

Abstract
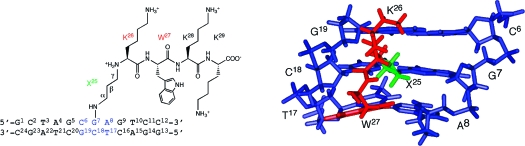
DNA−protein conjugates are potentially repaired via proteolytic digestion to DNA−peptide conjugates. The latter have been modeled with the amino-terminal lysine of the peptide KWKK conjugated via a trimethylene linkage to the N2-dG amine positioned in 5′-d(GCTAGCXAGTCC)-3′·5′-d(GGACTCGCTAGC)-3′ (X = N2-dG−trimethylene link−KWKK). This linkage is a surrogate for the reversible linkage formed by the γ-OH-1,N2-propanodeoxyguanosine (γ-OH-PdG) adduct. This conjugated KWKK stabilizes the DNA. Amino acids K26, W27, K28, and K29 are in the minor groove. The W27 indolyl group does not intercalate into the DNA. The G7N2 amine and the K26 N-terminal amine nitrogens are in the trans configuration with respect to the Cα or Cγ of the trimethylene tether, respectively. The structure of this DNA−KWKK conjugate is discussed in the context of its biological processing.
Cellular exposures to ionizing radiation, ultraviolet light, and a variety of chemicals and metals result in the formation of DNA−protein conjugates (DPCs).1 For a review, see Barker et al. (1). These bulky lesions are anticipated to interfere with both DNA replication and transcription, but there has been a paucity of data providing insights into the mechanisms of cellular tolerance and repair of these lesions. However, inhibition of nuclear proteosomal protein degradation reduces the repair of DPCs (2). Additionally, the proteolytically active 20S core of the 26S proteasome localizes to the nucleus (3,4), and inhibition of proteasome function with lactacystin results in the inhibition of repair of formaldehyde-induced DPCs, in normal, XP-A, and XP-F fibroblasts (2). These data are consistent with a scenario in which DPCs are targeted for proteolytic degradation. Interestingly, when topoisomerase I is conjugated to DNA in the presence of camptothecin, proteolysis occurs in a ubiquitin-dependent fashion, giving rise to a ladder of DNA−polypeptide products (5). Thus, an attractive mechanism for the repair of other DPCs involves the formation of DNA−peptide conjugates, as substrates for nucleotide excision repair (6−8).
One route to the preparation of site-specific DNA−peptide conjugates to probe the biological processing of these lesions was discovered when it was observed that peptides containing a single aromatic side chain flanked by basic residues, such as Lys-Trp-Lys (KWK), incise the phosphodiester backbones of DNA duplexes containing abasic sites. The reaction proceeds via a β-elimination mechanism involving nucleophilic attack of an amine at the C1′ carbon at the aldehyde of the abasic site (9−11). The mechanism involves formation of an imine (Schiff base) intermediate between the polypeptide and DNA that can be isolated upon reduction by sodium borohydride (12). Such studies have shown that the N-terminal α-amino groups exhibit lower pKa values and react with greater efficiencies than do the ε-amino groups of lysines (13). Similarly, chemical reducing agents have been used to probe the biochemical mechanisms of DNA glycosylase/AP lyases, such as the T4 pyrimidine dimer glycosylase (T4-pdg) (12,14,15). In that instance, the α-amino group of the N-terminal threonine catalyzed the β-elimination (12). The recognition specificity of the KWK peptide for abasic sites was attributed to the tryptophan moiety, which presumably contributed to stacking between bases adjacent to the abasic site. This model was supported by studies that demonstrated KWK binding to depurinated DNA was accompanied by an increase in fluorescence quenching (10).
More recently, DNA−peptide conjugates have been prepared from duplexes containing site-specific γ-hydroxy-1,N2-propano-2′-deoxyguanosine (γ-OH-PdG) lesions (16). When 2′-deoxycytosine is positioned opposite γ-OH-PdG in duplex DNA (17−20), the 1,N2-PdG ring opens to the N2-propylaldehyde (21−23). The aldehyde reacts with the primary amines in proteins and peptides to yield imine conjugates that can be isolated following chemical reduction (16). A similar reaction chemistry has been used to covalently link peptides to γ-OH-1,N2-propano-2′-deoxyadenosine (γ-OH-PdA) (24).
The biological processing of site-specific DNA−peptide conjugates has been reviewed (25). The mutagenic potentials of DNA−peptide conjugates generated by conjugating KWKK to γ-OH-PdG or γ-OH-PdA have been evaluated in a single-stranded pMS2 shuttle vector replicated in COS-7 cells (26). Replication bypass of KWKK conjugated to the γ-OH-PdG lesion resulted in mutations at an overall frequency of ∼8.4%, while the γ-OH-PdA-mediated KWKK conjugate induced mutations at only ∼0.4%. Thus, these conjugates are mutagenic, and their potential to cause mutations depends on the site of peptide attachment (26). The abilities of γ-OH-PdG or γ-OH-PdA DNA−KWKK conjugates to be bypassed by both human and Escherichia coli polymerases in vitro have been investigated (24). Human DNA polymerase κ catalyzed efficient, error-free bypass of peptides conjugated via γ-OH-PdG. These lesions blocked E. coli polymerases II, III, and V but not polymerase IV. Cells deficient in polymerase IV were extremely inefficient in replicating these lesions (27).
Presently, we have conjugated the amino-terminal lysine of the peptide KWKK via a trimethylene linkage to the N2-dG exocyclic amine site-specifically positioned in 5′-d(GCTAGCXAGTCC)-3′·5′-d(GGACTCGCTAGC)-3′ (X = N2-dG−trimethylene linkage−KWKK conjugate) (Scheme 1). The fully reduced trimethylene linkage provides a surrogate for the reversible linkage formed by the γ-OH-PdG adduct. We report that this conjugated peptide stabilizes the DNA duplex and that the trimethylene X25 and amino acids K26, W27, K28, and K29 orient in the minor groove. The tryptophan indolyl group does not intercalate into the duplex. The G7N2 amine nitrogen and the K26 N-terminal amine nitrogen are in the trans configuration with respect to the Cα or Cγ of the trimethylene tether, respectively.
Scheme 1. Numbering Scheme of the DNA−KWKK Conjugate, in Which the Trimethylene Tether Was Named as Residue X25.

Materials and Methods
DNA Containing the DNA−KWKK Conjugate
The site-specific DNA−KWKK conjugate was synthesized in 5′-d(GCTAGCXAGTCC)-3′ (X = N2-dG−KWKK) and purified as reported (26). The complementary strand 5′-d(GGACTCGCTAGC)-3′ was synthesized and purified by anion-exchange chromatography by the Midland Certified Reagent Co. (Midland, TX). Both oligodeoxynucleotides were characterized by MALDI-TOF mass spectrometry; their purities were assessed by capillary gel electrophoresis and HPLC. Both strands were desalted using Sephadex G-25 (Sigma-Aldrich, St. Louis, MO). The oligodeoxynucleotide concentrations were determined by UV absorption at 260 nm, using an extinction coefficient of 1.1 × 105 L·mol−1·cm−1. The DNA−KWKK conjugate was annealed with the complementary strand in 10 mM NaH2PO4, 100 mM NaCl, and 50 μM Na2EDTA (pH 7.0). The solution was heated to 95 °C for 10 min and then cooled to room temperature. The oligodeoxynucleotide duplex was purified using hydroxylapatite chromatography with a gradient from 10 to 200 mM NaH2PO4 in 100 mM NaCl and 50 μM Na2EDTA (pH 7.0) and desalted using Sephadex G-25.
