

Abstract
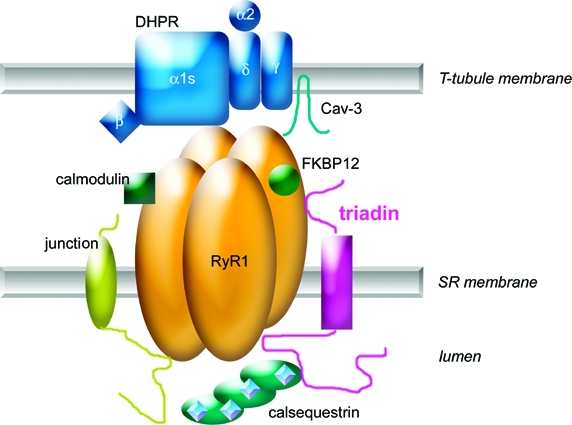
The triadin isoforms Trisk 95 and Trisk 51 are both components of the skeletal muscle calcium release complex. To investigate the specific role of Trisk 95 and Trisk 51 isoforms in muscle physiology, we overexpressed Trisk 95 or Trisk 51 using adenovirus-mediated gene transfer in skeletal muscle of newborn mice. Overexpression of either Trisk 95 or Trisk 51 alters the muscle fiber morphology, while leaving unchanged the expression of the ryanodine receptor, the dihydropyridine receptor, and calsequestrin. We also observe an aberrant expression of caveolin 3 in both Trisk 95- and Trisk 51-overexpressing skeletal muscles. Using a biochemical approach, we demonstrate that caveolin 3 is associated with the calcium release complex in skeletal muscle. Taking advantage of muscle and non-muscle cell culture models and triadin null mouse skeletal muscle, we further dissect the molecular organization of the caveolin 3-containing calcium release complex. Our data demonstrate that the association of caveolin 3 with the calcium release complex occurs via a direct interaction with the transmembrane domain of the ryanodine receptor. Taken together, these data suggest that caveolin 3-containing membrane domains and the calcium release complex are functionally linked and that Trisk 95 and Trisk 51 are instrumental to the regulation of this interaction, the integrity of which may be crucial for muscle physiology.
In skeletal muscle, the excitation−contraction (EC)1 coupling process takes place at the triads where T-tubules and the reticulum sarcoplasmic terminal cisternae are in close contact. EC coupling requires the expression at the triads of a multimeric calcium release complex (CRC) that includes the T-tubule voltage-dependent calcium channel dihydropyridine receptor (DHPR) and several sarcoplasmic proteins, namely, the calcium release channel ryanodine receptor (RyR) (1,2), the sarcoluminal Ca2+ binding calsequestrin (CSQ), and two of the four triadin isoforms identified to date in skeletal muscle, Trisk 95 (T95) and Trisk 51 (T51) (3,4). In vitro studies had identified a couple of regulatory RyR binding domains on triadin, and these regions are common to both Trisk 95 and Trisk 51 (5,6). Adenoviral-mediated overexpression of either Trisk 95 or Trisk 51 in primary cultures of skeletal muscle further demonstrated that only Trisk 95 plays a role in the regulation of the depolarization-induced calcium release mechanism (4), suggesting a specific role of Trisk 95 in EC coupling. Interestingly, triadin null mice exhibit a structural myopathy (7) with impaired depolarization-induced calcium release (7,8) indicating that the triadins are likely to be involved in the development of human myopathies for which a causative gene has not yet been identified. However, no modification has been yet identified in human triadins, both Trisk 95 and Trisk 51 being expressed in human skeletal muscle (9), and it is currently unknown whether overexpression of Trisk 95 and Trisk 51 would be detrimental for muscle function in vivo.
In this study, we use adenovirus-mediated gene transfer to overexpress Trisk 95 or Trisk 51 in mouse skeletal muscle as an alternative approach to investigating the function of these triadin isoforms. Herein, we show that overexpression of either Trisk 95 or Trisk 51 alters the muscle fiber morphology while leaving the expression of RyR, DHPR, and CSQ unchanged. We also observe that caveolin 3 (Cav-3), an essential structural component of caveolae involved in endocytosis and intracellular trafficking events (10), is aberrantly expressed in both Trisk 95- and Trisk 51-overexpressing skeletal muscles. We further demonstrate that Cav-3 is associated with the CRC via a direct interaction with the RyR, and we propose that the Trisk 95 and Trisk 51 level of expression is critical for the regulation of this interaction.
Materials and Methods
Animals
Wild-type mice (C57BL/6) were bred at the University of Iowa from stocks originally obtained from Jackson Laboratories (Bar Harbor, ME). Triadin null mice have been described previously (7) and were bred and maintained on a C57BL/6 background at Université Joseph Fourier. Animal care and procedures were approved and performed in accordance with the standards set forth by the Institutional Ethics Committee, the National Research Council Guide for the care and use of laboratory animals, the National Institutes of Health, and the Animal Care Use and Review Committee of the University of Iowa.
