

Abstract
Polyglutamine (polyQ) expansion within the ataxin-7 protein, a member of the STAGA [SPT3-TAF(II)31-GCN5L acetylase] and TFTC (GCN5 and TRRAP) chromatin remodeling complexes, causes the neurodegenerative disease spinocerebellar ataxia type 7 (SCA7). Proteolytic processing of ataxin-7 by caspase-7 generates N-terminal toxic polyQ-containing fragments that accumulate with disease progression and play an important role in SCA7 pathogenesis. To elucidate the basis for the toxicity of these fragments, we evaluated which posttranslational modifications of the N-terminal fragment of ataxin-7 modulate turnover and toxicity. Here, we show that mutating lysine 257 (K257), an amino acid adjacent to the caspase-7 cleavage site of ataxin-7 regulates turnover of the truncation product in a repeat-dependent manner. Modification of ataxin-7 K257 by acetylation promotes accumulation of the fragment, while unmodified ataxin-7 is degraded. The degradation of the caspase-7 cleavage product is mediated by macroautophagy in cell culture and primary neuron models of SCA7. Consistent with this, the fragment colocalizes with autophagic vesicle markers, and enhanced fragment accumulation increases in these lysosomal structures. We suggest that the levels of fragment accumulation within the cell is a key event in SCA7 neurodegeneration, and enhancing clearance of polyQ-containing fragments may be an effective target to reduce neurotoxicity in SCA7.
Introduction
Spinocerebellar ataxia type 7 (SCA7) is a dominantly inherited neurodegenerative disease characterized by late-onset degeneration of the cerebellum, brainstem, and retina. SCA7 is caused by expansion of the polyglutamine tract within the ataxin-7 protein (Lindblad et al., 1996; David et al., 1997; Del-Favero et al., 1998). SCA7 shares common properties of other polyglutamine expansion diseases, including Huntington's disease (HD), dentatorubral–pallidoluysian atrophy (DRPLA), and spinal bulbar muscular atrophy (SBMA), but a distinguishing clinical feature is blindness correlating with the transcriptional dysregulation of specific retinal genes (La Spada et al., 2001; Helmlinger et al., 2006). Ataxin-7 is a homolog of the yeast Sgf73p, a component of the SAGA (Spt/Ada/Gcn5/acetyltransferase) chromatin remodeling complex, and is now known to function in the human STAGA [SPT3-TAF(II)31-GCN5L acetylase] and TFTC (GCN5 and TRRAP) complexes (Sanders et al., 2002; McMahon et al., 2005), although its exact role remains unclear. Mutant polyglutamine expansion disrupts the normal function of ataxin-7 in these complexes (Palhan et al., 2005; Helmlinger et al., 2006) and confers additional gain-of-function properties, such as aggregation by polyQ-containing fragments (Holmberg et al., 1998).
Protein turnover is a crucial mechanism for maintaining cellular viability over time. Accordingly, disruption of proteolytic pathways occurs in polyglutamine expansion diseases, Alzheimer's disease and Parkinson's disease (for review, see Olzmann et al., 2008). The ubiquitin proteasome system (UPS) is one of two major proteolytic pathways. In this pathway, ubiquitin modification targets misfolded and short-lived proteins to the cytosolic 26S proteasome for degradation. polyQ aggregates are resistant to proteasomal degradation, and disrupt global UPS activity (Bennett et al., 2007; Tydlacka et al., 2008). In other studies, it was found that global UPS activity in vivo was not affected in the brains of polyQ disease transgenic mice (Bowman et al., 2005; Bett et al., 2009; Jeong et al., 2009). Ataxin-7 interacts with a portion of the 19S subunit (Matilla et al., 2001), and the polyQ-expanded form of ataxin-7 can inhibit proteasomal function (Wang et al., 2007). The second proteolytic pathway, autophagy, degrades whole organelles and cytoplasmic material. Three types of autophagy have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy (hereafter referred to as autophagy) can degrade polyQ-expanded fragments (Qin et al., 2003; Young et al., 2009). Upregulating autophagy can ameliorate polyQ-dependent toxicity in models of HD and SBMA (Yamamoto et al., 2006; Pandey et al., 2007; Sarkar et al., 2009).
The toxic effects of polyglutamine expansion are protein context dependent, and can be modulated by posttranslational modification. Proteolytic cleavage of these proteins by caspases generates short, polyglutamine-containing fragments with increased cellular toxicity (Wellington et al., 1998; Ellerby et al., 1999a,b). Proteolytic cleavage products are frequently found in aggregated inclusions observed in both in vitro and in vivo polyglutamine disease models (Ross, 1997; Zhou et al., 2003) and in postmortem tissue from patients (DiFiglia et al., 1997; Wellington et al., 2002). We have previously shown that ataxin-7 is cleaved by caspase-7 at amino acid positions D266 and D344 (Young et al., 2007). In SCA7 and HD mouse models, substitution of aspartate to alanine or asparagine at these sites blocks the formation of N-terminal truncation fragments and ameliorates disease symptoms (Graham et al., 2006) (S. J. Guyenet, A. R. La Spada, S. Mookerjee, A. Lin, S. K. Custer, S. F. Chen, B. L. Sopher, L. M. Ellerby, unpublished work).
Additionally, lysine modification by small ubiquitin-like protein (SUMO1) or acetylation has been shown to modulate protein accumulation and toxicity in HD, SCA1, DRPLA, and SBMA (Chan et al., 2002; Terashima et al., 2002; Steffan et al., 2004; Riley et al., 2005). A ubiquitin competitor, SUMO1, appears to stabilize proteins and decrease their degradation (Steffan et al., 2004). For SBMA, acetylation of polyQ-expanded androgen receptor impacts its aggregation and receptor trafficking (Thomas et al., 2004). In ataxin-7, we identified lysine 257 (K257), a highly conserved residue near the D266 caspase cleavage site, as an important modulator of fragment accumulation in vitro and in vivo. Through chemical disruption, we determined that autophagy mediates the turnover of the ataxin-7 fragment. Furthermore, the acetylase, CBP, and the deacetylase, HDAC7, regulated ataxin-7 turnover through acetylation of the fragment. The work presented here suggests that pathways that enhance the clearance of toxic fragments may effectively mitigate SCA7 pathogenesis.
Materials and Methods
Plasmid constructs.
Ataxin-7 cDNA containing either 10 or 92 CAG repeats in pcDNA3.1 (Invitrogen) was used in these studies as previously published (La Spada et al., 2001; Young et al., 2007). Site-directed mutagenesis was performed to generate K→R substitutions at amino acid position K-223 using the primers 5′-GCGCATCCTCATCAAGTTCCAAGTTGTTGAGATCACCCAAAGAGAAACTGCAGCTCAGGGGG-3′ and 5′-CCCCCTGAGCTGCAGTTTCTCTTTGGGTGATCTCAACAACTTGGAACTTGATGAGGATGCGC-3′; and at position K-257 using the primers 5′-CATGGTAGAATCATGACACCCTCTGTGAGAGTGGAAAAGATTCATCCGAGAATGTAAGGC-3′ and 5′-GCCTTACATCCATTCTCGGATGAATCTTTTCCACTCTCACAGAGGGTGTCATGATTCTACCATG-3′. Mutated nucleotides are underlined in bold. Mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. Primers were generated by Integrated DNA Technologies. All constructs were sequenced to confirm that the appropriate mutation was introduced and CAG repeat length was not altered. For coexpression studies, plasmids encoding GCN5 and HA-ubiquitin were purchased from Addgene (GCN5 catalog #14424; Ub catalog #18712). The CBP-MT construct was a kind gift from Dr. Aleksey Kazantsev (Harvard Medical School, Charlestown, MA). The HA-SUMO1 construct was from Dr. Zhao (University of Texas Southwestern Medical Center, Dallas, TX).