Thermal Stability of the DNA−KWKK Conjugate
The stability of the oligodeoxynucleotide duplex was measured in 10 mM NaH2PO4, 100 mM NaCl, and 50 μM Na2EDTA. The pH was adjusted as necessary with either dilute HCl or NaOH solutions. The sample was at 10 μM strand concentration. The melting transition was monitored using UV spectroscopy at 260 nm, and the Tm was calculated by the differentiation of the absorbance curve from 30 to 85 °C.
NMR
Experiments were performed on a Bruker Avance 800 spectrometer equipped with a cryogenic probe. The sample was at 0.8 mM strand concentration in 280 μL of 10 mM NaH2PO4, 100 mM NaCl, and 50 μM Na2EDTA (pH 7.0). For monitoring nonexchangeable protons, it was exchanged with D2O and suspended in 280 μL of 99.99% D2O. For observation of exchangeable protons the sample was dissolved in 280 μL of 9:1 H2O:D2O (v/v). The pH of the sample was adjusted as necessary with dilute DCl or NaOD solutions. The temperature was 25 °C for observation of nonexchangeable protons and 5 °C for observation of exchangeable protons. 1H chemical shifts were referenced to water. Data were processed using FELIX 2000 (Accelrys Inc., San Diego, CA) on Silicon Graphics workstations (Silicon Graphics Inc., Mountain View, CA). For all experiments, a relaxation delay of 1.5 s was used. 2D spectra were recorded with 512 real data in the t2 dimension and 2048 real data in the t1 dimension. For assignment of exchangeable protons, NOESY experiments used the watergate sequence for solvent suppression (28). The mixing time was 250 ms, and the spectrum was zero-filled to 1024 × 512 matrix. For assignment of nonexchangeable protons and the derivation of distance restraints, NOESY experiments used TPPI quadrature detection with mixing times of 60, 150, 200, and 250 ms, and the spectra were zero-filled during processing to create a matrix of 1024 × 1024 real points. DQF-COSY experiments were performed with TPPI quadrature detection and presaturation of the residual water during the relaxation delay; the spectra were recorded with 256 real data in the t2 dimension and 2048 real data in the t1 dimension and were zero-filled during processing to create a matrix of 1024 × 512 real points. 31P experiments were carried out on a Bruker Avance 600 spectrometer. Chemical shifts were not calibrated.
Distance and Torsion Angle Restraints
NOE-derived distances from cross-peak volumes measured at mixing 250 ms were calculated using MARDIGRAS (29,30). Isotropic correlation times of 2, 3, and 4 ns were used. Integrated cross-peak volumes were normalized from the intensities of protons with fixed intranuclear distances (i.e., cytosine H5−H6 and thymine CH3−H6 distances). The volume error was set to one-half the volume of the smallest peak. The RANDMARDI algorithm carried out 50 iterations for each set of data, randomizing peak volumes within limits specified by the input noise level (31). The standard deviation in particular distances served as the error bounds for the distances. Distance restraints were divided into classes and weighted according to the errors assessed in their measurements. Class 1, class 2, class 3, and class 4 restraints were calculated from completely resolved, slightly overlapped, medium overlapped, or heavily overlapped cross-peaks, respectively, that were at least 0.5 ppm from the water resonance or the diagonal line of the spectrum. Class 5 distances were calculated from all of the other cross-peaks. Empirical restraints preserved Watson−Crick hydrogen bonding and prevented propeller twisting between base pairs (32). NOEs that did not have a distance calculated by MARDIGRAS were assigned an estimated distance based on the peak intensities. Deoxyribose conformation was determined with the 3J coupling constants derived from DQF-COSY spectra (33). The backbone and sugar pucker torsion angle restraints were assigned from empirical data derived from B-DNA (34). The range of the dihedral angle restraints was ±30°.
Structural Refinement
The DNA−KWKK conjugates were constructed by bonding the trimethylene linkage Cα to G7N2 and Cγ to the N-terminal amine of K26, respectively, using Insight II (Accelrys Inc., San Diego, CA). Each nucleotide, amino acid, and the linker were considered as single residues. To provide the starting structures, duplexes were constructed with both A- and B-DNA conformations and were energy minimized by the conjugate gradient algorithm for 200 iterations without experimental restraints. Partial charges at the peptide-conjugated nucleotide were calculated using the SCF approach, utilizing the B3LYP/6-31G* function group with the program GAUSSIAN (35) with neutral total charge (Figure S1 in the Supporting Information). The rMD calculations were conducted with the AMBER parm99 force field (36). The generalized Born (GB) model (37,38) with parameters developed by Tsui and Case (39) was used for implicit water simulation. The cutoff radius for nonbonding interactions was 18 Å. The restraint energy function contained terms describing distance and torsion angle restraints, both in the form of square well potentials. Bond lengths involving hydrogens were fixed with the SHAKE algorithm (40).
First, a 1000-step energy minimization was performed with an iteration time of 1 fs without experimental restraints, followed by a 100000-iteration simulated annealing protocol with an integrator time step of 1 fs. The system was first heated to 600 K in 5000 iterations and kept at 600 K for 5000 iterations and then cooled to 100 K with a time constant of 8.0 ps in 80000 iterations; a final cooling procedure was applied to relax the system to 0 K with a time constant of 1.0 ps in 10000 iterations. Ten structures were converged initially from A- and B-type DNAs (five each). A force constant of 32.0 kcal·mol−1·Å−2 was used for all distance restraints. The lower and upper bounds for NOE restraints were obtained from MARDIGRAS calculations. The lower and upper bounds for base pair restraints were assigned empirically. The parabolic potential wells were set to 0.5 Å lower or higher than the lower and upper bounds, respectively. A force constant of 32.0 kcal·mol−1·rad−1 was used for all torsion angle restraints. The lower and upper bounds of torsion angle restraints were assigned empirically. The parabolic potential wells were set to 1.0° lower or higher than the lower and upper bounds, respectively. Convergence was assessed for structures having the lowest number of deviations from the experimental distance and dihedral restraints, lowest van der Waals energy, and the lowest overall energy. The structures were energy minimized for 250 iterations without restraints to obtain final structures.
CORMA (41) was utilized to estimate the NOE intensities from the structures refined from rMD calculations. Input volumes (intensities) were normalized from averaging the intensities of all protons. Random noise was added to all intensities to simulate spectral noise. An isotropic correlation time (τc) of 3 ns was used. The rotation of thymine CH3 groups was modeled using the three-jump site model (42). The sixth root residual (R1x) factor (43) was calculated for each structure. Helicoidal analyses were carried out with 3DNA (44).
Results
Characterization of the Conjugated DNA Duplex
Analysis of the DNA−KWKK conjugate by MALDI-TOF mass spectrometry verified the presence of the conjugated peptide. Further analysis by capillary gel electrophoresis and C-18 HPLC indicated that the duplex was of sufficient purity for spectroscopic analyses. To monitor the thermodynamic effect of linking the KWKK peptide to the γ-OH-PdG-modified duplex, thermal melting Tm values were determined for an unmodified control duplex, the γ-OH-PdG-modified duplex, and the duplex with the KWKK peptide conjugated with the γ-OH-PdG. Tm values for the peptide conjugate were determined to be 63, 63, and 58 °C at pH values of 5.3, 7.0, and 8.9, respectively. At neutral pH, linkage of the KWKK tetrapeptide to the DNA increased the Tm by 15 °C relative to the γ-OH-PdG lesion alone, which exhibited a Tm of 48 °C. At neutral pH, linkage of the KWKK tetrapeptide to the DNA also increased the Tm compared to the unmodified DNA, which exhibited a Tm of 60 °C. NMR analysis was conducted at pH values of 5.3, 7.0, and 8.9. At neutral pH, the spectroscopic data suggested the presence of multiple conformations in equilibrium. The NMR resonances at pH 5.3 matched the major set of resonances observed at pH 7.0. Therefore, NMR data used for structural refinement were collected at pH 5.3. Figure 1 displays 1H NMR spectra of the DNA−peptide conjugate at different temperatures. As the temperature increased, the base imino resonances broadened. The imino resonances of the terminal base pairs disappeared first and then the imino resonances from the penultimate base pairs. The imino resonances of nucleotides G7, T17, and G19 were the last to broaden.