Preparation of Antibodies and Microsomes
mAbs against CSQ (clone VIIID12, Affinity BioReagents) and Cav-3 (BD Transduction Laboratories) were used as described in the company datasheet. The sheep anti-DHPR α1 subunit was obtained from Upstate Biotechnology. Polyclonal antibodies against the RyR, the common N-terminal end of tradins, Trisk 95, and Trisk 51 were described previously (2,3,11). Guinea pig anti-Ca2+-ATPase was a gift from A. M. Lompré (12). Crude microsomes were prepared from 1-month-old mouse gastrocnemius muscle as previously described (13).
Viruses
The viruses were engineered and produced by the Gene Vector Production Network, at Genethon III (Evry, France). Three type 5 adenoviruses were used in this study, a control virus (AdV-DsRed) with the cDNA of the red fluorescent protein (DsRed), AdV-Trisk 95, an adenovirus with the full-length sequence of rat skeletal muscle T95 (EMBL AJ243304, 687 amino acids), and AdV-Trisk 51, an adenovirus with the full-length sequence of rat skeletal muscle T51 (EMBL AJ243303, 461 amino acids). All the transgenes were under the control of a CMV promoter.
In Vivo Infection
Injections of adenovirus into 3−4-day-old wild-type pups were performed as previously described (14), with some modification: the hamstring, quadriceps, calf, and tibialis anterior muscles of one leg were each injected percutaneously with 1010 pfu diluted in 10 μL of saline; the same muscles of the contralateral leg were each injected with an equal volume of saline. Pups were reintroduced to the mother and kept in quarantine for 5 days. All pups survived after injections. Muscles were harvested 30 days after injection, snap-frozen in liquid nitrogen-cooled isopentane, and stored at −80 °C until they were used.
Cell Culture and Cell Infection
Rat myogenic L6 cells (clone C5) or COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin. L6 cells were infected at a multiplicity of infection (MOI) of 200 with the adenoviruses encoding T95 (AdV-Trisk 95) or encoding T51 (AdV-Trisk 51). The cells were collected 48 h after infection.
Plasmids and Transfection
The transmembrane domain of human RyR1 (GenBank accession number J05200) corresponding to amino acids 4457−5038 was cloned in pEGFP-C1 (Clontech), to produce a 93 kDa fusion protein corresponding to the GFP fused with the channel part of RyR1. Human caveolin 3 (GenBank accession number AF036365) was cloned in pCDNA3.1 (Invitrogen). COS-7 cells were transfected with the two plasmids (2.5 μg of each plasmid for 8 × 105 cells) using ExGen 500 (Euromedex), and the cells were collected 24 h after transfection.
Immunoprecipitation
Five hundred micrograms of microsomes from rat skeletal muscle or 300 μg of cells was solubilized at 2 mg/mL in the presence of 1.6% CHAPS, 0.9 M NaCl, 0.1% phospholipids, 100 μM CaCl2, 50 μM EGTA, 20 mM Pipes (pH 7.1), and protease inhibitors (1 mM diisopropyl fluorophosphates and 100 μM phenylmethanesulfonyl fluoride), and immunoprecipitation was performed with antibodies against rat Trisk 95, rat Trisk 51, or nonimmune serum as described previously (2), using 10 mg of protein A immobilized on Sepharose 4B. All the immunoprecipitated proteins were then analyzed by Western blotting.
Western Blot
The presence of RyR, DHPR, Trisk 95, Trisk 51, calsequestrin, Ca2+-ATPase SERCA, or caveolin 3 in different samples was assayed by Western blotting, using a chemiluminescent reagent (Western lightning Chemiluminescence reagent plus, PerkinElmer Life Science). After electrophoretic separation on a 5−15% acrylamide gel, the proteins were electrotransferred to Immobilon P (Millipore) as previously described (3). The secondary antibodies were labeled with horseradish peroxidase (Jackson ImmunoResearch Laboratories). Quantitative analysis was performed using a Chemidoc XRS and Quantity One (Bio-Rad), and the amount of each protein was normalized to the amount in AdV-DsRed-infected muscles, after loading correction by Coomassie blue staining of myosin, as described previously (15).
