

Abstract
Central administration of insulin-like growth factor-I (IGF-I) attenuates sickness behavior in response to the cytokine inducer lipopolysaccharide. The present study was designed to determine the respective roles of the two main proinflammatory cytokines, tumor necrosis factorα (TNFα) and interleukin-1β (IL-1β), in these effects. Male CD1 mice were injected into the lateral ventricle (i.c.v.) of the brain with optimal amounts of either TNFα (50 ng) or IL-1β (2 ng) that induce sickness behavior. Behavioral responses to IGF-I (0, .1, and 1 μg) also given i.c.v. were measured at various time intervals before and after treatment with the two proinflammatory cytokines. Mice treated with TNFα and IL-1β lost body weight and displayed equivalent reductions in social exploration and instances of immobility. At the dose of .1 μg, IGF-I attenuated these signs of sickness in TNFα—but not in IL-1β-treated mice. At the dose of 1 μg, IGF-I attenuated IL-1β-induced immobility and the reduction in social exploration but had no effect on loss of body weight. These findings indicate that IGF-I is more potent in attenuating sickness behavior induced by TNFα than that caused by IL-1β, which is consistent with the relative specificity of the TNFα/IGF-I interactions in the brain.
Keywords: Insulin-like growth factor-I, Sickness behavior, IL-1β, TNFα, Mouse, Brain
1. Introduction
Insulin-like growth factor-I (IGF-I) is a multifunctional peptide that is essential for normal growth and development. Its actions are mediated primarily by the IGF-I receptor which is a transmembrane, ligand-activated tyrosine protein kinase highly homologous to the insulin receptor. Multiple signaling pathways are activated by the IGF-I receptor, including the mitogen-activated protein kinase and phosphatidylinositol 3′ kinase signaling pathways (Dupont et al., 2003; Vincent and Feldman, 2002). IGF-I promotes cell proliferation and inhibits cell death during both normal development and during stress and disease. These actions also occur in the central nervous system. Transgenic mice over-expressing IGF-I postnatally exhibit brain overgrowth characterized by increased neuron and oligodendrocyte number, as well as marked increases in myelination (D’Ercole et al., 2002). Mutant mice with ablated IGF-I and IGF-I receptor expression, as well as those with overexpression of the binding proteins to IGF-I that are capable of inhibiting IGF-I actions, exhibit brain growth retardation, decreases in glucose utilization (Cheng et al., 2000) and disruption in lipid and microtubule metabolism, leading to impaired neuronal somatic and dendritic growth (Cheng et al., 2003). These studies confirm a role for IGF-I in neural development, and indicate that IGF-I stimulates neurogenesis and synaptogenesis, facilitates oligodendrocyte development, promotes neuron and oligodendrocyte survival, and stimulates myelination. There is also evidence that IGF-I has neuroprotective effects following CNS and peripheral nerve injury (Guan et al., 2003; Zheng et al., 2000). In particular, there are important reciprocal interactions between IGF-I and proinflammatory cytokines that mediate neuroinflammation. For instance, overexpression of tumor necrosis factor-alpha (TNFα) causes brain growth retardation and neural damage that are associated with impaired IGF-I activity, particularly in the cerebellum (Ye et al., 2003). In the same manner, cytotoxic effects of TNFα on granular cerebellar neurons are associated with silencing of IGF-I survival signals (Venters et al., 1999). Reciprocally, addition of IGF-I can rescue neurons from TNFα-induced death (Kenchappa et al., 2004).
The inhibiting effects of supplemental IGF-I on neuroinflammation do not occur only in conditions of neuronal damage. There is evidence, for instance, that IGF-I can oppose the behaviorally depressing effects of the cytokine inducer lipopolysaccharide when both compounds are administered into the brain (Dantzer et al., 1999). These findings are important since they indicate that IGF-I can act as an anti-inflammatory cytokine in the brain. However, they do not provide any clue about the particular cytokine that is specifically targeted by IGF-I. In view of the findings previously outlined (Kenchappa et al., 2004; Venters et al., 1999; Ye et al., 2003), TNFα is a likely candidate. The present experiments were therefore carried out to specifically assess the antagonistic effects of IGF-I against the two main proinflammatory cytokines that mediate sickness behavior in the brain, TNFα and interleukin-1β (IL-1β) (Bluthé et al., 2000). The results show unequivocally that IGF-I more potently inhibits TNFα than IL-1β actions in the brain.