Cell culture, transfection, and harvesting.
HEK293T cells were cultured in DMEM (Mediatech) containing 1% penicillin/streptomycin (25 U/ml) and 10% heat-inactivated fetal bovine serum (DMEM complete) unless otherwise specified. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were seeded in six-well dishes at ∼2 × 106 cells per well. Each well was transfected with 4 μg of total DNA and 20 μl of Lipofectamine 2000 in 0.5 ml of DMEM lacking serum and penicillin/streptomycin. Following a 48 h incubation, cells were lysed directly in wells with 400 μl of 1× NuPAGE LDS sample buffer (Invitrogen) diluted in M-PER (Thermo Scientific) containing protease inhibitors (1 tablet/10 ml, Complete, Mini, Roche), phosphatase inhibitor cocktail II (100 μl/10 ml, Calbiochem), and 25 mmN-ethylmaleimide (Sigma). Histone deacetylase (HDAC) inhibitors were used as indicated: final concentrations of 50 μm Trichostatin A (Sigma), 30 μm Na-butyrate (Sigma), and 30 mm nicotinamide (Sigma) were added to cell media 10 min before harvesting in lysis buffer described above.
RNA interference.
Dharmacon On Target Plus SMARTpools (Thermo Scientific) were used for siRNA disruption of HDAC2 (M-003495), HDAC3 (M003496), HDAC4 (M-003496), HDAC5 (M-003498), HDAC6 (M-003499), HDAC7a (M-009330), and HDAC8 (M00350). HEK293T cells were seeded in six-well dishes at a density of 1 × 106 cells per well in 2.5 ml of DMEM complete medium. Cells were cotransfected with 1 μg of DNA and 40 μmol of RNA using 10 μl of Lipofectamine 2000 in 0.5 ml of DMEM according to the manufacturer's instructions. After 72 h incubation, cells were harvested as described above and analyzed by Western blotting.
Isolation and transfection of primary cerebellar granular neurons.
Cerebellums from 5–10 postnatal day 6 CD1 mouse pups were dissected into ice-cold HBSS (Invitrogen), and meningeal tissue was removed. The cerebellums were then transferred to a clean dish, mechanically disaggregated, and trypsinized for 20 min at 37°C in 0.08% trypsin made by diluting 1 ml of 0.25% trypsin into 2 ml of warmed HBSS. Cells were collected by centrifugation and resuspended in 5 ml of HBSS containing 12 mm MgSO4 and 5 U/μl DNase 1 (Invitrogen). Cells were triturated with three increasingly smaller-bore fire-polished glass pipettes, ∼1, 0.5, and 0.25 mm in diameter. Cells were then collected by centrifugation once more and resuspended in DMEM complete. Cells (4.5 × 106) were transfected with 2 μg of DNA using program G-013 on the Amaxa Nucleofector II with Amaxa Mouse Neuron Nucleofector kit. For immunocytochemistry, cells (5 × 105 per well) were plated on an eight-well poly-l-lysine-coated glass slide (BD Biosciences) and cultured for 2–4 d in Neurobasal A medium (Invitrogen) containing 2% B27 (Invitrogen) and 1% penicillin/streptomycin unless otherwise indicated. For Western blot analysis, 2 × 106 cells per well were plated in 12-well poly-l-lysine (Sigma)-coated polystyrene plates (Nunc). Following culturing, cells were either fixed in 2% paraformaldehyde (PFA) for imaging or harvested for Western blot analysis. All experiments and animal care were performed in accordance with the Buck Institute for Age Research Institutional Animal Care and Use Committee guidelines.
Western blot analysis and immunoprecipitation of ataxin-7.
Whole-cell lysates were sonicated at 40 mA in 5 × 5 s pulses. DTT was added to a final concentration of 10 mm and samples were heated to 95°C for 10 min. Ten or twenty-five micrograms of protein per sample were subject to SDS-PAGE using 4–12% NuPAGE Bis-Tris gels under reducing conditions in MES running buffer. Gels were run at constant 200 V for 1 h and transferred to 40 μm nitrocellulose membranes in 1× NuPAGE transfer buffer with 10% methanol at a constant 350 mA for 1 h. Membranes were incubated in TBS with 0.1% Tween 20 (TBS-T) and 5% milk for 1 h. Primary antibodies were diluted in TBS-T/5% milk at 1:10,000 for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (RDI-TRK5G4-6C5, Research Diagnostics); 1:1000 for ataxin-7 (PA1-749, Affinity Bioreagents, directed against N-terminal 17 aa of mouse and human ataxin-7), 1:200 for HDAC7 (12174, Abcam), and tubulin (Sigma); 1:500 for SUMO1 (40120, ActiveMotif); and 1:50 for lysine acetylation (9441, Cell Signaling Technology). Blots were developed using Pierce ECL or SuperSignal West Femto ECL reagents (Thermo Scientific). Band intensity was quantified using NIH ImageQuant TL v2005 or NIH ImageJ as indicated.
For immunoprecipitation, 500 μg of total 293T lysates diluted in M-PER were applied to G-Sepharose beads complexed to ataxin-7 antibody (Affinity Bioreagents PA1-749) or c-myc beads (Pierce). Following overnight incubation at 4°C, beads were washed three times in TBST and three times in 0.1 m Tris pH 7.4. Proteins were eluted into loading buffer by heating to 95°C for 10 min, followed by centrifugation to collect the eluted fraction. Western blotting was performed as described above.
Immunocytochemistry.
Cells were fixed by adding 8% paraformaldehyde directly to culture medium to a final concentration of 2%. Cells were incubated for 30 min with gentle agitation, washed 2 × 5 min with room temperature PBS, and stored in PBS at 4°C before staining. Cells were permeabilized in 0.25% Triton in 1× TBS for 15 min, washed 2 × 5 min in TBS, and blocked in 5% normal donkey serum (NDS) in TBS for 30 min. Primary antibody was diluted in 2.5% NDS in 1× TBS at 1:200 for ataxin-7 (La Spada et al., 2001; Garden et al., 2002; Young et al., 2007), 1:50 for LAMP-2 (clone M3/84, sc19991, Santa Cruz Biotechnology) and LC3 (M152-3, MBL), and 1:25 for ubiquitin (clone P4D1-A11, 05-944, Millipore) and incubated with cells overnight at 4°C. Cells were then washed 3 × 10 min in TBS, secondary antibody diluted in 2.5% NDS in 1× TBS (Alexa 488 anti-rabbit 1:250; Alexa 555 anti-mouse 1:500) was applied for 1 h at room temperature (RT) and washed, and cells were mounted with ProlongGold containing 4′,6′-diamidino-2-phenylindole (DAPI). Slides were cured overnight. Epifluorescence images were captured on a Nikon Eclipse E800. Filter cubes with the following specifications were used: red: excitation (Ex) 515-560, dichroic 565, emission (Em) 572-642; green: Ex 460-500, dichroic 505, Em 510-560; and blue: Ex 340-380, dichroic 400, Em 435-485. Confocal images were obtained using a Zeiss LSM 510 NLO microscope and Zeiss LSM Image Browser software. Laser settings used were 488 Ar, 543 He/Ne, and Chameleon Ultra tuned to 780 nm. Following capture, all images were background adjusted in Adobe Photoshop using identical parameters for each channel.