Immunofluorescence and Histological Analyses
Seven micrometer cryosections were prepared and analyzed by immunofluorescence or H&E staining as previously described (16). For immunofluorescent staining, sections were washed with phosphate-buffered saline (PBS), blocked with 3% bovine serum albumin (BSA) in PBS for 30 min at room temperature, and incubated with a primary antibody in a PBS/1% BSA mixture overnight at 4 °C. After incubation for 1 h at room temperature with an Alexa fluor 488-conjugated (Invitrogen) or Cy3-conjugated secondary antibody in a PBS/1% BSA mixture, the samples were mounted with PermaFluor (Beckman Coulter). Sections were observed under an MRC-600 laser scanning confocal microscope (Bio-Rad). Digitized images were captured under identical conditions. Images of H&E staining were photographed using a Leica DM RXA microscope equipped with an Olympus DP70 digital camera.
Results
Effect of Trisk 95 and Trisk 51 Overexpression on the Expression of the CRC Components
Trisk 95 and Trisk 51, the two major skeletal muscle triadin isoforms, were overexpressed in vivo using injection of adenovirus into newborn mice. As a control, the muscles were injected with an adenovirus encoding DsRed. To determine the effect of Trisk 95 or Trisk 51 overexpression on the expression of their calcium release complex’s partners, immunoblot analysis of crude muscle extracts was conducted 30 days after gene transfer (Figure 1A,B). Protein expression levels were normalized to the amounts measured in control muscles infected with AdV-DsRed (Figure 1A, lane 3). Both Trisk 95 and Trisk 51 were overexpressed by 60% (Figure 1A, lanes 1 and 2, and Figure 1B), but the expression levels of the RyR, the α1 subunit of DHPR, CSQ, and SERCA were not changed (Figure 1B). This experiment was conducted on muscles from different animals, with similar results (no modification of the expression level of the proteins assayed, except for that of the overexpressed triadin). Nevertheless, the level of overexpression depends on the infection level, which is not identical among all the animals. Using Western blot on muscle homogenate, the infected fibers are mixed with noninfected fibers, which constitute the majority of the fibers. To discriminate between infected and noninfected fibers and to evaluate more precisely the modification induced on the other proteins, we chose to analyze the expression of the CRC components in Trisk 95- and Trisk 51-overexpressing muscles by immunofluorescence. While the stainings for RyR, DHPR, and CSQ were unchanged in Trisk 95-infected fibers, we could observe a reduction in the level of Trisk 51 expression in Trisk 95-infected fibers (Figure 1C), an effect that was not observed in a Western blot. Likewise, only the level of Trisk 95 expression was reduced in Trisk 51-infected fibers (Figure 1D).
Figure 1.

Western blot and immunofluorescent analysis of the proteins of the calcium release complex in infected muscles. The experiments presented in panels A and B correspond to one representative experiment, which have been performed on three different infected muscles. The values have not been averaged among all the experiments as the infection level is not identical among all the infected muscles. (A) Expression of the components of the calcium release complex in skeletal muscle microsomes (20 μg) in Trisk 95-infected muscle (lane 1), Trisk 51-infected muscle (lane 2), or control DsRed-infected muscle (lane 3) as determined by Western blotting. (B) Level of proteins expressed as a fold of control DsRed-infected muscles, after loading correction using a quantity of myosin estimated by Coomassie blue staining. The quantification was performed on the Western blot presented in panel A. (C) Effect of Trisk 95 overexpression on proteins of the calcium release complex. Double immunofluorescent labeling was performed on transversal sections of muscle infected with AdV-T95 for 30 days, using antibodies directed against T95 and the α1s subunit of the DHPR (a−c), Trisk 95 and the RyR (d−f), T95 and CSQ (g−i), or T95 and T51 (j−l). The scale bar is 15 μm. (D) Effect of Trisk 51 overexpression on proteins of the calcium release complex. Double immunofluorescent labeling was performed on transversal sections of muscle infected with AdV-T51 for 30 days using antibodies directed against T51 and the α1s subunit of the DHPR (a−c), T51 and the RyR (d−f), T51 and CSQ (g−i), or T51 and T95 (j−l). The scale bar is 15 μm.
Effect of Trisk 95 and Trisk 51 Overexpression on the Muscle Histology
Histological analysis revealed no sign of muscle damage in control skeletal muscles injected with either saline (data not shown) or AdV-DsRed (Figure 2A, panel a). However, Trisk 95- and Trisk 51-infected skeletal muscles were characterized by the presence of fibrotic tissue, atrophic fibers, and regenerating fibers (Figure 2A, panels b and c). The extent of muscle damage was greater in T51-infected muscles than in T95-infected muscle, as assessed by the increased level of fibrosis and the presence of necrotic fibers. To specifically characterize Trisk 95- and Trisk 51-infected fibers, muscle sections were analyzed after immunostaining with caveolin 3 (Cav-3) as a plasma membrane marker (Figure 2B). In contrast to muscles infected with AdV-DsRed (Figure 2B, panels a−c and g−i), membrane staining revealed that the size of fibers infected by AdV-Trisk 95 and AdV-Trisk 51 was abnormal. Trisk 95-infected fibers were smaller than noninfected fibers (Figure 2B, panels d−f), and Trisk 51-infected fibers had a round shape (Figure 2B, panels j−l) and lost the contact between them. In contrast with the control AdV-DsRed-infected fibers (Figure 2B, insets), the expression pattern of Cav-3 was altered in fibers overexpressing Trisk 95 and Trisk 51. In these latter fibers, Cav-3 was shown to accumulate intracellularly and to colocalize with the triadins. As Cav-3 is expressed during the differentiation of the satellite cells in myotubes (17), it could be hypothesized that the aberrant staining of Cav-3 specifically occurs in regenerating fibers. However, staining of nuclei indicated that none of the cells displaying an intracellular staining of Cav-3 were regenerating cells (data not shown). Immunoblot analysis showed a similar amount of Cav-3 in crude extracts of infected muscle as compared to control samples from DsRed-infected muscles (Figure 2C,D), suggesting most probably a relocalization of Cav-3 upon triadin overexpression.