Figure 1.

1H NMR at different temperatures showing stabilities of the base pairs. W27 N1H was suppressed by the water suppression pulse, and the resonance was weak.
Assignments of the Nucleotide Protons
The nonexchangeable protons of nucleotides were assigned based upon the sequential connectivity of the base proton H6 or H8 dipolar couplings with H1′ sugar protons (45,46). A complete sequential NOESY connectivity was observed for both the KWKK-conjugate-containing and complementary strands (Figure S2 in the Supporting Information). Notably, the C6 H1′ → G7 H8 NOE was weaker than the other 5′-deoxyribose H1′ → 3′-purine H8 NOEs, suggesting a structural perturbation between nucleotides C6 and G7. Four additional cross-peaks in this region of the NOESY spectrum were assigned to the NOEs of tryptophan indolyl protons at W27 with nucleotide H1′ protons. Using standard strategies, the resonances of the H2′, H2′′, H3′, H4′, H5′, and H5′′ nucleoside protons were assigned, although it was not possible to assign all of the H5′, H5′′ protons unequivocally. The assignments are collected in Table S1 of the Supporting Information.
The resonances of base imino protons were assigned based on their sequential connectivities in NOESY spectra and were supported by NOEs to amino protons of Watson−Crick base pairs (Figure S3 in the Supporting Information) (47). Except for the terminal base pairs, NOE cross-peaks arising from Watson−Crick hydrogen bonding were observed, indicating the duplex conserved Watson−Crick base pairing. The DNA−KWKK conjugate exhibited a strong G7 N1H → G7N2H NOE, suggesting that the guanine amino proton was shielded from exchange with water. The G7N2H amino proton also exhibited NOEs to the trimethylene linkage protons, suggesting G7N2H was proximate to the trimethylene tether and peptide chain.
Assignments of Trimethylene Protons
The resonances of trimethylene (X25) protons were assigned based on their NOE correlations (Figure 2). An extra resonance appeared at 1.35 ppm and exhibited four strong NOE cross-peaks at δ 2.72, 2.56, 2.24, and 1.96 ppm. The resonance at δ 1.35 ppm was assigned to the geminal X25 Hβ protons. The NOE between resonances at δ 2.72 and δ 2.56 ppm was strong, and these resonances exhibited medium-intensity NOEs with resonances at δ 2.24 and δ 1.96 ppm. The NOE between the latter two resonances also was strong. The δ 2.72 and δ 2.56 ppm resonances exhibited weak NOEs to A8 H2 and were assigned to the geminal X25 Hα protons. The δ 2.24 and δ 1.96 ppm resonances exhibited NOEs to K26 Hα and were assigned to the geminal X25 Hγ protons. Since the resonances arising from the geminal Hβ protons could not be resolved, the X25 geminal Hα and Hγ protons were assigned by NOE correlations with A8 H2 and K26 Hα, respectively. The X25 Hα resonance at δ 2.56 ppm exhibited a stronger NOE to A8 H2 than did the resonance at δ 2.72 ppm, and the former was assigned as X25 Hα1, while the latter was assigned as X25 Hα2. Similarly, the X25 Hγ resonance at δ 2.24 ppm exhibited a stronger NOE to K26 Hα than did the resonance at δ 1.96 ppm, and the former was assigned as X25 Hγ1, and the latter was assigned to X25 Hγ2. The assignments are collected in Table S2 of the Supporting Information.
Figure 2.

Assignment of trimethylene linkage protons. The X25 Hα protons exhibited NOEs to A8 H2, whereas the X25 Hγ protons displayed NOEs to K26 Hα. Both exhibited strong NOEs to the geminal X25 Hβ protons. The NOESY data were collected at 250 ms mixing time.
Assignments of Amino Acid Protons
The assignments of the tryptophan W27 nonexchangeable protons are shown in Figure 3A. The geminal Hβ resonances exhibited strong NOEs to Hα. The intensity of the Hα → Hβ NOE and the 3J-coupling constants of the Hα → Hβ correlation observed in the DQF-COSY spectrum were used to determine the configurations of the Hβ protons. The Hα → Hβ1 NOE correlation was weaker and the 3J-coupling constant was greater, suggesting Hα and Hβ1 were in the trans configuration, whereas Hα and Hβ2 were in the cis configuration. The Hα and Hβ protons exhibited NOEs to the indolyl protons H2 and H4. The H4 protons exhibited strong NOE and J-couplings with H5, whereas H2 did not have interactions with H5. The NOESY and COSY H5 → H6 and H6 → H7 correlations determined the assignments of the H6 and H7 resonances. The indolyl imino proton appeared at 10.5 ppm (Figure 1). It had strong NOEs to H2 and H7. The assignments are collected in Table S2 of the Supporting Information.
Figure 3.

Assignment of KWKK protons. (A) Trp protons. W27 H4 exhibited strong NOEs to Hα, Hβ, and H5, whereas W27 H2 did not exhibit NOEs to the aromatic protons. (B) Lys protons. The assignment of K26 is shown as an example. The NOESY data were collected at 60 ms mixing time.
Three sets of lysine Hα → Hβ NOE correlations were observed in the NOESY spectrum (Figure 3B). K26 Hα exhibited NOE correlations with the X25 Hγ protons. The lysine K28 proton resonances were sharp, while the K29 proton resonances were broad. The K28 and K29 Hα proton resonances were distinguished on the basis of NOEs to the amide protons (Figure 4). K28 Hα had NOE correlations with W27 NαH and K28 NαH, whereas K29 Hα exhibited NOEs to K28 NαH and K29 NαH. The assignments of the remaining lysine proton resonances were based on the sequential connectivity from Hα → Hβs → Hγs → Hδs → Hεs in NOESY and COSY spectra. Figure 3B demonstrates the assignments for the K26 proton resonances. The geminal K26 Hγ protons exhibited strong NOEs to the Hβ and Hδ protons, and a strong Hε → Hδ NOE was used to assign the Hε proton resonances. As for the W27 Hβ protons, the geminal protons were assigned based on NOE intensities and 3J-coupling constants.
Figure 4.

Assignments of peptide amide protons on the basis of their NOEs with Hα and Hβ. The NOESY data were collected at 250 ms mixing time.
The peptide amide protons exhibited NHi → Hαi−1 and NHi → Hαi NOEs (subscript i represents residue number) to the Hα protons (Figure 4). In addition, the amide protons also exhibited NOEs to the Hβ and Hγ protons. For structure refinement distance restraints were estimated based on the intensities of these NOEs. The N-terminal amine proton at K26 was not observed, presumably due to its exchange with water.
Chemical Shift Perturbations
The chemical shifts of the base aromatic H6/H8 protons and deoxyribose H1′ protons were compared with the corresponding unmodified duplex (Figure S4 in the Supporting Information). Small chemical shift perturbations were observed at the peptide-conjugate region of the duplex. The chemical shifts of the amino acid nonexchangeable protons were also compared with those of the free KWKK peptide, which was assumed to exist as a random coil. All Hα protons had shifts ≥0.18 ppm when the peptide was conjugated with DNA. The chemical shift perturbations of the Hβ protons upon conjugation to the DNA were notable. The K26 and K28 side chain protons also shifted significantly. K29 exhibited broad resonances. The side chain resonances were shifted <0.1 ppm. The remarkable chemical shift differences for K28 as compared to the free KWKK peptide suggested K28 adopted an ordered conformation. However, no interresidue NOE was found for the nonexchangeable protons of either K28 or K29. The chemical shift perturbations of the W27 indolyl resonances were ∼0.1 ppm.