Production and characterization of transgenic mice.
The MoPrP-Flag-(1X)-SCA7(10/92Q)-myc(1X) expression vectors were derived as follows. A 0.9 kb chloramphenicol resistance cassette was PCR-cloned into the NotI and XhoI restriction sites in an SCA7 cDNA. This cloning step fused the N-terminal 143 bp and C-terminal 355 bp of SCA7 coding sequence to the ends of the selection cassette (deleting the intervening 2.4 kb of SCA7 sequence). This modified SCA7 cDNA was PCR-amplified using primers that were engineered to insert an N-terminal FLAG and a C-terminal myc epitope tag (5′-GCGGAGGTCGACGCCACCATGGACTACAAAGACGATGACGACAAGCTTATGTCGGAGCGGGCCGCGGATG-3′ and 5′-GAGGTGGTCGACTCAGCTATTCAGATCCTCTTCTGAGATGAGTTTTTGTTCGGGACGTGCCTTTGGCTGATGAAG-3′). This 1.5 kb PCR product was digested with SalI and cloned into the XhoI site of MoPrP.Xho (Borchelt et al., 1996), yielding a new vector designated PrP-SCA7-Not/Xba (#435-3). The chloramphenicol resistance cassette was liberated from this vector using the flanking SCA7 restriction sites (AatII and XhoI), and the intervening AatII–XhoI SCA7 sequence was cloned into this vector restoring the full-length SCA7 coding sequence. SCA7 cDNA sequences containing a wild-type 10Q and a mutant 92Q yielded a set of vectors designated 410-3 and 411-3, respectively.
Translational competence of the transgenic constructs was verified by transient transfection into HEK293T cells using Superfect (Qiagen). The vector backbone was removed by Pf1F I digestion and then microinjected into oocyte pronuclei. Mice were in a C57BL/6J background. Founder mice were identified by PCR analysis of tail DNA. SCA7 genotyping primers used (5′-CCGAGAATGGATGGCACACT-3′ and 5′-TGCGGTGGTTGCTGAGAGT-3′) amplify a 440 bp fragment of human SCA7. Southern and Western blot analysis were performed as previously described (La Spada et al., 2001) and expression for PrP-mycFlag-SCA7-10Q-myc line A305 and PrP-Flag-SCA7-92Q-myc line B306 used in these studies were similar. Similar to our previous characterized SCA7-92Q-6076 transgenic line (La Spada et al., 2001), PrP-mycFlag-SCA7-92Q-line 306 overexpresses ataxin-7 at ∼2.0-fold endogenous levels, displays retinal pathology by 12 weeks of age, and has a shortened lifespan (9 months).
Immunohistochemistry of SCA7 mice.
SCA7 mice were perfused with PBS and then 4% PFA in PBS (20 ml each). Paraffin-embedded sections were washed with xylene for 2 × 5 min to deparaffinize the tissue. Sections were rehydrated in consecutive ethanol washes (100% to 70%) before resuspension in 1× Tris-buffered saline for 15 min. All washes were performed at room temperature. For antigen retrieval, sections were microwaved in 10 mm citrate buffer, pH 6.0, for 5 min at 40% power in a 1100 W microwave oven (Sanyo). Sections were allowed to cool in the same buffer for 20 min at RT, then transferred to 1× TBS for 10 min. For immunostaining, sections were blocked in 10% normal donkey serum in 1× TBS for 1 h at RT with chicken anti-mouse IgG at 1:500. Primary antibody was diluted in 1% BSA in 1× TBS as described above and incubated overnight at 4°C. All subsequent steps were performed at RT. Slices were washed 3 × 10 min in 1× TBS. Secondary antibody was diluted 1:500 in 1% BSA in 1× TBS and applied to slices for 1 h. Ataxin-7 antibody K (1:200) was labeled with Alexa 488 donkey anti-rabbit, and LC3 (1:50) was labeled with Alexa 555 donkey anti-mouse. Following incubation in secondary antibody, slices were washed 3 × 20 min in 1× TBS and mounted in Prolong Gold with DAPI.
Results
Predicted modification targets cluster around caspase-7 cleavage sites
We have previously found that caspase-7 cleavage of ataxin-7 at D266 is an important determinant of cellular toxicity (Young et al., 2007) and in the progression of SCA7 in transgenic mice (Guyenet et al., unpublished results). In other polyglutamine diseases, the interplay between ubiquitination and SUMOylation of target lysines can strongly influence intracellular protein accumulation and toxicity (Steffan et al., 2004). To determine whether posttranslational modification plays a role in ataxin-7 turnover, we analyzed the ataxin-7 protein for modification consensus sequences (Fig. 1). For SUMOylation, we used both SUMOplot (Abgent) and SUMOsp (http://sumosp.biocuckoo.org/). SUMOplot is a strong predictor of consensus sequences, while SUMOpre incorporates mammalian phylogenetic similarity into its scoring algorithm to weight the consensus sites that are of particular evolutionary significance in mammals. Of the initial predictions, the two most highly predicted sites, K223 and K257, were both proximal to the D266 caspase-7 cleavage site (Fig. 1A,B). These residues are also adjacent to predicted phosphorylation sites at S224 and S255 (Fig. 1C), suggesting the potential for functional interaction between them. In addition, since acetylation/deacetylation–SUMOylation switch has been shown to regulate transcription factors/repressors such as MEF2 or HIC (Stankovic-Valentin et al., 2007), ataxin-7 lysines could be targeted by both SUMOylation and acetylation.
Figure 1.
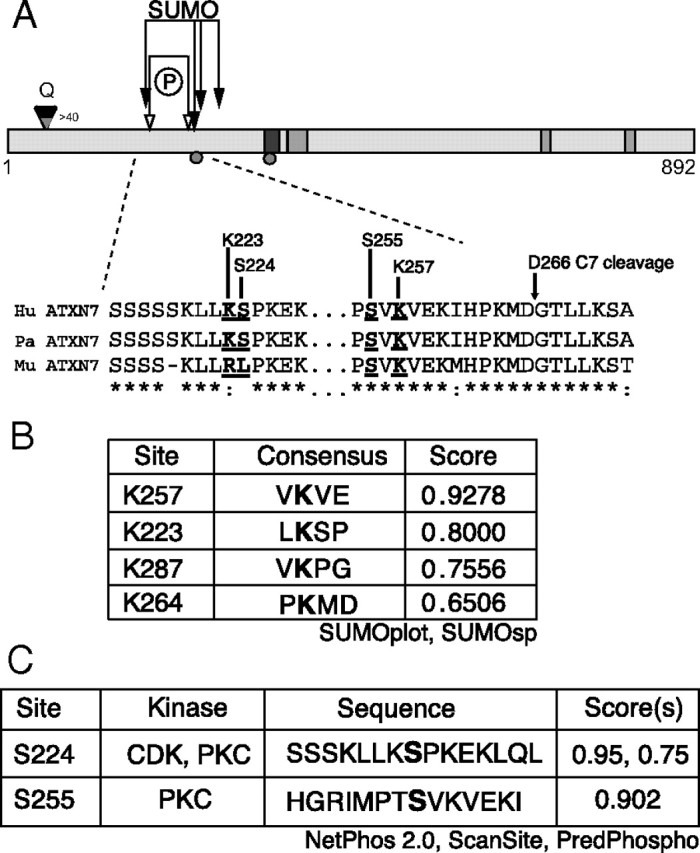
Schematic of the ataxin-7 structure. A, The 892 aa protein contains a nuclear export signal (NES; black rectangle) and three nuclear localization signals (NLS; gray rectangle). The diamond indicates the location of the polyglutamine stretch, and the caspase cleavage sites at Asp-266 and Asp-344 are indicated by circles. Arrows indicate sites of putative PTMs: phosphorylation (open) and SUMOylation (closed). Lysine 257 is indicated with dropped arrow. B, SUMOylation predictions using SUMOplot (Abgent) and SUMOsp (http://sumosp.biocuckoo.org/). Predicted targets shown in bold. C, Phosphorylation predictions using NetPhos2.0 (www.cbs.dtu.dk/services/NetPhos/), ScanSite (http://scansite.mit.edu), and PredPhospho (http://www.nih.go.kr/phosphovariant/html/seq_input_predphospho2.htm). Predicted targets are shown in bold.