Figure 2.

Alteration of muscle morphology in infected muscles. (A) Histological analysis by Hematoxylin-eosin staining of gastrocnemius infected for 30 days with AdV-DsRed (CTRL, panel a), AdV-Trisk 95 (inf T95, panel b), or AdV-Trisk 51 (inf T51, panel c). Only in muscle infected with T95 or T51 is the presence of fibrotic tissue observed, as well as a few atrophic fibers (asterisks), necrotic fibers stained in pink (inf T51 only), and a few regenerating fibers, characterized by their centrally located nuclei (arrows). The scale bar is 50 μm. (B) Double immunofluorescent labeling with anti-Cav-3 and anti-T95 antibodies (a−f) or anti-Cav-3 and anti-T51 (g−l) antibodies was performed on muscle sections from CTRL DsRed-infected muscle (a−c and g−i), T95-infected muscle (d−f), or T51-infected muscle (j−l). Cav-3 and T95 (insets of panels a−c and d−f) or Cav-3 and T51 (insets of panels g−i and j−l) intracellular staining was compared by confocal microscopy. (C) The expression level of Cav-3 was analyzed by Western blotting on T95-infected muscle (lane 1), T51-infected muscle (lane 2), or DsRed-infected muscle (lane 3). Five micrograms of mouse skeletal microsomes was loaded in each lane. (D) The quantification of the amount of caveolin 3 in each lane of panel C was performed, and the results were compared to the amount in DsRed-infected muscle (CTRL), after correction of loading by myosin quantification.
Association of Cav-3 with the Component of the Skeletal Muscle CRC
In vivo modification of triadin expression results in modification of Cav-3, indicating a functional link between the two proteins. We studied their possible association by immunoprecipitation in rat skeletal muscle (Figure 3A). Using isoform specific antibodies, both Trisk 95 and Trisk 51 co-immunoprecipitated Cav-3 (Figure 3A, lanes 2 and 3). Trisk 95 and Trisk 51 also co-immunoprecipitated each other and the RyR, which indicates that Cav-3 is associated with the CRC.
Figure 3.

Association of Cav-3 with the CRC and the triadins. (A) Co-immunoprecipitation on rat skeletal muscle. Immunoprecipitation was performed on 500 μg of rat skeletal muscle with antibodies against Trisk 95 (lane 2), with antibodies against Trisk 51 (lane 3), with preimmune serum (lane 4), or with antibodies against the DHPR (lane 5). The immunoprecipitated proteins were then analyzed by Western blotting with antibodies against the RyR, against the DHPR, against the N-terminal end of triadin, or against Cav-3. Lane 1 contained 5 μg of rat skeletal muscle microsomes as a control. (B) Co-immunoprecipitation in L6 cells. Immunoprecipitation was performed on 300 μg of Trisk 95-infected cells (lane 2), Trisk 51-infected cells (lane 3), or control cells (lane 4), with antibodies specific for T95 or T51. The immunoprecipitated proteins were then analyzed by Western blotting (lanes 5−8) with the anti-RyR, anti-Nter triadin, or anti-Cav 3. Lane 1 contained 5 μg of rat skeletal muscle microsomes as a control. The RyR is absent from L6 cells (infected and control, lanes 2−4) and is not co-immunoprecipited with triadin isoforms (lanes 5−8).
An association between the DHPR and Cav-3 was proposed in skeletal muscle (18) and shown in cardiac muscle (19). Thus, it is possible that the molecular complex between the triadins and Cav-3 requires a direct interaction of Cav-3 with the DHPR. To test the direct interaction between the DHPR and Cav-3, we analyzed the co-immunoprecipitation in rat skeletal muscle of Cav-3 with DHPR or the triadins (Figure 3A). The DHPR efficiently co-immunoprecipitated Cav-3 and the RyR (Figure 3A, lane 5), but not the triadins. Conversely, both Trisk 95 and Trisk 51 co-immunoprecipitated Cav-3 but not the DHPR (Figure 3A, lanes 2 and 3), suggesting that the association of triadin and Cav-3 is not DHPR-dependent.