Deoxyribose and Phosphodiester Backbone Angle Conformations
Evaluation of the DQF-COSY spectrum revealed that for all nucleotides the pseudorotation of the deoxyribose rings was either C1′-exo or C2′-endo. The 31P resonances exhibited a small chemical shift dispersion, centered in the spectral region characteristic of B-form DNA duplexes. The individual resonances were not assigned.
Location of the Trimethylene Linkage and Amino Acids
NOE cross-peaks associated with the trimethylene linkage and the amino acid protons are shown in Figure 5. The trimethylene (X25) and amino acids K26 and W27 exhibited NOEs only with the minor groove protons A8 H2, G9 H1′, G9 H4′, and C18 H1′. No interresidue NOEs were observed for K28 and K29. Table 1 lists the NOEs between the nucleotides, trimethylene linkage, and the amino acids that were converted to distance restraints used for the structure refinement.
Figure 5.

NOEs between nucleotides, the trimethylene linkage, and the KWKK amino acids indicating the minor groove orientation of the peptide.
Table 1. Interresidue NOEs between Nucleotide, Trimethylene, and Amino Acids Used for Structure Refinement.
| atom | NOEs |
|---|---|
| A8 H2 | W27 H7 (s); W27 H6 (w); X25 Hα1 (m); X25 Hα2 (m); X25 Hβ (m); X25 Hγ2 (w) |
| A8 H1′ | X25 Hα1 (w); X25 Hα2 (w); X25 Hβ (w); W27 H2 (m) |
| G9 H5′ | W27 H2 (m); W27 H4 (w); W27 H5 (w) |
| G19 H1′ | X25 Hβ (w) |
| G19 H4′ | K26 Hβ1 (m); K26 Hβ2 (m); K26 Hγ (m); K26 Hδ (w) |
| K26 Hα | X25 Hβ (m); X25 Hγ1 (m); X25 Hγ2 (s) |
| K26 Hβ2 | X25 Hγ2 (w) |
| W27 H2 | G9 H4′ (m); G9 H5′′ (m); X25 Hβ (w); X25 Hγ2 (w) |
| W27 H6 | C18 H1′ (w); C18 H4′ (m); C18 H5′ (w) |
| W27 H7 | G9 H1′ (m); C18 H1′ (m); X25 Hα1 (w); X25 Hα2 (w); X25 Hβ (w) |
Structure Refinement
A total of 447 distance restraints, including 258 intranucleotide and 189 internucleotide restraints, were calculated from the intensities of NOE cross-peaks using MARDIGRAS. These included 34 restraints between nucleotides, the trimethylene linkage, and the amino acids. For the adenine H2 protons, which exhibit long T1 relaxation times, the calculated distances, especially involving A8 H2, were examined. These distances were close to or shorter than the lower bound of the corresponding NOE distance restraints. A total of 31 distance restraints were derived from the NOE intensities of the amide correlations and were used to refine the conformations of K28 and K29. Since the NMR data suggested that the DNA duplex maintained a B-type structure, a total of 52 empirical distance restraints arising from Watson−Crick base pairing interactions were used, as were 200 empirical torsion angle restraints that were applied to refine the nonterminal nucleotides. The restraints used for the structure refinement are summarized in Table 2 and detailed in Table S3 of the Supporting Information.
Table 2. rMD Restraints and Statistical Analysis of rMD Converged Structures of the DNA−Peptide Conjugate.
| total restraints for rMD calculation | 730 |
| experimental NOE distance restraints | 447 |
| intraresidue NOE restraints | 258 |
| interresidue NOE restraints | 189 |
| NOEs between DNTP, tether, and AAa | 34 |
| estimated NOE restraints of amide protons | 31 |
| empirical base pair restraints | 52 |
| empirical torsion angle restraints | 200 |
| backbone torsion angle restraints | 100 |
| sugar torsion angle restraints | 100 |
| structure statisticsb | |
| NMR R-factor (R1x) (×10−2) | 6.61 |
| intraresidue NOEs | 5.77 |
| interresidue NOEs | 7.98 |
| rmsd of refined structures | 0.59 |
| rmsd without K28 and K29 | 0.48 |
DNT means nucleotides; AA means amino acids.
Mixing time used to calculate R1x was 250 ms. R1x = ∑∣(a0)i1/6 − (ac)i1/6∣/∣(a0)i1/6∣, where (a0) and (ac) are the intensities of observed (nonzero) and calculated NOE cross-peaks, respectively.
The rMD calculations for the DNA−KWKK conjugate were performed from initial A- and B-form starting structures. Ten final structures, five each for A- and B-DNA starting structures, with lowest energies, were obtained. All structures, overlaid in Figure 6, converged as indicated by pairwise rmsd comparisons (Table 2). However, the rMD calculations did not converge for K28 and K29. Lysine K28 was likely ordered, but insufficient restraints were available to determine its location (Figure 6). Lysine K29 was undoubtedly disordered. The accuracies of the emergent structures were evaluated by comparison of theoretical NOE intensities calculated by CORMA for the refined structure to the experimental NOE intensities to yield sixth root residuals (R1x) (Figure S5 in the Supporting Information). The overall residuals, as well as the residuals for intra- or interresidue NOEs, were consistently less than 0.1 (Table 2). Except for K29 whose resonances were broad, R1x values for each residue were less than 0.15. Thus, the refined structures provided accurate depictions of the NMR data. For example, the G7 H8 → C6 H1′ NOE was weak (Figure S2 in the Supporting Information), and the distance between G7 H8 and C6 H1′ was 4.6 Å in the refined structure, consistent with the weak NOE.
Figure 6.

Superposition of the refined structures calculated from either A- or B-DNA starting structures. In the structural refinement, K28 and K29 did not converge, which was attributed to the lack of interresidue distance restraints.
Structure of the DNA−KWKK Conjugate
The DNA−KWKK conjugate maintained a B-DNA type structure, and all nucleotides maintained the anti conformation about the glycosyl bond. Watson−Crick hydrogen bonding was conserved at all base pairs. Figure 7 shows an expanded view of the KWKK-conjugated region. The G7N2 amine nitrogen and the K26 N-terminal nitrogen were in the trans configuration with respect to the Cα or Cγ of the trimethylene tether, respectively. The amino acids X25, K26, and W27 were oriented in the minor groove. The base stacking at the conjugated region together with the W27 indolyl group is shown in Figure 8. The W27 indolyl group was not intercalated into the duplex. The backbone torsion angles in the refined structure are provided in Table S4 of the Supporting Information.
Figure 7.
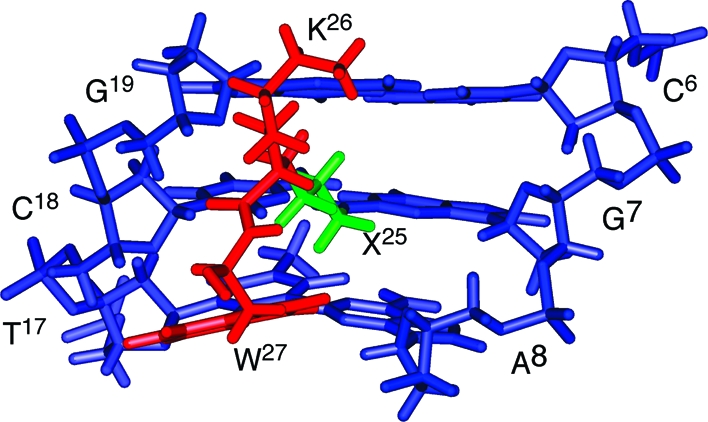
An expanded view of the average structure from the minor groove. Blue, green, and red sticks represent oligodeoxynucleotide, trimethylene linkage, and peptide, respectively.
Figure 8.