To demonstrate that ataxin-7 is modified by SUMO, c-myc-tagged ataxin-7 was immunopurified and incubated with the E1 and E2 ligases required for SUMOylation in addition to SUMO-1 (+) or a nonconjugatable SUMO-1 mutant (−). Consistent with our predictions, ataxin-7 is modified by SUMOylation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The major band migrated with an apparent molecular weight of 100 kDa (120 kDa for ataxin-7-92Q), whereas the slower migrating band showed an ∼14 kDa increase in molecular mass, consistent with the addition of a single SUMO moiety. Indeed, the same bands were observed in 293T cells cotransfected with myc-ataxin-7 and SUMO-1 expression vectors (data not shown). Using a similar immunoprecipitation strategy, we also found that ataxin-7 and fragments expressed in 293T cells are immunoreactive to anti-acetylation antibodies (Fig. 2A). In addition, we found that ataxin-7-immunoprecipitated cellular lysates from MoPrP-flag-SCA7-10Q-myc or MoPrP-flag-SCA7-92Q-myc transgenic mice are immunoreactive to anti-acetylation antibodies (Fig. 2B), suggesting that the modification occurs in vivo.
Figure 2.
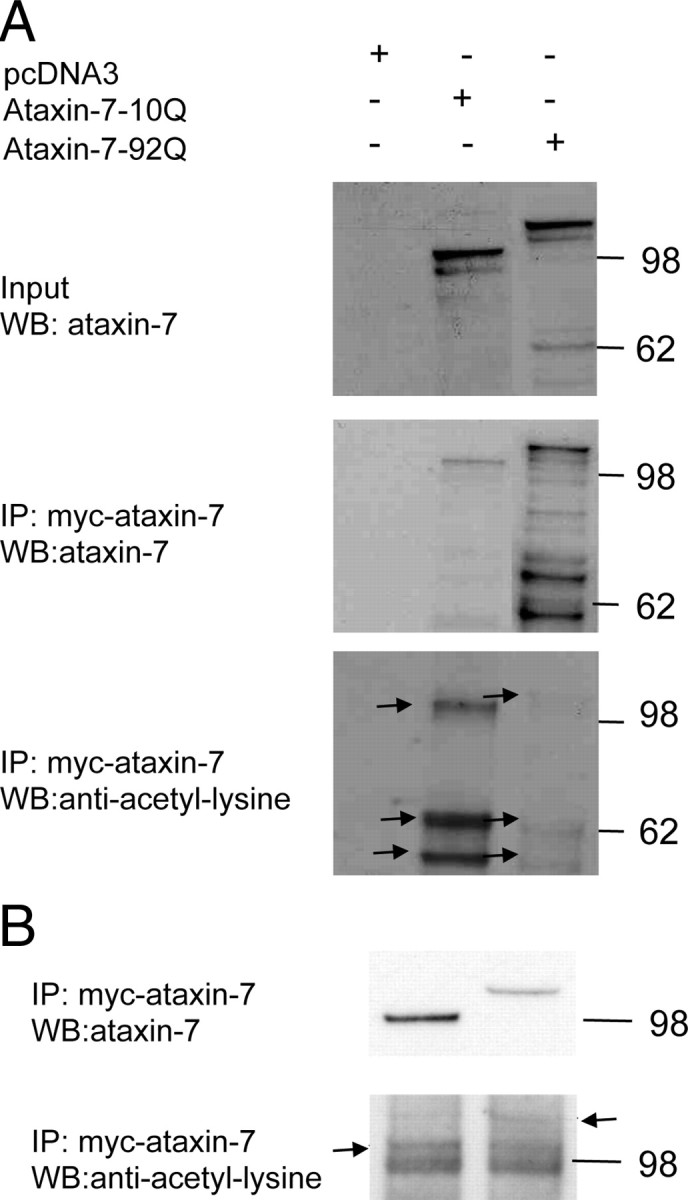
Acetylation of full-length ataxin-7. A, Myc-tagged full-length ataxin-7-10Q, ataxin-7-92Q, and empty vector were expressed in 293T cells. Immunoprecipitation followed by immunoblot analysis were carried out using cell lysates transfected with ataxin-7. Blots were probed with monoclonal acetyl lysine antibody (lower panel). The arrowheads represent ataxin-7-10Q or ataxin-7-92Q full-length and fragments acetylated. B, MoPrP-flag-SCA7-10Q-myc and MoPrP-flag-SCA7-92Q-myc transgenic cerebellar mouse lysates were immunoprecipitated with anti-ataxin-7. Western blots of the immunoprecipitated lysates were probed with anti-ataxin-7 and with monoclonal anti-acetyl lysine antibody.
Lysine 257 regulates turnover of the 1-266 ataxin-7 cleavage fragment
To determine whether modification of the predicted ataxin-7 SUMOylation sites K223 or K257 are important for regulating the stability of an ataxin-7 cleavage fragment, we made single lysine-to-arginine substitutions at these residues in a truncated ataxin-7-10Q or 92Q construct expressing amino acids 1-266. This protein is identical to the fragment generated by caspase-7 cleavage at D266 of ataxin-7. Substitution of arginine (R) for lysine (K) maintains size and charge characteristics, but is not a substrate for posttranslational modification. Constructs expressing the ataxin-7-10Q (1-266), ataxin-7-10Q (1-266) K223R, ataxin-7-10Q (1-266) K257R, ataxin-7-92Q (1-266), ataxin-7-92Q (1-266) K223R, and ataxin-7-92Q (1-266) K257R fragments were transiently transfected into HEK293T cells, and protein was analyzed by Western blot analysis (Fig. 3A,B).
Figure 3.