To test whether Cav-3 and the triadins are able to associate directly, we studied their association in the L6 rat muscle cell line that does not express the known CRC proteins but expresses Cav-3. L6 cells were infected with AdV-Trisk 95 or AdV-Trisk 51 to induce the expression of each triadin isoform (Figure 3B, lanes 2−4). The absence of the RyR and the presence of Cav-3 were confirmed in these cells (Figure 3B, lanes 2−4). Direct association of Cav-3 with either triadin was studied by co-immunoprecipitation with the anti-T95 and anti-T51 antibodies. Although Trisk 95 and Trisk 51 were efficiently immunoprecipitated, Cav-3 was co-immunoprecipitated with none (Figure 3B, lanes 5−7), indicating that Cav-3 is not engaged in a direct interaction with any of the triadins. As both triadins are linked in skeletal muscle, as demonstrated by their co-immunoprecipitation (Figure 3A), it is possible that the simultaneous presence of Trisk 95 and Trisk 51 is required for association with Cav-3. To test this hypothesis, L6 cells were co-infected with Adv-Trisk 95 and AdV-Trisk 51. However, Cav-3 was not co-immunoprecipitated with either anti-triadin isoform antibody (data not shown), suggesting that the presence of the two isoforms is not sufficient for the association with Cav-3
RyR1 Is Associated with Cav-3
An association between RyR2 and Cav-3 was shown in cardiac muscle (20). To test whether Cav-3 directly interacts with RyR1 in skeletal muscle, we took advantage of the triadin null mouse model (7) to analyze the co-immunoprecipitation of Cav-3 with RyR1 in the absence of triadin. As shown in Figure 4A, Cav-3 was efficiently co-immunoprecipitated with the RyR in both WT (Figure 4A, lane 1) and triadin null skeletal muscle microsomes (Figure 4A, lane 3), demonstrating that triadin was not necessary for this association and that the RyR−Cav-3 interaction could be direct. To confirm the direct interaction of the RyR and Cav-3, immunoprecipitation experiments were performed in a non-muscle cell line. COS-7 cells were cotransfected either with Cav-3 and GFP-RyR transmembrane domain (last 582 amino acids fused in frame with GFP on its N-terminal part) or with Cav-3 and GFP alone. The anti-GFP antibody efficiently co-immunoprecipitated the RyR and Cav-3 (Figure 4B, lane 6) but failed to co-immunoprecipitate Cav-3 in the presence of GFP alone (Figure 4B, lane 5), confirming the direct interaction between Cav-3 and the RyR1 transmembrane domain.
Figure 4.

Co-immunoprecipitation of Cav-3 with the RyR. (A) Immunoprecipitation was performed on mouse skeletal muscle from a WT or triadin KO mouse with antibodies against RyR (lanes 1 and 3) or with preimmune serum (lanes 2 and 4). The immunoprecipitated proteins were then analyzed by Western blotting with antibodies against the RyR or Cav-3. (B) Immunoprecipitation was performed with anti-GFP antibodies on COS-7 cells cotransfected with GFP and Cav-3 (lane 5) or GFP-RyR and Cav-3 (lane 6). The immunoprecipitated proteins were then analyzed by Western blotting with antibodies against GFP or Cav-3.
Discussion
In vivo overexpression of the triadin isoform Trisk 95 or Trisk 51 was induced by adenoviral-mediated gene transfer in the hindlimb muscles of newborn mice. Overexpression of one triadin isoform induced a reduction in the expression level of the second one. The different triadin isoforms are issued of the alternative splicing of the same gene (9), and even if the mechanisms driving the specific expression of each isoform are unknown, it is clear that the two triadins Trisk 95 and Trisk 51 not only are always associated together, as seen in the co-immunoprecipitation experiments, but also mutually regulate their expression, to keep the total amount of triadin in the skeletal muscle almost constant. The expression of the main components of the calcium release complex, i.e., RyR, DHPR, the Ca2+-ATPase SERCA, and CSQ, was not affected. However, an interesting finding of our study was the alteration of Cav-3 expression in triadin-infected fibers. Using a biochemical approach, we demonstrated the existence of a molecular complex, including the triadins and Cav-3. Using differential immunoprecipitations in WT and triadin null skeletal muscle, as well as in muscle and non-muscle cells, we further demonstrated that Cav-3 was associated with the CRC via a direct interaction with the transmembrane domain of RyR1. Our data also suggest that two different populations of CRC exist in the skeletal muscle triads, one involving a DHPR−RyR−Cav-3 complex and one involving a Trisk 95−Trisk 51−RyR−Cav-3 complex, as shown in Figure 3. This could be related to the early electronic microscopy study demonstrating that only half of the RyR was coupled to the DHPR (1), and one could imagine that the DHPR and triadin are mutually excluding each other in the CRC, the RyR uncoupled to DHPR interacting with triadin.