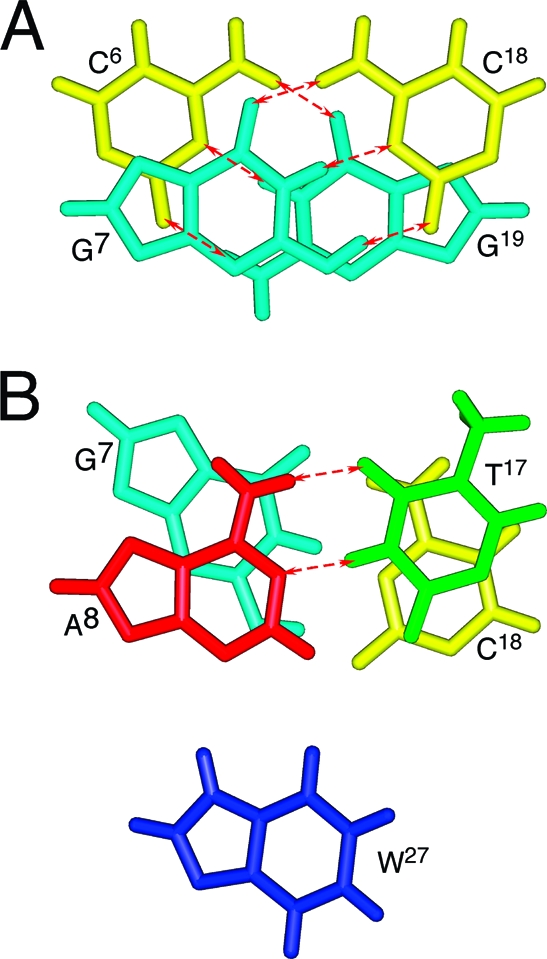
Base stacking of the average structure showing Watson−Crick hydrogen bonds at the KWKK conjugate region. Red arrows represent the Watson−Crick hydrogen bonds.
Discussion
If not repaired, DPCs interfere with DNA processing (1). An attractive mechanism for repair involves the initial formation of DNA−peptide conjugates via proteolytic digestion (2−4), which serve as substrates for nucleotide excision repair (6,7,25). This is supported by observations showing that inhibition of nuclear proteosomal protein degradation reduces the repair of DPCs (2), that the proteolytically active 20S core of the 26S proteasome localizes to the nucleus (3,4), and that inhibition of proteasome function with lactacystin results in the inhibition of repair of formaldehyde-induced DPCs, in normal, XP-A, and XP-F fibroblasts (2). One widely used DNA−peptide conjugate involves conjugation of KWKK to N2-dG via the γ-OH-PdG lesion, followed by chemical reduction to yield the reduced conjugate (16). Because its biological processing has been examined, the structure of this DNA−peptide conjugate is of interest.
Structure of the Reduced DNA−Peptide Conjugate
In this sequence, the KWKK peptide orients in the minor groove, and the DNA duplex maintains a B-type conformation (Figure 6). Amino acids K26, W27, and K28 form an ordered structure, which is consistent with their sharp NMR resonances and the significant chemical shift perturbations observed for these amino acids. In contrast, the N-terminal lysine K29 remains disordered and exposed to solvent. Its interactions with the DNA, if any, are weak. The W27 indolyl group is not intercalated (Figures 7 and 8). Thus, this N2-dG peptide conjugate differs from peptide conjugates formed via a β-elimination mechanism involving nucleophilic attack of the N-terminal amine at the C1′ carbon at the aldehyde of abasic sites (9−11). The targeting of the KWK peptide for abasic sites was attributed to stacking of the tryptophan residue between bases adjacent to the abasic site. This was supported by studies demonstrating that KWK binding to depurinated DNA was accompanied by increased fluorescence quenching (10).
The trimethylene tether of the DNA−KWKK conjugate has three potentially stable conformations: the W-, U-, and skew U-conformers (48,49). The refined structure demonstrates that the trimethylene tether prefers the W-conformer. This places the G7N2 amino nitrogen and the K26 N-terminal amino nitrogen in the trans conformation with respect to the Cα or Cγ of the tether, respectively. The adoption of the W-conformer appears to be favored because it minimizes the steric interactions of the tether with G7 and K26.
The thermal stability of this KWKK conjugate is notable since there is no evidence for tryptophan intercalation. It seems plausible that electrostatic interactions involving the ε-NH2 groups of the lysines, which are anticipated to be protonated at neutral pH, contribute to the thermal stability. The formation of salt bridges between the ε-NH3+ groups and the DNA phosphates does not seem to occur. The DNA 31P resonances do not exhibit large chemical shift perturbations, and the chemical shifts of lysine Hε protons remain similar to those in the free peptide KWKK. Moreover, in isothermal rMD calculations, all peptide amino groups remain >5 Å away from any DNA phosphodiester.
This DNA−KWKK conjugate provides clues as to the thermodynamic and structural properties that might lead to other stable minor groove DNA−peptide conjugates. The failure of the tryptophan moiety to intercalate implies that aromatic amino acids such as Phe, Trp, and Tyr are not necessary to stabilize minor groove peptide conjugates. On the other hand, the favorable electrostatic interactions involving the ε-NH3+ groups of lysines suggest that the presence of arginines in the peptide might also stabilize the minor groove peptide conjugate. Notably, the peptide RWRR also forms a stable conjugate with DNA containing the 1,N2-γ-OH-PdG adduct (16). Honig and co-workers (50) suggested that it is energetically less costly to remove the charged arginine guanidinium group from water than it is to remove the smaller amino group of a lysine (50). This argues that the RWRR conjugate might be of greater thermal stability than is the KWKK conjugate. Citing the nucleosome core particle, the same group (50) also proposed that the binding of arginines to regions of high negative electrostatic potential in narrow minor grooves (51), a consequence of isoelectric focusing (52), provides an important mode for protein−DNA recognition (50). Monovalent cations localize in the minor groove of narrow A-tracts in DNA (53−57). Other DNA-binding proteins that contain positively charged amino acids are reported to form DPCs (58,59). The interactions between oligodeoxynucleotide and the KWKK peptide observed for this conjugate are anticipated to be present in the native reversible DNA−peptide conjugates induced by the 1,N2-γ-OH-PdG adduct.
Comparison to Formaldehyde-Induced DPCs
DPCs induced by the formaldehyde-dG adduct (2,60−66) contain a methylene tether (dG-N2-CH2-N−protein). They are unstable and have half-life times of several hours in vitro(2). Compared to the present DPC containing the trimethylene tether, those induced by the formaldehyde−dG adduct locate the peptide closer to the duplex. This might allow three to four amino acids to be accommodated in the minor groove. The structures of saturated aldehyde-induced DPCs that contain a methylene tether may also be modeled by the trimethylene conjugate.
Biological Implications
Repair of DNA−peptide cross-links has been investigated using purified NER proteins from both prokaryotic (6,8) and human systems (67). These investigations consistently revealed that peptide lesions, less than or equal to 12 amino acids in length, were excellent substrates for these excision repair complexes. In both systems, rates of incision were roughly comparable with other efficiently recognized substrates, such as polycyclic aromatic hydrocarbon adducts or (6−4) photoproducts. These robust catalytic efficiencies are observed on DNAs containing either abasic site-linked peptides (8,67) or γ-OH-PdG-linked peptides (6) even though these lesions stabilize, rather than destabilize duplex DNA. These data are similar to prior studies that investigated the structure−activity relationship of psoralen monoadducted DNAs and the UvrABC complex (68−70). Although the psoralen monoadduct stabilized the DNA duplex, the UvrABC system recognized and incised it with a very high efficiency, while in contrast, it was a very poor substrate for the human NER complex (70).