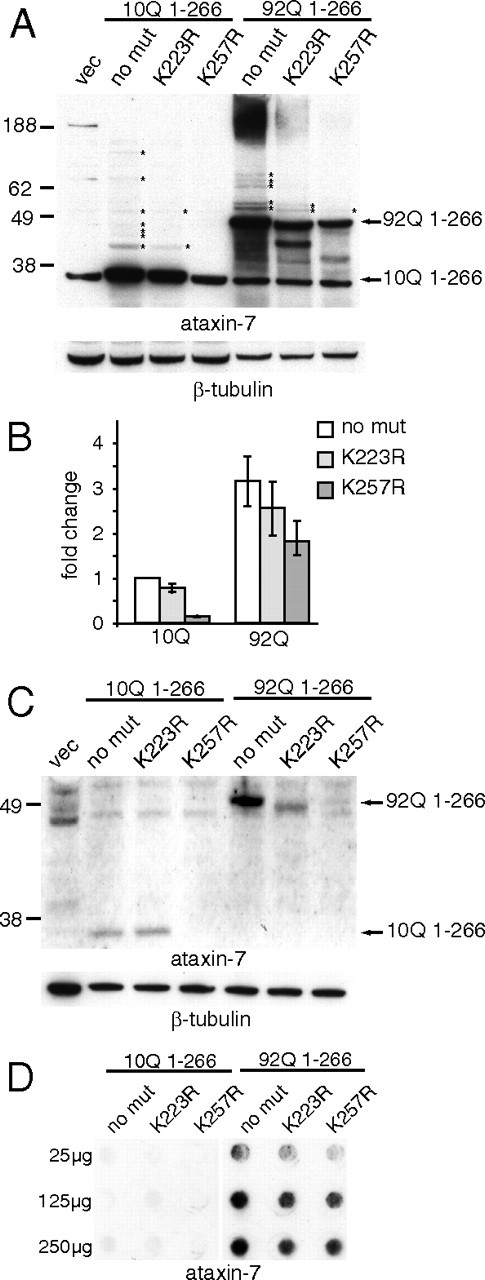
Mutations at lysine 257 regulate turnover of ataxin-7 fragment in 293T and primary cerebellar granule cells. A, Whole-cell lysates of HEK293T cells overexpressing ataxin-7 constructs was subjected to 4–12% SDS-PAGE and Western blotting on nitrocellulose using the antibodies indicated. β-Tubulin was used as a loading control. A nonspecific band at 36 kDa is present in all lanes at the same molecular weight as ataxin-10Q (1-266) protein. B, Quantitation of Western blots in A using NIH ImageJ. Values are represented as the fold change relative to ataxin-7-10Q (1-266) and normalized to β-tubulin. C, Whole-cell lysates of primary cerebellar neurons overexpressing ataxin-7 constructs was subjected to 4–12% SDS-PAGE and Western blotting on nitrocellulose using the antibodies indicated. β-Tubulin was used as a loading control. D, Filter trapping of insoluble fragments on cellulose acetate using 25–250 μg of insoluble fraction-enriched protein lysate from HEK293T cells, indicating that loss of soluble protein in A and B reflects loss of total protein and not accumulating insoluble aggregates.
As shown in Figure 3A, these mutations result in loss of the shifted bands (14 kDa increments) that represent modified forms of ataxin-7-10Q or -92Q proteins (denoted with asterisks), as well as the high molecular weight material in the lanes with the ataxin-7-92Q proteins consistent with aggregation. In addition, protein levels of the ataxin-7 K257R mutants are significantly decreased, indicating that modification at this site regulates the stability or turnover of the ataxin-7 (1-266) fragment. Moreover, this effect is polyQ repeat dependent, conferring a much larger (∼6-fold) decrease in the context of 10Q than 92Q (<2-fold) (Fig. 3B). These results were also observed in primary cerebellar neurons transiently transfected with the ataxin-7 fragment (Fig. 3C), indicating that the molecular consequences of the K257R mutation are consistent across cell types and validating the use of HEK293T cells for further molecular characterization. Of note, in primary cerebellar neurons, the clearance of the ataxin-7-92Q fragment is impaired when compared with the ataxin-7-10Q fragment, and mutation at K257 promotes clearance of both proteins.
To confirm that the reduction in ataxin-7 (1-266) K257R protein reflected loss of total protein and not the accumulation of insoluble aggregates, we performed filter trapping of HEK293T lysate from cells expressing ataxin-7-10Q and ataxin-7-92Q, with either no lysine substitutions, K223R, or K257R, onto cellulose acetate membranes. As shown in Figure 3D, we found that the 10Q-containing fragments do not aggregate, consistent with our findings from Western blot analysis, and the 92Q-containing fragments aggregate proportionately to the amount of soluble protein for each mutant, indicating that K257R mutation confers loss of total protein and not simply reduction of soluble protein. Finally, this modulation appears to be specific to the 1-266 fragment, as the K257R mutation does not measurably alter the turnover of full-length ataxin-7 (data not shown).
Macroautophagy mediates ataxin-7 fragment clearance
Since the K257R mutation appears to substantially decrease the stability of the 1-266 aa ataxin-7 fragment, we were interested in identifying whether degradation was mediated by the ubiquitin proteasome system (UPS) or through autophagy. To identify the mechanism of degradation for the K257R mutant fragments, we chemically disrupted these pathways using epoxomicin and 3-methyladenine (3-MA), respectively (Fig. 4). Epoxomicin binds to subunits of the 26S proteasome and selectively disrupts its activity, while 3-MA blocks autophagy by inhibiting class III PI3K activity, preventing autophagosome formation. HEK293T cells were transiently transfected with the N-terminal 1-266 fragments of the ataxin-7, ataxin-7-10Q (1-266), ataxin-7-10Q (1-266) K223R, ataxin-7-10Q (1-266) K257R, ataxin-7-92Q (1-266), ataxin-7-92Q (1-266) K223R, or ataxin-7-92Q (1-266) K257R and treated with either epoxomicin (25 μm) or 3-MA (10 mm) 48 h after transfection. As shown in Figure 4, A and B, Western blot analysis demonstrated that UPS inhibition had a negligible effect on ataxin-7-10Q or -92Q protein levels, while disruption of macroautophagy resulted in an increase in ataxin-7. Addition of 3-MA resulted in a significant increase in the steady-state levels of ataxin-7-10Q (1-266), ataxin-7-10Q (1-266) K223R, ataxin-7-10Q (1-266) K257R, ataxin-7-92Q (1-266) K223R, or ataxin-7-92Q (1-266) K257R. In particular, the levels of ataxin-7 (1-266) K257R or K223R proteins were similar to the unmodified ataxin-7 fragment. Interestingly, ataxin-7-92Q (1-266) levels were not altered by either 3-MA or epoxomicin.
Figure 4.

Macroautophagy promotes ataxin-7 fragment degradation. A, B, HEK293T cells overexpressing ataxin-7-10Q (1-266) (A) and ataxin-7-92Q (1-266) (B) were treated with 25 μm epoxomicin (epox) or 10 mm 3-MA for 3 h before harvesting 48 h after transfection. Whole-cell lysate was subjected to 4–12% SDS-PAGE and Western blotting with antibodies indicated. GAPDH was used as a loading control. Arrows indicate ataxin (1-266) fragment. The images are representative of N = 3. ANOVA analysis of ataxin-7 and mutants compared with ataxin-7 with MA gave a p value of <0.05 for all comparisons except ataxin-7-92Q with 3-MA.
We also found that blocking macroautophagy in primary neurons expressing ataxin-7 resulted in similar increases of ataxin-7 protein levels (data not shown). These findings support a primary role for autophagy, but not proteasomal degradation, in the turnover of the ataxin-7 fragments. Relevant to the gain-of-function properties for polyglutamine expansions, the ataxin-7-92Q (1-266) was resistant to degradation by autophagy. Since mutation at either K223 or K257 resulted in a protein susceptible to degradation, it is likely that posttranslational modification at these sites plays a critical role in blocking degradation of the mutant fragment.