Triadin (Trisk 95 or Trisk 51) overexpression results in an increased level of intracellular Cav-3 staining, which is indicative of an in vivo functional link between the two proteins. As we cannot demonstrate Cav-3 overexpression equivalent to triadin overexpression, this increased level of intracellular Cav-3 labeling is most probably due to Cav-3 relocalization. Our finding that Cav-3 is functionally linked to the proteins of the CRC raises the question of the physiological relevance of the association between the caveolae and the SR. Cav-3 was proposed to play a role in calcium homeostasis (21−23), and previous studies showed that the IP3R and SERCA, both involved in calcium homeostasis (24), were detected within the caveolae of several different cell types, including muscle cells (21,22). More recently, Li et al. (24) showed the presence of microdomains enriched in cholesterol, sphingolipids, and Cav-3 in the SR membrane. Assuming that the caveolae−SR contacts are partly responsible for the control of the cytoplasmic free Ca2+ concentrations in very precisely defined spaces (25), the CRC involving triadins could be located at those sites. Previous studies showed that caveolae are sites of extracellular Ca2+ entry (26) and suggested that caveolae are involved in refilling depleted intracellular calcium stores, a mechanism called store-operated calcium entry (SOCE). Caveolae could then act as intermediates between the SR and the plasma membrane. We have previously shown that Trisk 95 can modulate the mechanism coupling the depletion of intracellular Ca2+ stores to extracellular Ca2+ entry. The overexpression of Trisk 95 decreases the level of this Ca2+ entry in myotubes (27). It would therefore be possible that the overexpression of Trisk 95 disturbs the SOCE by its effect on Cav-3. The relocalization of Cav-3 from the plasma membrane to intracellular compartments following the overexpression of Trisk 95 would directly disturb the Ca2+ entry by caveolae and would therefore reduce the level of store-operated Ca2+ entry.
Both Cav-3 null mice and transgenic mice overexpressing Cav-3 display a dystrophic phenotype (17). A likely explanation for the Cav-3 null mice phenotype is the alteration of the intracellular trafficking and targeting of membrane proteins and protein complexes, such as dysferlin (2,28) and the dystrophin glycoprotein complex (17). Noteworthy is the fact that Cav-3 null skeletal muscle also exhibits abnormal DHPR and RyR staining, suggesting that Cav-3 is involved in triad formation during muscle development (29,30). Trisk 95- and Trisk 51-overexpressing muscles exhibited obvious signs of pathology that included central nucleated fibers and heterogeneous fiber sizes that resemble those of dystrophic muscles. Given that Trisk 95 and Trisk 51 seem to mutually regulate their expression level and given that triadin null mice exhibit muscle weakness (7), it is tempting to speculate that a precise expression level of the triadins is crucial for normal muscle function (31); an excess of triadin could be as deleterious as a lack of triadin on the muscle physiology. However, while Trisk 95 overexpression was shown to block EC coupling in skeletal muscle (4), Trisk 51 overexpression has no effect on EC coupling (4). Therefore, it is unlikely that the muscle pathology observed in triadin-overexpressing muscle results entirely from EC coupling dysfunction. In this context, one could speculate that Trisk 95 or Trisk 51 overexpression perturbs the traffic of proteins forming the Ca2+ release machinery by Cav-3 trapping, thus resulting in the abnormal muscle phenotype observed here. A mechanism of Cav-3 trapping has been proposed for the mutant form of Cav-3, P104L, responsible for some cases of myopathy (LGMD1C) (32,33); therefore, this mechanism has already been shown to be responsible for a pathology. Although no disease-causing mutation has been identified yet in the triadin gene, it is possible that modification of the triadin expression level underlies a new pathophysiological mechanism for human myopathies, possibly involving Cav-3 alteration.
Acknowledgments
We thank Genethon (Evry, France) for the production of the adenoviruses used in this study.
This work was supported by grants from Association Française contre les Myopathies (AFM), from Groupement d'Intêret Scientifique (GIS)-Maladies Rares, from Agence Nationale de la Recherche (ANR-Maladies Rares), from Société Française de Myologie (SFM), and from Fondation Ducoin.
Footnotes
Abbreviations: Cav-3, caveolin 3; CRC, calcium release complex; CSQ, calsequestrin; DHPR, dihydropyridine receptor; EC, excitation−contraction; KO, knockout; RyR, ryanodine receptor; SERCA, sarco-endoplasmic reticulum Ca2+-ATPase; T95, Trisk 95; T51, Trisk 51; WT, wild type.
