 signaling. J Biol Chem. 2005;280:13879–87. doi: 10.1074/jbc.M412551200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.