2. Methods
2.1. Animals
All experiments were performed on adult male CD1 Crl:CD-1 (ICR) IGS mice (8-week-old, mean body weight: 29.8 g, when tested). Mice were obtained from a colony raised in the laboratory and maintained in groups of five in transparent polycarbonate cages (26.6 × 21.4 × 14.3 cm). They were maintained under standard colony conditions on corncob litter (U.A.R., Epinay-sur-Orge, France) in a temperature- (23 ± 1 °C) and humidity (40%)-controlled room and on a 12–12 h light/dark cycle (lights off at 09:00 h) with free access to feed and water.
For social exploration tests, juvenile male mice (21–28 days of age) of the Crl:CD1(ICR)BR strain served as social stimuli. They were housed in groups of 10 in transparent polycarbonate cages (42.6 × 27.8 × 15.4 cm) and in a different room. Behavioral tests were initiated during the dark phase, with strict adherence to the guidelines for experimentation in animals that were approved by the Institutional Animal Care and Use Committee. Every effort was made to minimize animal numbers and suffering. To habituate mice to the handling procedure to be used during the experiments, mice were handled daily for 5 days prior to initiation of treatments.
2.2. Surgical procedures
For intracerebroventricular (i.c.v.) injections, a stainless-steel guide cannula (23-gauge, 7 mm length) was implanted unilaterally 1 mm above the lateral ventricle. For this purpose, mice were anesthetized by intraperitoneal (i.p.) administration of .01 ml/g of a mixture of ketamine (6.6 mg/kg, Imalgene 1000, Rhône Mérieux, Lyon, France) and xylazine (.9 mg/kg, Bayer Pharma, Puteaux, France). Mice were secured in a Kopf stereotaxic instrument (Tujunga, CA, USA). Coordinates for the guide cannula were .6 mm posterior to bregma, 1.6 mm lateral, and 2.0 mm below the skull surface at the point of entry (Paxinos and Franklin, 2001). A 2-week recovery period was allowed before initiation of the treatments. At the conclusion of the experiment, cannula placement was verified by injecting a solution of India ink into the guide cannula and sectioning the brain after removal from the skull.
2.3. Behavioral observations
Mice were isolated 24 h before the experiment in transparent polycarbonate cages (26.6 × 21.4 × 14.3 cm). All behavioral observations were carried out during the dark phase, between 09:00 and 17:00 h, using a video camera under red light illumination. Sickness behavior induced by IL-1β or TNFα was assessed by three criteria: (a) decrease in duration of social exploration directed toward a juvenile introduced for 4 min into the home cage of the test animal; (b) appearance of immobility of the test animal during the observation session; and (c) reduction in body weight. All of these procedures used to measure sickness have been validated in mice (Bluthé et al., 1991b). Briefly, behavioral observations took place in the isolation cage of the test animal. During each exposure, a trained observer unaware of the treatment recorded total duration of social exploration of the juvenile by using pre-set keys on the keyboard of a microcomputer. Social exploration consisted mainly of anogenital sniffing and included also nosing, sniffing, and grooming behaviors directed toward the juvenile. Immediately after the first behavioral test mice were weighed, then the resident mouse was injected i.c.v. with the treatments under study and retested with different juveniles 1.5, 3, and 6 h after treatments, then weighed on a top-loading balance accurate to .1 g at 24 h after injections. Behavior was monitored at least 1.5 h after i.c.v. treatment administration, since this latency is necessary for development of the characteristic behavioral alterations of sickness (Kent et al., 1992).
2.4. Treatments
Recombinant rat IL-1β (biological activity: 317 IU/μg, National Biological Standards and Control, Potters Bar, UK) (2 ng/mouse, i.c.v.) was selected on the basis of previous experiments carried out in CD1 mice (Cremona et al., 1998). Activity of the recombinant rat IL-1β preparation used in this study had already been characterized in the rat system (Anforth et al., 1998), and this cytokine is fully biologically active in the mouse system as demonstrated by its ability to induce the whole spectrum of clinical signs of sickness (Cremona et al., 1998). The dose of recombinant murine TNFα (R&D systems, 50 ng/mouse, i.c.v.) used was selected from a dose-effect relationship of this cytokine on social exploration in CD1 mice so as to produce a decrease in social exploration which was similar to that induced by IL-1β (Bluthé, unpublished data). Based on previous experiments showing the ability of 1 μg recombinant human IGF-I (Intergen, NY, USA) injected i.c.v. to attenuate behavioral effects of i.c.v. LPS in mice (Dantzer et al., 1999), the present experiments were carried out with two doses of IGF-I (.1 and 1 μg/mouse). All substances were dissolved in apyrogenic 0.9% saline and administered to non-anesthetized animals in a volume of 2 μl by gravity over 90 s using a 30-gauge needle. Each mouse received only one combination of treatments.