Overexpression of lysine modifiers alters ataxin-7 accumulation
Several covalent posttranslational modifications, such as ubiquitination, SUMOylation, methylation, and acetylation can target lysine residues. Posttranslational modifiers of ataxin-7 stabilization could be mediated by small ubiquitin-like protein (SUMO-1). SUMO-1 is one of a family of small ubiquitin like proteins (SUMO-1-4) described to participate in a multitude of cellular functions including transcription, nuclear translocation, and inflammation. SUMO-1 directly modifies proteins using three steps similar to ubiquitination using the E1 equivalent activating complex (SAE I and II) and E2 also known as Ubc9. Another enzymatic pathway that modifies lysines is histone acetylases (HATs). Transcriptional regulatory proteins, including GCN5, PCAF, p300, CBP, and TAFII250, possess intrinsic histone acetyltransferase HAT activity acetylase. The role of lysine acetylation in regulating nuclear proteins and transcriptional factors has been well established. Histone acetylation affects chromatin structure and gene expression; tumor suppressor protein p53 can be acetylated at multiple sites, and different acetylation modifications have distinct effects on p53 function. Relevant to our studies, ataxin-7 is in the acetyltransferase complex, STAGA, which contains the acetyltransferase GCN5.
To characterize the contribution of possible lysine modification, including ubiquitination, SUMOylation, and acetylation, to ataxin-7 stability and turnover, we cotransfected ataxin-7-10Q or -92Q (1-266) with constructs expressing ubiquitin, SUMO1, GCN5, or CBP (Fig. 5A). We found that only one of the lysine-modifying enzymes, CBP, when coexpressed, led to significant increases in ataxin-7 accumulation, while GCN5 showed a modest decrease. This suggests that acetylation may play a role in stabilizing the ataxin-7 fragments. Unexpectedly, ubiquitin or SUMOylation coexpression did not alter or increased the ataxin-7 fragment levels. Our initial prediction was that, like huntingtin, ubiquitin overexpression would speed degradation of ataxin-7, while SUMO1 overexpression might result in the stabilization of the protein (Bence et al., 2001; Steffan et al., 2004; Bennett et al., 2007).
Figure 5.
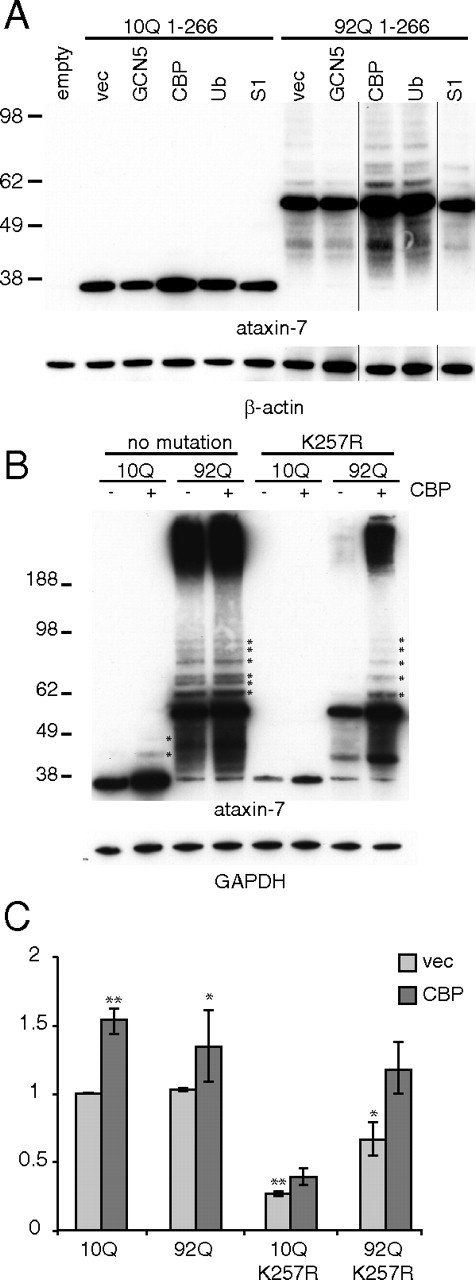
Effects of lysine modifier coexpression on ataxin-7 fragment accumulation. A, Ataxin-7-10Q or -92Q (1-266) and the indicated acetylase were transiently transfected into HEK293T cells. Lysate was collected directly into sample buffer at 48 h and analyzed by Western blot analysis with the indicated antibody. β-Actin was used a loading control. B, Ataxin-7-10Q (1-266), ataxin-7-92Q (1-266), ataxin-7-10Q (1-266) K257R, or ataxin-7-92Q (1-266) K257R and the indicated acetylase were transiently transfected into HEK293T cells. Lysate was collected directly into sample buffer at 48 h and analyzed by Western blot analysis with the indicated antibody. GAPDH was used as a loading control. Asterisks indicate possible SUMOylation or ubiquitination promoted by acetylation. C, Quantification of band intensity in B was performed using NIH ImageQuant TL v2005 and normalized to GAPDH (ANOVA analysis was performed, *p < 0.05, **p < 0.01). The analysis includes only the soluble ataxin-7 forms. We note that the increase in ataxin-7-10Q was 1.5-fold with CBP compared with ataxin-7-10Q alone. The ataxin-10Q K257R increased 1.2-fold with CBP compared with ataxin-10Q K257R. The N terminus of ataxin-7 may have other acetylation sites that account for this small increase.
Since CBP coexpression with ataxin-7 increased protein levels, we investigated the effect of CBP on the steady-state levels of ataxin-7 (1-266) and the K257 mutant. As shown in Figure 5, B and C, we found that the ataxin-7-10Q K257R did not reach the same levels as the ataxin-7-10Q (1-266) with CBP coexpression, suggesting that acetylation of ataxin-7 at this site contributes to ataxin-7 stabilization. The ataxin-7-92Q K257R gave a similar effect when both the soluble and insoluble fractions were accounted for (Fig. 5B).
HDAC7 knockdown modifies ataxin-7 fragment turnover
HDACs maintain the acetylation levels of histone and nonhistone proteins. Mammalian HDACs are classified into four classes: type I, type IIa,b, type III, and type IV. Class I and II HDACs have an active site zinc coordinated by histidine and aspartate residues. The class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) are widely expressed in most tissues and are localized in the nucleus (contain import/export nuclear localization signals). Class IIa HDACs (HDAC4, HDAC5, HDAC7, and HDAC9) contain a bipartite structure with an N-terminal domain that interacts with transcription factors and class I HDAC3. Since we found lysine acetylation influences clearance of the ataxin-7 fragments, we investigated whether modulation of deacetylation activity by siRNA knockdown of HDACs altered ataxin-7 fragment levels. To assess the effect of HDAC activity on ataxin-7 stability and clearance, we screened cotransfection of siRNA to HDAC2–HDAC8 with ataxin-7-10Q (1-266) or ataxin-7-92Q (1-266) fragments (data not shown). Among the seven HDACs tested, we found that HDAC7 modulates the turnover of ataxin-7. Upon knockdown of HDAC7, we observed an increase ataxin-7-10Q (1-266) or ataxin-7-92Q (1-266) fragments levels compared with controls (Fig. 6). Thus, cotransfection experiments with HDAC7 siRNA resulted in the accumulation of ataxin-7.
Figure 6.