References
- Block B. A.; Imagawa T.; Campbell K. P.; Franzini-Armstrong C. (1988) Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107, 2587–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty I.; Robert M.; Villaz M.; Lai Y.; De Jongh K. S.; Catterall W. A.; Ronjat M. (1994) Biochemical Evidence for a complex involving Dihydropyridine Receptor and Ryanodine Receptor in Triad Junctions of Skeletal Muscle. Proc. Natl. Acad. Sci. U.S.A. 91, 2270–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty I.; Thevenon D.; Scotto C.; Groh S.; Sainnier S.; Robert M.; Grunwald D.; Villaz M. (2000) Cloning and characterization of a new isoform of skeletal muscle triadin. J. Biol. Chem. 275, 8206–8212. [DOI] [PubMed] [Google Scholar]
- Smida Rezgui S.; Vassilopoulos S.; Brocard J.; Platel J. C.; Bouron A.; Arnoult C.; Oddoux S.; Garcia L.; De Waard M.; Marty I. (2005) Triadin (TRISK 95) over-expression blocks excitation-contraction coupling in rat skeletal myotubes. J. Biol. Chem. 280, 39302–39308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh S.; Marty I.; Ottolia M.; Prestipino G.; Chapel A.; Villaz M.; Ronjat M. (1999) Functional interaction of the cytoplasmic domain of triadin with the skeletal ryanodine receptor. J. Biol. Chem. 274, 12278–12283. [DOI] [PubMed] [Google Scholar]
- Ohkura M.; Furukawa K.; Fujimori H.; Kuruma A.; Kawano S.; Hiraoka M.; Kuniyasu A.; Nakayama H.; Ohizumi Y. (1998) Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochemistry 37, 12987–12993. [DOI] [PubMed] [Google Scholar]
- Oddoux S.; Brocard J.; Schweitzer A.; Szentesi P.; Giannesini B.; Brocard J.; Fauré J.; Pernet-Gallay K.; Bendahan D.; Lunardi J.; Csernoch L.; Marty I. (2009) Triadin deletion induces impaired skeletal muscle function. J. Biol. Chem. 284, 34918–34929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; Franzini-Armstrong C.; Lopez J. R.; Jones L. R.; Kobayashi Y. M.; Wang Y.; Kerrick W. G.; Caswell A. H.; Potter J. D.; Miller T.; Allen P. D.; Perez C. F. (2007) Triadins modulate intracellular Ca2+ homeostasis but are not essential for excitation-contraction coupling in skeletal muscle. J. Biol. Chem. 282, 7864–7874. [DOI] [PubMed] [Google Scholar]
- Thevenon D.; Smida-Rezgui S.; Chevessier F.; Groh S.; Henry-Berger J.; Romero N. B.; Villaz V.; De Waard M.; Marty I. (2003) Human skeletal muscle triadin: Gene organization and cloning of the major isoform, Trisk 51. Biochem. Biophys. Res. Commun. 303, 669–675. [DOI] [PubMed] [Google Scholar]
- Williams T. M.; Lisanti M. P. (2004) The caveolin proteins. Genome Biol. 5, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty I.; Robert M.; Ronjat M.; Bally I.; Arlaud G.; Villaz M. (1995) Localization of the N-terminal and C-terminal ends of triadin with respect to the sarcoplasmic reticulum membrane of rabbit skeletal muscle. Biochem. J. 307, 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enouf J.; Lompre A. M.; Bredoux R.; Bourdeau N.; de La Bastie D.; Levy-Toledano S. (1988) Different sensitivity to trypsin of the human platelet plasma and intracellular membrane Ca2+ pumps. J. Biol. Chem. 263, 13922–13929. [PubMed] [Google Scholar]
- Vassilopoulos S.; Thevenon D.; Smida Rezgui S.; Brocard J.; Chapel A.; Lacampagne A.; Lunardi J.; DeWaard M.; Marty I. (2005) Triadins are not triad-specific proteins: Two new skeletal muscle triadins possibly involved in the architecture of sarcoplasmic reticulum. J. Biol. Chem. 280, 28601–28609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allamand V.; Donahue K. M.; Straub V.; Davisson R. L.; Davidson B. L.; Campbell K. P. (2000) Early adenovirus-mediated gene transfer effectively prevents muscular dystrophy in α-sarcoglycan-deficient mice. Gene Ther. 7, 1385–1391. [DOI] [PubMed] [Google Scholar]
- Monnier N.; Marty I.; Faure J.; Castiglioni C.; Desnuelles C.; Sacconi S.; Estournet B.; Ferreiro A.; Romero N.; Laquerriere A.; Lazaro L.; Martin J. J.; Morava E.; Rossi A.; Van der Kooi A.; Verschuuren C.; Lunardi J. (2008) Null mutations causing depletion of the type 1 ryanodine receptor (RYR1) are commonly associated with recessive structural congenital myopathies with cores. Hum. Mutat. 29, 670–678. [DOI] [PubMed] [Google Scholar]