To date, repair of DNA−peptide cross-links in which the peptide linkage is via an acrolein-mediated dG has only been analyzed using the prokaryotic NER system (6). A direct comparison of the relative kinetics of UvrABC incision measured using either KWKK− or KFHEKHHSHRGY−DNA conjugates or the T4-pdg DPC, conjugated via either abasic site or γ-OH-PdG chemistry, revealed that all of the UvrABC proteins were necessary to catalyze the incision reactions (6). The incision kinetics of the KFHEKHHSHRGY−DNA conjugate at either the abasic or γ-OH-PdG sites were significantly greater than that of DNA containing a fluorescein-conjugated dT (7). However, the KFHEKHHSHRGY conjugate attached via an abasic site was incised three times more efficiently than when conjugated via γ-OH-PdG. Thus, it seems likely that structural and biophysical differences arising due to the length of the peptide conjugates, or their sites of attachment to DNA, are likely to modulate their recognition and repair by the nucleotide excision repair apparatus. If not repaired, these DNA−KWKK conjugates are mutagenic, and their potential to cause mutations depends on the site of peptide attachment. Compared to the reduced N2-propyl alcohol adduct, a surrogate for the reversible N2-propylaldehyde rearrangement product arising from the γ-OH-PdG adduct (21−23), this DNA−KWKK conjugate increases miscoding (8). Replication bypass in the COS-7 site-specific mutagenesis assay results in mutations at a frequency of ∼8.4%, particularly G → T transversions (26). DNA polymerase κ, which bypasses several minor groove dG adducts in an error-free manner (71−73), also catalyzes efficient, error-free bypass of this KWKK conjugate (8). The E. coli orthologue of pol κ, pol IV, pauses opposite and three nucleotides beyond the site of this lesion, with incorporation being accurate. In contrast, pol ν, an A-family polymerase involved in the bypass of bulky major groove lesions, is blocked by this KWKK conjugate (74). Neither the E. coli replicative polymerase, pol III, nor the damage-inducible polymerases, pol II and pol V, efficiently incorporate nucleotides opposite this DNA−KWKK conjugate (8). Enal-induced DPCs can also involve the amino groups of dC and dA. The DNA−KWKK conjugate arising from the 1,N6-dA acrolein-induced adduct is less mutagenic than that arising from this DNA−KWKK conjugate (8). Moreover, it results in fewer mutations compared to the γ-OH-PdA lesion (8). DNA pol ν catalyzes efficient high-fidelity bypass of the conjugate arising from the γ-OH-PdA adduct (74). The dA (and dC) conjugates presumably place the KWKK in the more spacious major groove whereas this γ-OH-PdG conjugate locates the peptide in the minor groove. The observation that the mutagenic potential depends on the site of peptide attachment suggests that the structural consequences of the corresponding dA (and dC) conjugates may differentially impact DNA processing, as compared to the presently studied lesion.
Conclusions
The structure of an oligodeoxynucleotide duplex containing the KWKK peptide conjugated site-specifically via a fully reduced trimethylene tether, a model for the native linkage in the presence of the γ-OH-PdG adduct, revealed that the peptide was located in the minor groove. Significantly, the thermal stability of the DNA duplex was increased. This was attributed to electrostatic interactions involving the protonated lysine ε-NH3+ groups, rather than intercalation of the tryptophan moiety into the duplex. Amino acids K26, W27, and K28 were ordered and located in the minor groove, whereas K29 was disordered and likely exposed to solvent.
Acknowledgments
Dr. Markus Voehler assisted with NMR experiments.
Supporting Information Available
Table S1, chemical shifts of the nucleotide protons; Table S2, chemical shifts of the trimethylene tether and amino acid protons; Table S3, NOE distance restraints used for the structure refinement; Table S4, backbone torsion angles of the refined structure; Figure S1, partial charges of the trimethylene tether obtained from DFT calculations; Figure S2, NOE connectivity of nucleobase H6/H8 protons to deoxyribose H1′ protons; Figure S3, assignment of nucleotide exchangeable protons; Figure S4, chemical shift perturbations of the DNA−peptide conjugate; Figure S5, sixth root CORMA residuals (R1x) of the DNA−peptide conjugate. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health (NIH) Grants PO1 ES-05355 (I.D.K., C.J.R., R.S.L., and M.P.S.) and RO1 CA-106858 and RO1 ES-04091 (A.K.M. and R.S.L.). Funding for the NMR spectrometers was supplied by Vanderbilt University, the Vanderbilt Center in Molecular Toxicology (P30 ES-000267), and by NIH Shared Instrumentation Grant RR-005805. The Vanderbilt Ingram Cancer Center is supported by NIH Grant P30 CA068485.
The structural coordinates have been deposited in the Protein Data Bank (ID: 2KV6).
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: DPC, DNA−protein conjugate; γ-OH-PdG, γ-hydroxy-1,N2-propanodeoxyguanosine; NOESY, nuclear Overhauser effect spectroscopy; COSY, correlation spectroscopy; DQF-COSY, double-quantum-filtered COSY; NOE, nuclear Overhauser effect; rMD, restrained molecular dynamics.
Supplementary Material
References
- Barker S.; Weinfeld M.; Murray D. (2005) DNA-protein crosslinks: Their induction, repair, and biological consequences. Mutat. Res. 589, 111–135. [DOI] [PubMed] [Google Scholar]
- Quievryn G.; Zhitkovich A. (2000) Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 21, 1573–1580. [PubMed] [Google Scholar]
- Brooks P.; Fuertes G.; Murray R. Z.; Bose S.; Knecht E.; Rechsteiner M. C.; Hendil K. B.; Tanaka K.; Dyson J.; Rivett J. (2000) Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem. J. 346, 155–161. [PMC free article] [PubMed] [Google Scholar]
- Lafarga M.; Berciano M. T.; Pena E.; Mayo I.; Castano J. G.; Bohmann D.; Rodrigues J. P.; Tavanez J. P.; Carmo-Fonseca M. (2002) Clastosome: A subtype of nuclear body enriched in 19S and 20S proteasomes, ubiquitin, and protein substrates of proteasome. Mol. Biol. Cell 13, 2771–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai S. D.; Liu L. F.; Vazquez-Abad D.; D’Arpa P. (1997) Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J. Biol. Chem. 272, 24159–24164. [DOI] [PubMed] [Google Scholar]
- Minko I. G.; Kurtz A. J.; Croteau D. L.; Van Houten B.; Harris T. M.; Lloyd R. S. (2005) Initiation of repair of DNA-polypeptide cross-links by the UvrABC nuclease. Biochemistry 44, 3000–3009. [DOI] [PubMed] [Google Scholar]
- Truglio J. J.; Croteau D. L.; Skorvaga M.; DellaVecchia M. J.; Theis K.; Mandavilli B. S.; Van Houten B.; Kisker C. (2004) Interactions between UvrA and UvrB: The role of UvrB’s domain 2 in nucleotide excision repair. EMBO J. 23, 2498–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko I. G.; Zou Y.; Lloyd R. S. (2002) Incision of DNA-protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. Proc. Natl. Acad. Sci. U.S.A. 99, 1905–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre J.; Laval J. (1981) Specific nicking of DNA at apurinic sites by peptides containing aromatic residues. J. Biol. Chem. 256, 10217–10220. [PubMed] [Google Scholar]