2.5. Experimental protocols
The experiments were carried out in two successive series. In a first series, mice were pretreated i.c.v. with IGF-I (.1 μg/mouse, i.c.v.) or saline immediately before being treated with either IL-1β, TNFα or saline (5–7 mice per group). In a second series carried out to assess the effects of a higher dose of IGF-I on IL-1β-induced sickness behavior, two doses of IGF-I (.1 and 1 μg/mouse, i.c.v.) were injected to different mice injected with saline or IL-1β (6–9 mice/group).
2.6. Statistical analysis
Social exploration and body weight during the first social presentation were analyzed by a one-way ANOVA. Duration of social investigation was analyzed by a 3-way ANOVA with pre-treatment (saline or IGF-I) and treatment (saline, IL-1β or TNFα) as between-subject factors and time (0, 1.5, 3, and 6 h) as a within-subject factor. Because immobility was never observed in the absence of cytokine treatment, duration of immobility was analyzed only when it was present, i.e., at 1.5, 3, and 6 h in the first series of experiments and at 1.5 and 3 h in the second series of experiments. Statistical analysis was carried out with a 3-way ANOVA with pre-treatment (saline or IGF-I) and treatment (IL-1β or TNFα) as between-subject factors and time (1.5, 3, and 6 h) as a within-subject factor. The effects of treatments on changes in body weight over a post-treatment 24-h period were assessed using a two-way ANOVA (pre-treatment × treatment). Post hoc comparisons of individual group means were carried out by the protected least significant difference test (LSD).
In the second series of experiments, social exploration and immobility were analyzed by a two-way ANOVA with treatment as a between-subject factor and time as a within subject factor. The effects of treatments on changes in body weight over a 24-h post-treatment period were assessed using a one-way ANOVA. Post hoc comparisons of individual group means were carried out by the protected least significant difference test (LSD).
3. Results
3.1. IGF-I attenuates-TNFα-but not IL-1β-induced sickness behavior
Baseline of social exploration (time 0 in Fig. 1A) ranged from 136 to 172 s, and did not differ according to pre-treatment and treatment [F (5, 33) = 1.4, p = .26]. IL-1β and TNFα reduced duration of social exploration in a time-dependent manner [treatment: F (2, 28) = 96.0, p < .001; time: F (3, 84) = 60.4, p < .001; and treatment × time: F (6, 84) = 35.1, p < .001]. IGF-I pre-treatment (.1 μg/mouse) had not effect alone but significantly increased duration of social exploration [F (1, 28) = 4.0, p < .05] and this effect was dependent on the treatment [F (2, 28) = 5.8, p < .01] and the treatment × time interaction [F (6, 84) = 2.0, p < .05]. Post hoc comparisons of individual group means revealed that IGF-I attenuated the TNFα-induced reduction in social exploration 1.5 and 3 h after pre-treatment whereas IGF-I has no effect on IL-1β-induced sickness behavior (Fig. 1A).
Fig. 1.
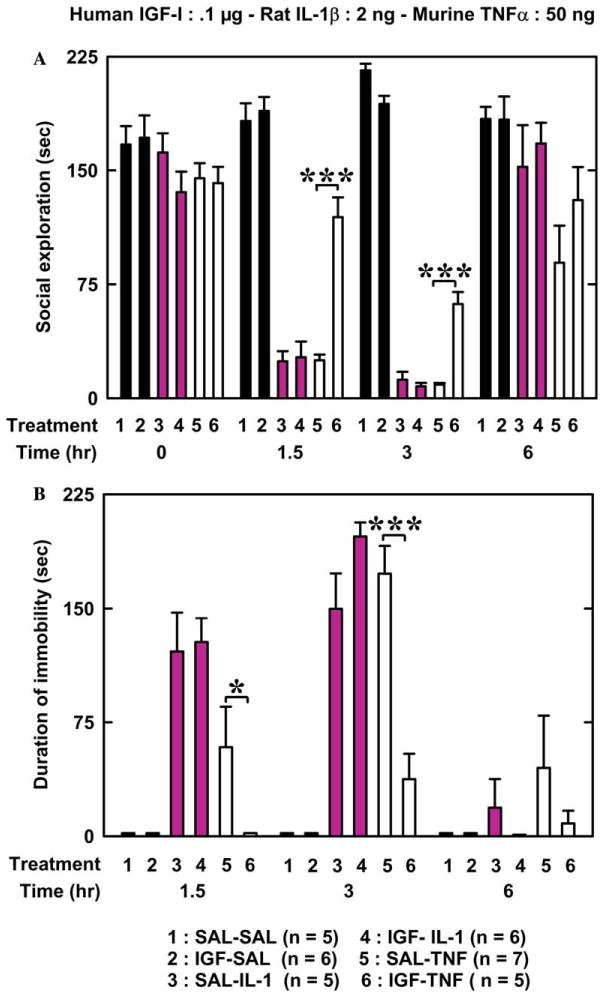
A low dose of IGF-I attenuates sickness behavior induced by TNFα but not by IL-1β. IGF-I or physiological saline (SAL) was injected i.c.v. just before SAL, IL-1β or TNFα. The figure represents duration of social exploration (A) and immobility (B) measured immediately before the treatments and at various time intervals afterwards. [(A): ***p < .001, for SAL-TNF vs IGF-TNF; (B) *p < .05 and ***p < .001 for SAL-TNF vs IGF-TNF]. Values are means ± SEM.