- Kobuke K.; Piccolo F.; Garringer K. W.; Moore S. A.; Sweezer E.; Yang B.; Campbell K. P. (2008) A common disease-associated missense mutation in α-sarcoglycan fails to cause muscular dystrophy in mice. Hum. Mol. Genet. 17, 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F.; Engelman J. A.; Volonte D.; Zhang X. L.; Minetti C.; Li M.; Hou H. Jr.; Kneitz B.; Edelmann W.; Lisanti M. P. (2001) Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and T-tubule abnormalities. J. Biol. Chem. 276, 21425–21433. [DOI] [PubMed] [Google Scholar]
- Couchoux H.; Allard B.; Legrand C.; Jacquemond V.; Berthier C. (2007) Loss of caveolin-3 induced by the dystrophy-associated P104L mutation impairs L-type calcium channel function in mouse skeletal muscle cells. J. Physiol. 580, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balijepalli R. C.; Foell J. D.; Hall D. D.; Hell J. W.; Kamp T. J. (2006) Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc. Natl. Acad. Sci. U.S.A. 103, 7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B. P.; Patel H. H.; Roth D.; Lai N. C.; Niesman I. R.; Farquhar M. G.; Insel P. A. (2005) G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J. Biol. Chem. 280, 31036–31044. [DOI] [PubMed] [Google Scholar]
- Fujimoto T.; Nakade S.; Miyawaki A.; Mikoshiba K.; Ogawa K. (1992) Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. J. Cell Biol. 119, 1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T. (1993) Calcium pump of the plasma membrane is localized in caveolae. J. Cell Biol. 120, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki M.; Anderson R. G. (2003) Function of caveolae in Ca2+ entry and Ca2+-dependent signal transduction. Traffic 4, 717–723. [DOI] [PubMed] [Google Scholar]
- Li C.; Duan W.; Yang F.; Zhang X. (2006) Caveolin-3-anchored microdomains at the rabbit sarcoplasmic reticulum membranes. Biochem. Biophys. Res. Commun. 344, 1135–1140. [DOI] [PubMed] [Google Scholar]
- Lohn M.; Furstenau M.; Sagach V.; Elger M.; Schulze W.; Luft F. C.; Haller H.; Gollasch M. (2000) Ignition of calcium sparks in arterial and cardiac muscle through caveolae. Circ. Res. 87, 1034–1039. [DOI] [PubMed] [Google Scholar]
- Isshiki M.; Ying Y. S.; Fujita T.; Anderson R. G. (2002) A molecular sensor detects signal transduction from caveolae in living cells. J. Biol. Chem. 277, 43389–43398. [DOI] [PubMed] [Google Scholar]
- Vassilopoulos S.; Brocard J.; Garcia L.; Marty I.; Bouron A. (2007) Retrograde regulation of store-operated calcium channels by the ryanodine receptor-associated protein triadin 95 in rat skeletal myotubes. Cell Calcium 41, 179–185. [DOI] [PubMed] [Google Scholar]
- Ampong B. N.; Imamura M.; Matsumiya T.; Yoshida M.; Takeda S. (2005) Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol. 24, 134–144. [PubMed] [Google Scholar]
- Parton R. G.; Way M.; Zorzi N.; Stang E. (1997) Caveolin-3 associates with developing T-tubules during muscle differentiation. J. Cell Biol. 136, 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Deviez D. J.; Martin S.; Laval S. H.; Lo H. P.; Cooper S. T.; North K. N.; Bushby K.; Parton R. G. (2006) Aberrant dysferlin trafficking in cells lacking caveolin or expressing dystrophy mutants of caveolin-3. Hum. Mol. Genet. 15, 129–142. [DOI] [PubMed] [Google Scholar]
- Marty I.; Fauré J.; Fourest-Lieuvin A.; Vassilopoulos S.; Oddoux S.; Brocard J. (2009) Triadin: What possible function 20 years later?. J. Physiol. 587, 3117–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minetti C.; Sotgia F.; Bruno C.; Scartezzini P.; Broda P.; Bado M.; Masetti E.; Mazzocco M.; Egeo A.; Donati M. A.; Volonte D.; Galbiati F.; Cordone G.; Bricarelli F. D.; Lisanti M. P.; Zara F. (1998) Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 18, 365–368. [DOI] [PubMed] [Google Scholar]
- Galbiati F.; Volonte D.; Minetti C.; Chu J. B.; Lisanti M. P. (1999) Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J. Biol. Chem. 274, 25632–25641. [DOI] [PubMed] [Google Scholar]