- Behmoaras T.; Toulme J. J.; Helene C. (1981) A tryptophan-containing peptide recognizes and cleaves DNA at apurinic sites. Nature 292, 858–859. [DOI] [PubMed] [Google Scholar]
- Mazumder A.; Gerlt J. A.; Absalon M. J.; Stubbe J.; Cunningham R. P.; Withka J.; Bolton P. H. (1991) Stereochemical studies of the β-elimination reactions at aldehydic abasic sites in DNA: Endonuclease III from Escherichia coli, sodium hydroxide, and Lys-Trp-Lys. Biochemistry 30, 1119–1126. [DOI] [PubMed] [Google Scholar]
- Dodson M. L.; Schrock R. D. III; Lloyd R. S. (1993) Evidence for an imino intermediate in the T4 endonuclease V reaction. Biochemistry 32, 8284–8290. [DOI] [PubMed] [Google Scholar]
- Kurtz A. J.; Dodson M. L.; Lloyd R. S. (2002) Evidence for multiple imino intermediates and identification of reactive nucleophiles in peptide-catalyzed β-elimination at abasic sites. Biochemistry 41, 7054–7064. [DOI] [PubMed] [Google Scholar]
- Schrock R. D. III; Lloyd R. S. (1991) Reductive methylation of the amino terminus of endonuclease V eradicates catalytic activities. Evidence for an essential role of the amino terminus in the chemical mechanisms of catalysis. J. Biol. Chem. 266, 17631–17639. [PubMed] [Google Scholar]
- McCullough A. K.; Sanchez A.; Dodson M. L.; Marapaka P.; Taylor J. S.; Lloyd R. S. (2001) The reaction mechanism of DNA glycosylase/AP lyases at abasic sites. Biochemistry 40, 561–568. [DOI] [PubMed] [Google Scholar]
- Kurtz A. J.; Lloyd R. S. (2003) 1,N2-deoxyguanosine adducts of acrolein, crotonaldehyde, and trans-4-hydroxynonenal cross-link to peptides via Schiff base linkage. J. Biol. Chem. 278, 5970–5976. [DOI] [PubMed] [Google Scholar]
- Chung F. L.; Young R.; Hecht S. S. (1984) Formation of cyclic 1,N2-propanodeoxyguanosine adducts in DNA upon reaction with acrolein or crotonaldehyde. Cancer Res. 44, 990–995. [PubMed] [Google Scholar]
- Chung F. L.; Young R.; Hecht S. S. (1989) Detection of cyclic 1,N2-propanodeoxyguanosine adducts in DNA of rats treated with N-nitrosopyrrolidine and mice treated with crotonaldehyde. Carcinogenesis 10, 1291–1297. [DOI] [PubMed] [Google Scholar]
- Chung F. L.; Chen H. J.; Nath R. G. (1996) Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis 17, 2105–2111. [DOI] [PubMed] [Google Scholar]
- Chung F. L.; Nath R. G.; Ocando J.; Nishikawa A.; Zhang L. (2000) Deoxyguanosine adducts of t-4-hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: Detection and potential sources. Cancer Res. 60, 1507–1511. [PubMed] [Google Scholar]
- de los Santos C.; Zaliznyak T.; Johnson F. (2001) NMR characterization of a DNA duplex containing the major acrolein-derived deoxyguanosine adduct γ-OH-1,N2-propano-2′-deoxyguanosine. J. Biol. Chem. 276, 9077–9082. [DOI] [PubMed] [Google Scholar]
- Cho Y. J.; Kim H. Y.; Huang H.; Slutsky A.; Minko I. G.; Wang H.; Nechev L. V.; Kozekov I. D.; Kozekova A.; Tamura P.; Jacob J.; Voehler M.; Harris T. M.; Lloyd R. S.; Rizzo C. J.; Stone M. P. (2005) Spectroscopic characterization of interstrand carbinolamine cross-links formed in the 5′-CpG-3′ sequence by the acrolein-derived γ-OH-1,N2-propano-2′-deoxyguanosine DNA adduct. J. Am. Chem. Soc. 127, 17686–17696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y. J.; Wang H.; Kozekov I. D.; Kozekova A.; Kurtz A. J.; Jacob J.; Voehler M.; Smith J.; Harris T. M.; Rizzo C. J.; Lloyd R. S.; Stone M. P. (2006) Orientation of the crotonaldehyde-derived N2-[3-oxo-1(S)-methyl-propyl]-dG DNA adduct hinders interstrand cross-link formation in the 5′-CpG-3′ sequence. Chem. Res. Toxicol. 19, 1019–1029. [DOI] [PubMed] [Google Scholar]
- Minko I. G.; Yamanaka K.; Kozekov I. D.; Kozekova A.; Indiani C.; O’Donnell M. E.; Jiang Q.; Goodman M. F.; Rizzo C. J.; Lloyd R. S. (2008) Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 21, 1983–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko I. G.; Kozekov I. D.; Harris T. M.; Rizzo C. J.; Lloyd R. S.; Stone M. P. (2009) Chemistry and biology of DNA containing 1,N2-deoxyguanosine adducts of the α,β-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem. Res. Toxicol. 22, 759–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko I. G.; Kozekov I. D.; Kozekova A.; Harris T. M.; Rizzo C. J.; Lloyd R. S. (2008) Mutagenic potential of DNA-peptide crosslinks mediated by acrolein-derived DNA adducts. Mutat. Res. 637, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari A.; Minko I. G.; Harbut M. B.; Finkel S. E.; Goodman M. F.; Lloyd R. S. (2008) Replication bypass of interstrand cross-link intermediates by Escherichia coli DNA polymerase IV. J. Biol. Chem. 283, 27433–27437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665. [DOI] [PubMed] [Google Scholar]
- Borgias B. A.; James T. L. (1990) MARDIGRAS—A procedure for matrix analysis of relaxation for discerning geometry of an aqueous structure. J. Magn. Reson. 87, 475–487. [Google Scholar]
- Liu H.; Tonelli M.; James T. L. (1996) Correcting NOESY cross-peak intensities for partial relaxation effects enabling accurate distance information. J. Magn. Reson. B 111, 85–89. [DOI] [PubMed] [Google Scholar]
- Liu H.; Spielmann H. P.; Ulyanov N. B.; Wemmer D. E.; James T. L. (1995) Interproton distance bounds from 2D NOE intensities: Effect of experimental noise and peak integration errors. J. Biomol. NMR 6, 390–402. [DOI] [PubMed] [Google Scholar]
- Kouchakdjian M.; Marinelli E.; Gao X.; Johnson F.; Grollman A.; Patel D. (1989) NMR studies of exocyclic 1,N2-propanodeoxyguanosine adducts (X) opposite purines in DNA duplexes: Protonated X(syn):A(anti) pairing (acidic pH) and X(syn):G(anti) pairing (neutral pH) at the lesion site. Biochemistry 28, 5647–5657. [DOI] [PubMed] [Google Scholar]
- Rinkel L. J.; Altona C. (1987) Conformational analysis of the deoxyribofuranose ring in DNA by means of sums of proton-proton coupling constants: A graphical method. J. Biomol. Struct. Dyn. 4, 621–649. [DOI] [PubMed] [Google Scholar]
- Arnott S.; Hukins D. W. L. (1972) Optimised parameters for A-DNA and B-DNA. Biochem. Biophys. Res. Commun. 47, 1504–1509. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.,. et al. (1998) GAUSSIAN 98, 1998 ed., Gaussian, Inc., Pittsburgh, PA.
- Case D. A., Pearlman D. A., Caldwell J. W., Cheatham T. E. III, Wang J., Ross W. S., Simmerling C. L., Darden T. A., Merz K. M., Stanton R. V., Cheng A. L., Vincent J. J., Crowley M., Tsui V., Gohlke H., Radmer R. J., Duan Y., Pitera J., Massova I., Seibel G. L., Singh U. C., Weiner P. K., and Kollman P. A. (2002) AMBER, 7.0 ed., University of California, San Francisco, CA.