Saline- and IGF-I-treated mice always remained active, which is why immobility data for these animals are equal to 0 in Fig. 1B. IL-1β and TNFα increased immobility [F (1, 19) = 12.9, p < .01], an effect that was most marked at 3 h post-injection [F (2, 38) = 61.5, p < .001]. IGF-I pre-treatment decreased duration of immobility [F (1, 19) = 5.7, p < .05] in a treatment [pre-treatment × treatment: F (1, 19) = 10.6, p < .01] and time [pre-treatment × time: F (2, 38) = 14.4, p < .001] dependent manner. The pre-treatment × treatment × time interaction was significant [F (2, 38) = 7.6, p < .001]. Post hoc comparisons of individual group means revealed that IGF-I attenuated TNFα-induced immobility 1.5 and 3 h after injection but had no effect on IL-1β-induced immobility (Fig. 1B).
Initial body weight ranged from 28.6 to 31.0 g and did not differ according to treatments at initiation of the experiment (time 0) [F (5, 33) = 1.33, p = .28]. There was no significant effect of pre-treatment and treatment factors and pre-treatment × treatment interaction on body weight change at 24 h. [F (1, 28) = 3.44, p < .08; F (2, 28) = 1.19, p = .32; and F (2, 28) = 2.7, p < .09, respectively] (Fig. 2).
Fig. 2.
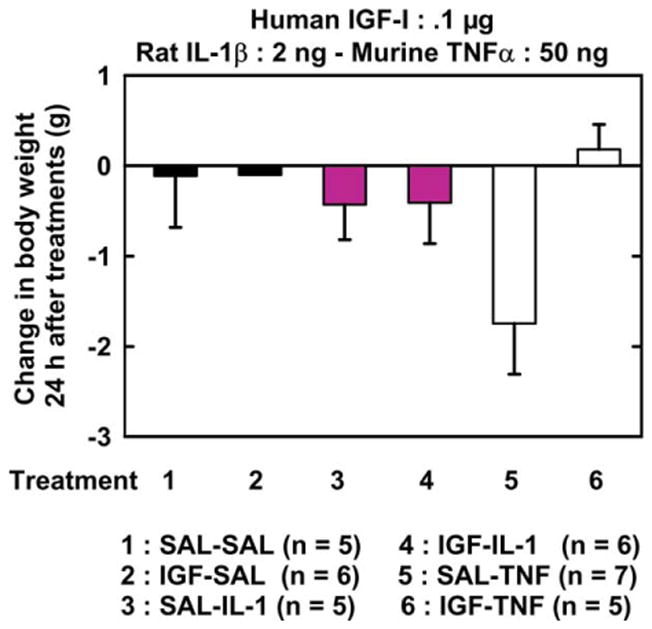
IGF-I does not alter the effects of IL-1β and TNFα on changes in body weight. IGF-I or SAL was injected i.c.v. just before i.c.v. IL-1β, TNFα or SAL. Body weight change was calculated as the difference between body weight at 24 h post-treatment and body weight immediately before treatments. Values are means ± SEM.
3.2. IGF-I at a higher dose attenuates IL-1β-induced sickness behavior
The duration of social exploration ranged from 153 to 159 s at time 0 and did not differ between treatments [F (3,26) = .054, p = .98]. Duration of social exploration varied according to treatment in a time-dependent manner [treatment: F (3,23) = 6.75, p < .01; time: F(3,69) = 44.4, p < .001; and treatment × time F (9,69) = 6.8, p < .001]. Post hoc comparisons of individual groups revealed that the highest dose of IGF-I (1 μg/mouse) attenuated IL-1β-induced decrease in social exploration 3 h after injection whereas IGF-I at the lower dose (.1 μg/mouse) potentiated the IL-1β-induced decrease in social exploration 1.5 h after injection (Fig. 3A).