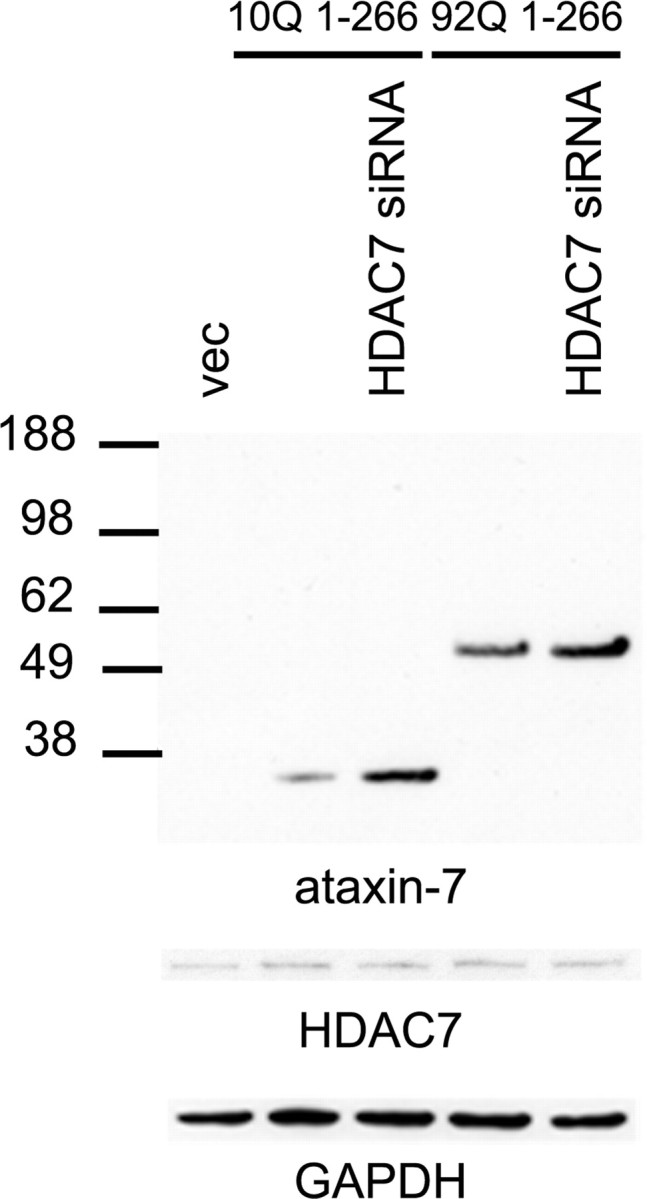
Effects of HDAC7 on the turnover of ataxin-7. A direct effect of HDAC7 was confirmed by siRNA knockdown, which increased ataxin-7 levels in both ataxin-7-10Q (1-266)- and ataxin-7-92Q (1-266)-transfected cultures compared with controls with nontargeted siRNA. GAPDH was used as a loading control. HDAC7 levels are indicated for each condition.
Colocalization of ataxin-7 with degradation markers
Next we performed immunofluorescence analysis on primary cerebellar neurons overexpressing the 10Q or 92Q ataxin-7 1-266 fragment with no mutation, or the K223R, or K257R mutation (Fig. 7A). One question relevant to our study is whether the K257R mutant, which is rapidly degraded by autophagy, has altered localization. As reported previously, we find that ataxin-7 displays a primarily nuclear localization with some cytoplasmic staining. We found that while the K223R mutant appears to localize similarly to the nonmutant fragment, the K257R mutant displays primarily a cytoplasmic localization in addition to increased levels of endogenous LAMP-2, a lysosomal marker and protein involved in autophagy.
Figure 7.
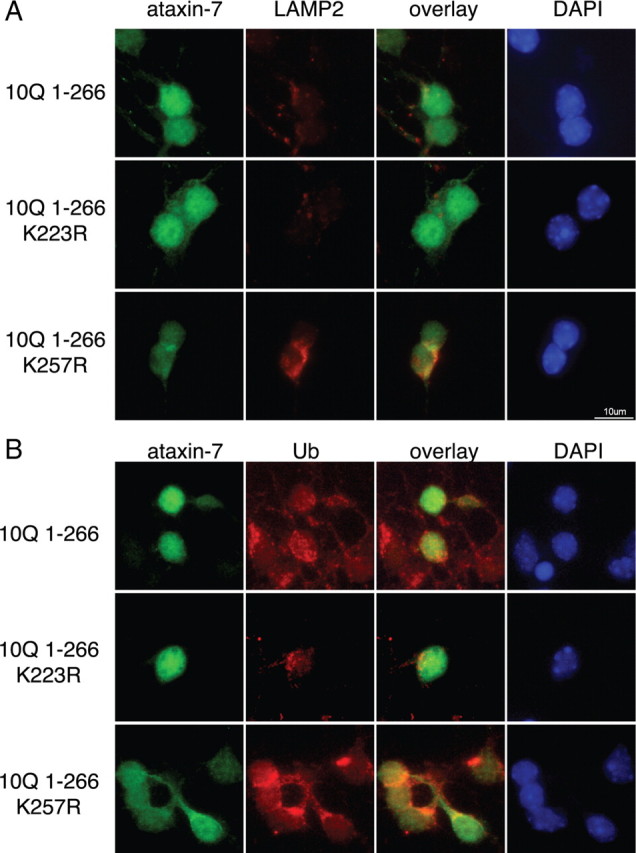
Expression of ataxin-7 and colocalization with endogenous lysosomal marker LAMP-2 and ubiquitin in primary cerebellar cultures. A, Ataxin-7-10Q (1-266), ataxin-7-10Q (1-266) K223R, or ataxin-7-10Q (1-266) K257R was transfected in primary cerebellar cultures, fixed, and stained with ataxin-7 antibody complexed to Alexa 488 (green), LAMP-2 antibody complexed to Alexa 555 (red), and DAPI (blue). B, Ataxin-7-10Q (1-266), ataxin-7-10Q (1-266) K223R, or ataxin-7-10Q (1-266) K257R was transfected in primary cerebellar cultures, fixed, and stained with ataxin-7 antibody complexed to Alexa 488 (green), ubiquitin antibody complexed to Alexa 555 (red), and DAPI (blue). Scale bar indicates 10 μm.
Additionally, we performed costaining of cells overexpressing ataxin-7 and ubiquitin to determine whether ubiquitin-positive staining was similarly altered in the presence of the ataxin-7 K257R mutant (Fig. 7B). Consistent with the localization observed in Figure 7A, we observed altered ataxin-7-K257R fragment localization in the cytoplasm, coincident with cytoplasmic ubiquitin staining. Interestingly, in cells expressing the ataxin-7-K257R fragment, the ubiquitin staining appears more intense and dispersed than in the other ataxin-7-expressing cells.
Induction of autophagy in PrP-SCA7-92Q mice
Previous characterization of SCA7 transgenic models has demonstrated the production of proteolytic cleavage products (Zander et al., 2001; Garden et al., 2002; Young et al., 2007). To evaluate whether autophagy is relevant to disease pathogenesis, we characterized a newly generated MoPrP-flag-SCA7-10Q-myc and MoPrP-flag-SCA7-92Q-myc transgenic mouse model (La Spada et al., 2001) (Guyenet et al., unpublished results) for LC3 expression and the presence of punctate LC3-positive structures. The appearance of LC3-positive puncta is indicative of the induction of autophagy. As previously noted, MoPrP-flag-SCA7-92Q-myc transgenic mice have a dramatic neurological phenotype and premature death and accumulate proteolytic fragments (Guyenet et al., unpublished results). As expected, expression of the polyglutamine-expanded ataxin-7 causes severe Purkinje cell degeneration (Fig. 8A). Unlike our previous model, we detected robust expression of ataxin-7 in the Purkinje cells (Fig. 8A). Using an antibody to LC3, immunostaining of cerebellum from MoPrP-flag-SCA7-92Q-myc mice demonstrated an increase in the expression of LC3 (Fig. 8A) as well as increased LC3-positive structures (Fig. 8B) in the Purkinje cells when compared with MoPrP-flag-SCA7-10Q-myc transgenic mice or nontransgenic littermates. Quantification of LC3-positive puncta revealed the MoPrP-flag-SCA7-92Q-myc Purkinje cells had at least five puncta per cell, while the MoPrP-flag-SCA7-10Q-myc did not (Fig. 8C). This suggests that the autophagy is induced in the MoPrP-flag-SCA7-92Q-myc transgenic mice.