- Hawkins G. D.; Cramer C. J.; Truhlar D. G. (1995) Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 246, 122–129. [Google Scholar]
- Hawkins G. D.; Cramer C. J.; Truhlar D. G. (1996) Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 100, 19824–19839. [Google Scholar]
- Tsui V.; Case D. A. (2000) Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 56, 275–291. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Keepers J. W.; James T. L. (1984) A theoretical study of distance determination from NMR. Two-dimensional nuclear Overhauser effect spectra. J. Magn. Reson. 57, 404–426. [Google Scholar]
- Liu Y.; Zhao D.; Altman R.; Jardetzky O. (1992) A systematic comparison of three structure determination methods from NMR data: Dependence upon quality and quantity of data. J. Biomol. NMR 2, 373–388. [DOI] [PubMed] [Google Scholar]
- Thomas P. D.; Basus V. J.; James T. L. (1991) Protein solution structure determination using distances from two-dimensional nuclear Overhauser effect experiments: Effect of approximations on the accuracy of derived structures. Proc. Natl. Acad. Sci. U.S.A. 88, 1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X. J.; Olson W. K. (2003) 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 31, 5108–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid B. R. (1987) Sequence-specific assignments and their use in NMR studies of DNA structure. Q. Rev. Biophys. 20, 2–28. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Shapiro L.; Hare D. (1987) DNA and RNA: NMR studies of conformations and dynamics in solution. Q. Rev. Biophys. 20, 35–112. [DOI] [PubMed] [Google Scholar]
- Boelens R.; Scheek R. M.; Dijkstra K.; Kaptein R. (1985) Sequential assignment of imino- and amino-proton resonances in 1H NMR spectra of oligonucleotides by two-dimensional NMR spectroscopy. Application to a lac operator fragment. J. Magn. Reson. 62, 378–386. [Google Scholar]
- Dooley P. A.; Tsarouhtsis D.; Korbel G. A.; Nechev L. V.; Shearer J.; Zegar I. S.; Harris C. M.; Stone M. P.; Harris T. M. (2001) Structural studies of an oligodeoxynucleotide containing a trimethylene interstrand cross-link in a 5′-(CpG) motif: Model of a malondialdehyde cross-link. J. Am. Chem. Soc. 123, 1730–1739. [DOI] [PubMed] [Google Scholar]
- Dooley P. A.; Zhang M.; Korbel G. A.; Nechev L. V.; Harris C. M.; Stone M. P.; Harris T. M. (2003) NMR determination of the conformation of a trimethylene interstrand cross-link in an oligodeoxynucleotide duplex containing a 5′-d(GpC) motif. J. Am. Chem. Soc. 125, 62–72. [DOI] [PubMed] [Google Scholar]
- Rohs R.; West S. M.; Sosinsky A.; Liu P.; Mann R. S.; Honig B. (2009) The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram B.; Sharp K. A.; Honig B. (1989) The electrostatic potential of B-DNA. Biopolymers 28, 975–993. [DOI] [PubMed] [Google Scholar]
- Honig B.; Nicholls A. (1995) Classical electrostatics in biology and chemistry. Science 268, 1144–1149. [DOI] [PubMed] [Google Scholar]
- Hud N. V.; Sklenar V.; Feigon J. (1999) Localization of ammonium ions in the minor groove of DNA duplexes in solution and the origin of DNA A-tract bending. J. Mol. Biol. 286, 651–660. [DOI] [PubMed] [Google Scholar]
- Howerton S. B.; Sines C. C.; VanDerveer D.; Williams L. D. (2001) Locating monovalent cations in the grooves of B-DNA. Biochemistry 40, 10023–10031. [DOI] [PubMed] [Google Scholar]
- Hamelberg D.; Williams L. D.; Wilson W. D. (2001) Influence of the dynamic positions of cations on the structure of the DNA minor groove: Sequence-dependent effects. J. Am. Chem. Soc. 123, 7745–7755. [DOI] [PubMed] [Google Scholar]
- Hud N. V.; Polak M. (2001) DNA-cation interactions: The major and minor grooves are flexible ionophores. Curr. Opin. Struct. Biol. 11, 293–301. [DOI] [PubMed] [Google Scholar]
- Howerton S. B.; Nagpal A.; Williams L. D. (2003) Surprising roles of electrostatic interactions in DNA-ligand complexes. Biopolymers 69, 87–99. [DOI] [PubMed] [Google Scholar]
- O’Connor P. M.; Fox B. W. (1989) Isolation and characterization of proteins cross-linked to DNA by the antitumor agent methylene dimethanesulfonate and its hydrolytic product formaldehyde. J. Biol. Chem. 264, 6391–6397. [PubMed] [Google Scholar]
- Tolstonog G. V.; Mothes E.; Shoeman R. L.; Traub P. (2001) Isolation of SDS-stable complexes of the intermediate filament protein vimentin with repetitive, mobile, nuclear matrix attachment region, and mitochondrial DNA sequence elements from cultured mouse and human fibroblasts. DNA Cell Biol. 20, 531–554. [DOI] [PubMed] [Google Scholar]
- Cosma G. N.; Wilhite A. S.; Marchok A. C. (1988) The detection of DNA-protein cross-links in rat tracheal implants exposed in vivo to benzo[a]pyrene and formaldehyde. Cancer Lett. 42, 13–21. [DOI] [PubMed] [Google Scholar]
- Ma T. H.; Harris M. M. (1988) Review of the genotoxicity of formaldehyde. Mutat. Res. 196, 37–59. [DOI] [PubMed] [Google Scholar]
- Heck H. D.; Casanova M.; Starr T. B. (1990) Formaldehyde toxicity—New understanding. Crit. Rev. Toxicol. 20, 397–426. [DOI] [PubMed] [Google Scholar]
- Hubal E. A.; Schlosser P. M.; Conolly R. B.; Kimbell J. S. (1997) Comparison of inhaled formaldehyde dosimetry predictions with DNA-protein cross-link measurements in the rat nasal passages. Toxicol. Appl. Pharmacol. 143, 47–55. [DOI] [PubMed] [Google Scholar]
- Shaham J.; Bomstein Y.; Gurvich R.; Rashkovsky M.; Kaufman Z. (2003) DNA-protein crosslinks and p53 protein expression in relation to occupational exposure to formaldehyde. Occup. Environ. Med. 60, 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Li C. M.; Lu Z.; Ding S.; Yang X.; Mo J. (2006) Studies on formation and repair of formaldehyde-damaged DNA by detection of DNA-protein crosslinks and DNA breaks. Front. Biosci. 11, 991–997. [DOI] [PubMed] [Google Scholar]
- Schmid O.; Speit G. (2007) Genotoxic effects induced by formaldehyde in human blood and implications for the interpretation of biomonitoring studies. Mutagenesis 22, 69–74. [DOI] [PubMed] [Google Scholar]
- Reardon J. T.; Sancar A. (2006) Repair of DNA-polypeptide crosslinks by human excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 103, 4056–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Hearst J. E. (1986) Thermostability of double-stranded deoxyribonucleic acids: Effects of covalent additions of a psoralen. Biochemistry 25, 5895–5902. [DOI] [PubMed] [Google Scholar]
- Isaacs R. J.; Spielmann H. P. (2004) A model for initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA Repair (Amsterdam) 3, 455–464. [DOI] [PubMed] [Google Scholar]
- Huang J. C.; Hsu D. S.; Kazantsev A.; Sancar A. (1994) Substrate spectrum of human excinuclease: Repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc. Natl. Acad. Sci. U.S.A. 91, 12213–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avkin S.; Goldsmith M.; Velasco-Miguel S.; Geacintov N.; Friedberg E. C.; Livneh Z. (2004) Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: The role of DNA polymerase κ. J. Biol. Chem. 279, 53298–53305. [DOI] [PubMed] [Google Scholar]
- Bi X.; Slater D. M.; Ohmori H.; Vaziri C. (2005) DNA polymerase κ is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J. Biol. Chem. 280, 22343–22355. [DOI] [PubMed] [Google Scholar]
- Choi J. Y.; Angel K. C.; Guengerich F. P. (2006) Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase κ. J. Biol. Chem. 281, 21062–21072. [DOI] [PubMed] [Google Scholar]
- Yamanaka K.; Minko I. G.; Takata K.; Kolbanovskiy A.; Kozekov I. D.; Wood R. D.; Rizzo C. J.; Lloyd R. S. (2010) Novel enzymatic function of DNA polymerase ν in translesion DNA synthesis past major groove DNA-peptide and DNA-DNA cross-links. Chem. Res. Toxicol. 23, 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.