Fig. 3.
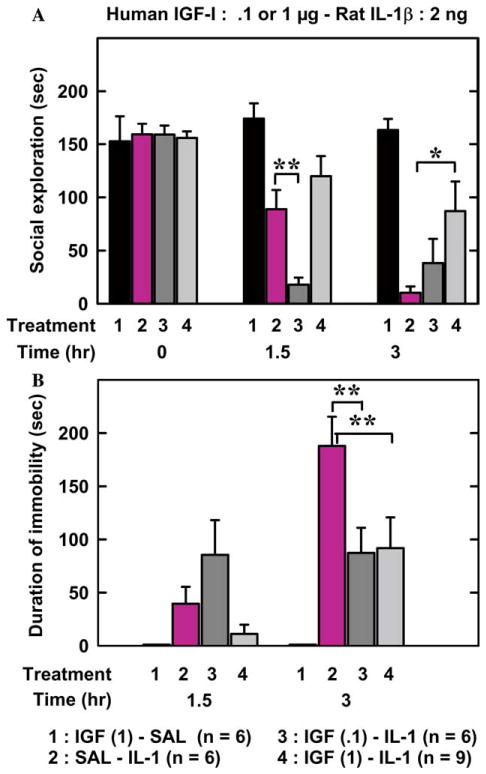
Increasing the dose of IGF-I attenuates IL-1β-induced sickness behavior. SAL or IGF-I (.1 or 1 μg) was injected i.c.v. just before SAL or IL-1 injected via the same route. The figure represents duration of social exploration (A) and immobility (B) measured immediately before the treatments and at various time intervals thereafter. [(A): **p < .01 for SAL-IL-1 vs IGF (.1)-IL-1 and *p < .05 for SAL-IL-1 vs IGF (1)-IL-1; (B): **p < .01 for SAL-IL-1 vs both IGF (.1)-IL-1 and IGF (1)-IL-1]. Values are means ± SEM.
IGF-I-SAL-treated mice always remained active, which is why immobility data for these animals are equal to 0 in Fig. 3B. Duration of immobility varied according to time [F (1, 18) = 29.9, p < .001] and treatments [F (2, 18) = 7.6, p < .01]. Post hoc comparisons of individual groups revealed that the two doses of IGF-I attenuated immobility-induced by IL-1β at 3 h after treatments (Fig. 3B).
Body weight ranged from 30.5 to 31.7 g and did not differ according to treatments on time 0 [F (3, 23) = .32, p = .81]. Body weight changes differed according to treatments [F (3, 23) = 4.27, p < .05]. Post hoc comparisons of individual group means revealed that IL-1β depressed body weight and IGF-I did not abrogate this effect (p < .05) (Fig. 4).
Fig. 4.
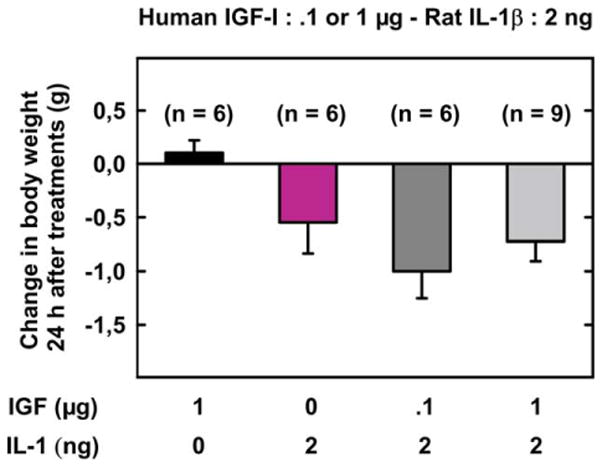
Effects of IGF-I on IL-1β-induced changes in body weight. IGF-I (.1 or 1 μg) or SAL was injected i.c.v. just before SAL or IL-1β injected via the same route. Body weight change was calculated as the difference between body weight at 24 h post-treatment and body weight immediately before treatments. Values are means ± SEM.
4. Discussion
The present results demonstrate that IGF-I administered i.c.v. is much more potent in inhibiting centrally injected TNFα than IL-1β, as measured by sickness behavior. These results can be interpreted to suggest that the beneficial effects of IGF-I in the brain are mainly related to its antagonism of TNFα activity. When injected into the brain, both TNFα and IL-1β reduce social exploration, induce immobility, and decrease body weight. In terms of dose-effect relationship, IL-1β is much more potent than TNFα (Bluthé et al., 1994). We have previously shown that the behaviorally depressing effects of TNFα are mediated by the production of IL-1β (Bluthé et al., 1991a) and that TNFα plays a major role in LPS-induced sickness behavior only when IL-1β is deficient (Bluthé et al., 2000). It is therefore not surprising that IGF-I has only partial antagonistic effects on TNFα-induced sickness behavior at the lower dose tested in this study since this same dose of IGF-I has no effect on IL-1β-induced sickness behavior.
Previous studies on the ability of IGF-I to antagonize proinflammatory cytokines on various cell types have not carefully considered a possible differential effect of this growth factor according to the nature of the cytokine. For instance, IGF-I decreased both the basal and the cytokine-stimulated degradation of proteoglycan in explanted cartilage exposed to TNFα or to IL-1β (Tyler, 1989). IGF-I antagonized inhibitory and cytotoxic effects of IL-1β in islets of Langerhans (Mabley et al., 1997). There was apparently no difference in the sensitivity of IL-1β versus that of TNFα to these effects (Hill et al., 1999). In the same manner, IGF-I was able to antagonize IL-1β and TNFα induction of nitric oxide synthase in vascular smooth muscle cells (Schini et al., 1994). However, all these experiments have been conducted in vitro with doses of IGF-I largely in excess. In vivo, IGF-I has been shown to decrease induction of both TNFα and IL-1β in septic shock (Inoue et al., 1995) and in burn wounds (Spies et al., 2001). In this last case, the effects were obtained by local IGF-I gene transfer. No similar experiments have been done in the brain. The main finding of the present experiments is the higher activity of IGF-I against TNFα-rather than IL-Iβ-induced sickness behavior. This is not an artifact due to a difference in baseline levels of sickness behavior since the doses of TNFα and IL-1β have been carefully selected for inducing similar depressing effects on behavior. The effects of TNFα are actually more prolonged than those of IL-1β, indicating that TNFα tends to be more potent at the dose tested than IL-1β. The alleviated effects of IGF-I on TNFα-induced decrease in social exploration and locomotor activity were not reflected in its effect on TNFα-induced body weight loss. Whether this is due to a higher inter-individual variation in this variable or to different mechanisms cannot be determined at this stage. However, this type of dissociation between treatment effects on the behavioral vs metabolic effects of cytokines has already been observed (Bluthé et al., 1995).
TNFα causes sickness behavior by inducing synthesis of IL-1β (Bluthé et al., 1991a). Since IGF-I ameliorates sickness behavior induced by TNFα but not that caused by IL-1β, it appears likely that IGF-I acts by inhibiting the ability of TNFα to induce synthesis of IL-1β. There are two receptors for TNFα, and we have already established that activation of TNF-R1 is adequate for inducing sickness behavior (Bluthé et al., 1994). Furthermore, we have shown that a partial agonist of the murine TNF-R1 also induces sickness behavior (Bluthé et al., 1991a; Bluthé et al., 1994). It is therefore likely that IGF-I somehow antagonizes activation of TNF-R1 that leads to induction of IL-1 synthesis. A protein known as “Factor Associated with N-SMase” (FAN) binds directly to only the p55 receptor and links TNFα to activation of neutral sphingomyelinase (Adam-Klages et al., 1996; Adam-Klages et al., 1998). FAN has been shown to bind to only two receptors, p55 and CD40, a member of the TNF-R superfamily (Segui et al., 1999). Although the TNF-R1 and IL-1R share several signaling pathways (Wu and Arron, 2003), FAN has not been shown to associate with the IL-1β receptor. It is possible that IGF-I interacts downstream of TNF-R1 with FAN, which might explain why IGF-I is more active in ameliorating TNFα-rather than IL-1β-induced sickness behavior. FAN knockout mice could be used to conclusively determine if TNFα-activated FAN is responsible for inducing sickness behavior caused by central TNFα.
In conclusion, the present results show that IGF-I antagonizes in a relatively specific manner TNFα behavioral actions in the brain. They therefore open important new avenues for studying further the mechanisms of interaction of this growth factor with this pivotal cytokine.
Acknowledgments
The technical assistance of Guy Olivié is gratefully acknowledged. Dr. Robert H. McCusker carefully edited the manuscript. This research was supported by grants to R.D. (NIH-MH 71349) and K.W.K. (NIH-MH 51569, NIH-AI 50442, Hatch 200386 and private gifts).
References
- Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Kronke M. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- Adam-Klages S, Schwandner R, Adam D, Kreder D, Bernardo K, Kronke M. Distinct adapter proteins mediate acid versus neutral sphingomyelinase activation through the p55 receptor for tumor necrosis factor. J Leukoc Biol. 1998;63:678–682. doi: 10.1002/jlb.63.6.678. [DOI] [PubMed] [Google Scholar]
- Anforth HR, Bluthé RM, Bristow A, Hopkins S, Lenczowski MJ, Luheshi G, Lundkvist J, Michaud B, Mistry Y, Van Dam AM, Zhen C, Dantzer R, Poole S, Rothwell NJ, Tilders FJ, Wollman EE. Biological activity and brain actions of recombinant rat interleukin-1alpha and interleukin-1beta. Eur Cytokine Netw. 1998;9:279–288. [PubMed] [Google Scholar]
- Bluthé RM, Beaudu C, Kelley KW, Dantzer R. Differential effects of IL-1ra on sickness behavior and weight loss induced by IL-1 in rats. Brain Res. 1995;677:171–176. doi: 10.1016/0006-8993(95)00194-u. [DOI] [PubMed] [Google Scholar]
- Bluthé RM, Dantzer R, Kelley KW. Interleukin-1 mediates behavioural but not metabolic effects of tumor necrosis factor alpha in mice. Eur J Pharmacol. 1991a;209:281–283. doi: 10.1016/0014-2999(91)90184-r. [DOI] [PubMed] [Google Scholar]
- Bluthé RM, Parnet P, Dantzer R, Kelley KW. Interleukin-1 receptor antagonist blocks effects of IL-1α and IL-1β on social behaviour and body weight in mice. Neurosci Res Commun. 1991b;9:151–158. [Google Scholar]
- Bluthé RM, Layé S, Michaud B, Combe C, Dantzer R, Parnet P. Role of interleukin-1beta and tumour necrosis factor-alpha in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. Eur J Neurosci. 2000;12:4447–4456. [PubMed] [Google Scholar]
- Bluthé RM, Pawlowski M, Suarez S, Parnet P, Pittman Q, Kelley KW, Dantzer R. Synergy between tumor necrosis factor alpha and interleukin-1 in the induction of sickness behavior in mice. Psychoneuroendocrinology. 1994;19:197–207. doi: 10.1016/0306-4530(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Cheng CM, Mervis RF, Niu SL, Salem N, Jr, Witters LA, Tseng V, Reinhardt R, Bondy CA. Insulin-like growth factor 1 is essential for normal dendritic growth. J Neurosci Res. 2003;73:1–9. doi: 10.1002/jnr.10634. [DOI] [PubMed] [Google Scholar]
- Cheng CM, Reinhardt RR, Lee WH, Joncas G, Patel SC, Bondy CA. Insulin-like growth factor 1 regulates developing brain glucose metabolism. Proc Natl Acad Sci USA. 2000;97:10236–10241. doi: 10.1073/pnas.170008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona S, Goujon E, Kelley KW, Dantzer R, Parnet P. Brain type I but not type II IL-1 receptors mediate the effects of IL-1 beta on behavior in mice. Am J Physiol. 1998;274:R735–R740. doi: 10.1152/ajpregu.1998.274.3.R735. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Gheusi G, Johnson RW, Kelley KW. Central administration of insulin-like growth factor-1 inhibits lipopolysaccharide-induced sickness behavior in mice. NeuroReport. 1999;10:289–292. doi: 10.1097/00001756-199902050-00015. [DOI] [PubMed] [Google Scholar]
- D’Ercole AJ, Ye P, O’Kusky JR. Mutant mouse models of insulin-like growth factor actions in the central nervous system. Neuropeptides. 2002;36:209–220. doi: 10.1054/npep.2002.0893. [DOI] [PubMed] [Google Scholar]
- Dupont J, Dunn SE, Barrett JC, LeRoith D. Microarray analysis and identification of novel molecules involved in insulin-like growth factor-1 receptor signaling and gene expression. Recent Prog Horm Res. 2003;58:325–342. doi: 10.1210/rp.58.1.325. [DOI] [PubMed] [Google Scholar]
- Guan J, Bennet L, Gluckman PD, Gunn AJ. Insulin-like growth factor-1 and post-ischemic brain injury. Prog Neurobiol. 2003;70:443–462. doi: 10.1016/j.pneurobio.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Hill DJ, Petrik J, Arany E, McDonald TJ, Delovitch TL. Insulin-like growth factors prevent cytokine-mediated cell death in isolated islets of Langerhans from pre-diabetic non-obese diabetic mice. J Endocrinol. 1999;161:153–165. doi: 10.1677/joe.0.1610153. [DOI] [PubMed] [Google Scholar]
- Inoue T, Saito H, Fukushima R, Inaba T, Lin MT, Fukatsu K, Muto T. Growth hormone and insulin-like growth factor I enhance host defense in a murine sepsis model. Arch Surg. 1995;130:1115–1122. doi: 10.1001/archsurg.1995.01430100093018. [DOI] [PubMed] [Google Scholar]
- Kenchappa P, Yadav A, Singh G, Nandana S, Banerjee K. Rescue of TNFalpha-inhibited neuronal cells by IGF-1 involves Akt and c-Jun N-terminal kinases. J Neurosci Res. 2004;76:466–474. doi: 10.1002/jnr.20081. [DOI] [PubMed] [Google Scholar]
- Kent S, Bluthé RM, Dantzer R, Hardwick AJ, Kelley KW, Rothwell NJ, Vannice JL. Different receptor mechanisms mediate the pyrogenic and behavioral effects of interleukin 1. Proc Natl Acad Sci USA. 1992;89:9117–9120. doi: 10.1073/pnas.89.19.9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabley JG, Belin V, John N, Green IC. Insulin-like growth factor I reverses interleukin-1beta inhibition of insulin secretion, induction of nitric oxide synthase and cytokine-mediated apoptosis in rat islets of Langerhans. FEBS Lett. 1997;417:235–238. doi: 10.1016/s0014-5793(97)01291-x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. 2. (Deluxe) Academic Press; New York: 2001. [Google Scholar]
- Schini VB, Catovsky S, Schray-Utz B, Busse R, Vanhoutte PM. Insulin-like growth factor I inhibits induction of nitric oxide synthase in vascular smooth muscle cells. Circ Res. 1994;74:24–32. doi: 10.1161/01.res.74.1.24. [DOI] [PubMed] [Google Scholar]
- Segui B, Andrieu-Abadie N, Adam-Klages S, Meilhac O, Kreder D, Garcia V, Bruno AP, Jaffrezou JP, Salvayre R, Kronke M, Levade T. CD40 signals apoptosis through FAN-regulated activation of the sphingomyelin-ceramide pathway. J Biol Chem. 1999;274:37251–37258. doi: 10.1074/jbc.274.52.37251. [DOI] [PubMed] [Google Scholar]
- Spies M, Nesic O, Barrow RE, Perez-Polo JR, Herndon DN. Liposomal IGF-1 gene transfer modulates pro- and anti-inflammatory cytokine mRNA expression in the burn wound. Gene Ther. 2001;8:1409–1415. doi: 10.1038/sj.gt.3301543. [DOI] [PubMed] [Google Scholar]
- Tyler JA. Insulin-like growth factor 1 can decrease degradation and promote synthesis of proteoglycan in cartilage exposed to cytokines. Biochem J. 1989;260:543–548. doi: 10.1042/bj2600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venters HD, Tang Q, Liu Q, VanHoy RW, Dantzer R, Kelley KW. A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc Natl Acad Sci USA. 1999;96:9879–9884. doi: 10.1073/pnas.96.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vincent AM, Feldman EL. Control of cell survival by IGF signaling pathways. Growth Horm IGF Res. 2002;12:193–197. doi: 10.1016/s1096-6374(02)00017-5. [DOI] [PubMed] [Google Scholar]
- Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Bioessays. 2003;25:1096–1105. doi: 10.1002/bies.10352. [DOI] [PubMed] [Google Scholar]
- Ye P, Price W, Kassiotis G, Kollias G, D’Ercole AJ. Tumor necrosis factor-alpha regulation of insulin-like growth factor-I, type 1 IGF receptor, and IGF binding protein expression in cerebellum of transgenic mice. J Neurosci Res. 2003;71:721–731. doi: 10.1002/jnr.10512. [DOI] [PubMed] [Google Scholar]
- Zheng WH, Kar S, Dore S, Quirion R. Insulin-like growth factor-1 (IGF-1): a neuroprotective trophic factor acting via the Akt kinase pathway. J Neural Transm Suppl. 2000:261–272. doi: 10.1007/978-3-7091-6301-6_17. [DOI] [PubMed] [Google Scholar]