Figure 8.
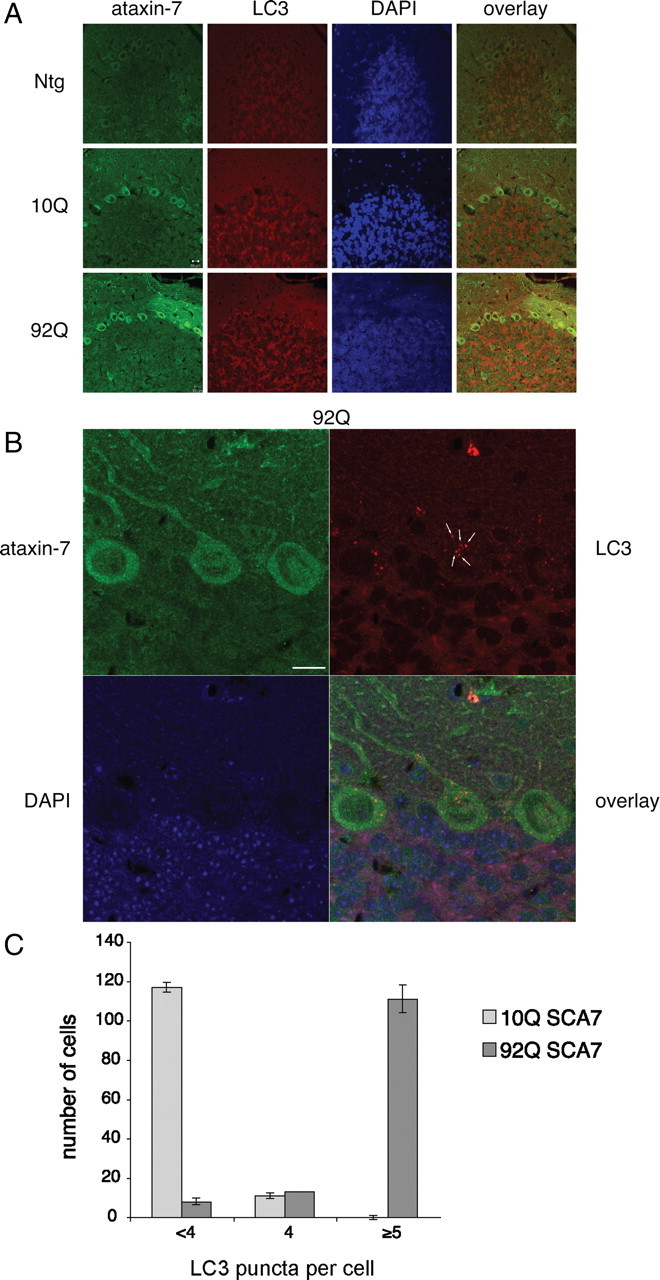
Immunohistochemical staining of 9-month-old MoPrP-flag-SCA7-10Q-myc and MoPrP-flag-SCA7-92Q-myc transgenic mouse model and littermate control with ataxin-7 and LC3. A, The cerebellum reveals intense ataxin-7 cytoplasmic and nuclear staining in the granule cell layer, molecular layer, and Purkinje cells of the SCA7-10Q and SCA7-92Q mice. Ataxin-7 staining reveals decrease in dendritic arbors of the Purkinje cells of the MoPrP-flag-SCA7-92Q-myc transgenic mice when compared with MoPrP-flag-SCA7-10Q-myc mice or nontransgenic littermate controls. LC3 staining reveals an increase in staining in the molecular, granule, and Purkinje cell layer for the MoPrP-flag-SCA7-92Q-myc transgenic mice when compared with MoPrP-flag-SCA7-10Q-myc mice or nontransgenic (Ntg) littermate controls. B, Further analysis of transgenic SCA7 mice show increases in LC3-positive structures (white arrows) in the Purkinje cells for the MoPrP-flag-SCA7-92Q-myc transgenic mice. Scale bars indicate 10 μm. Images were acquired on the confocal microscope as specified in Materials and Methods, Immunocytochemistry. C, Quantification of the LC3-positive structures in the Purkinje cells for the MoPrP-flag-SCA7-92Q-myc transgenic mice when compared with MoPrP-flag-SCA7-10Q-myc mice. Puncta per cell were counted in 100–200 Purkinje cells per 7 μm slice (N = 3).
Discussion
Identifying posttranslational modifications of polyglutamine proteins and understanding their downstream effect has become increasingly important in determining the mechanisms of polyglutamine disease pathogenesis. There are multiple examples in HD and the polyglutamine-dependent SCAs demonstrating that phosphorylation, ubiquitination, SUMOylation, and acetylation affect key behaviors of these proteins that are often altered in disease, including the following: (1) localization into the nucleus (Riley et al., 2005), mediated by phosphorylation-dependent SUMOylation; and (2) protein stability and accumulation, mediated by competitive binding of ubiquitin and SUMO (Steffan et al., 2004). In this study, we were interested in the turnover of polyglutamine-containing ataxin-7 fragments following cleavage by caspase-7, specifically, whether their accumulation could be modulated by posttranslational modification. The clearance of toxic protein forms is an important factor in ameliorating the so-called “proteinopathies,” neurodegenerative diseases caused by protein misfolding and aggregation (for review, see Williams et al., 2006). In this study, we provide evidence that Lys-257, near the Asp-266 cleavage fragment C terminus (Young et al., 2007), is a modification target that alters ataxin-7 fragment accumulation in a repeat-dependent manner.
One of the most striking findings of this study is that a proteolytic cleavage product of caspases, in this case the substrate is ataxin-7, is specifically regulated by autophagy. This mechanism of regulation may have broader implications in the regulation of cell death pathways by caspases. It is possible that caspase cleavage products are not indiscriminately degraded but that in some cases the process is regulated by posttranslational modification. This form of regulation may have implications for caspase cleavage products that act as amplifiers of the cell death process or as dominant-negatives to inactivate survival complexes. Since CBP, but not GCN5, had an impact on ataxin-7 fragment turnover, we suggest that acetylation/deacetylation of proteolytic fragments generated during cell death may represent an important level of regulation by specific HATs/HDACs for accelerating or attenuating apoptosis. Consistent with the role of acetylation in regulating cell death, Ku70 acetylation promotes the dissociation of Bax from Ku70 and promotes cell death (Li et al., 2007). Further caspase cleavage of HDAC3 results in cytoplasmic accumulation, acetylation of nuclear targets, and concurrently enhanced cell death (Escaffit et al., 2007).
The role of autophagy in ameliorating disease-induced toxicity has been explored in a number of proteinopathy models, including Alzheimer's disease and Parkinson's disease, as well as the polyglutamine models HD and SCA3 (Yamamoto et al., 2006; Bilen and Bonini, 2007). The general proteinopathy model posits that misfolded protein aggregates, polyglutamine-containing or otherwise, are refractory to UPS-mediated degradation and that the resulting intracellular accumulation triggers the numerous downstream effects that lead to cell dysfunction and death. Since autophagy is a large-scale, generally nonselective process (excluding chaperone-mediated autophagy), the misfolded aggregates that are no longer subject to UPS-mediated proteolysis are still available for autophagic degradation. Thus, increased activation of autophagy may compensate for proteasomal malfunction, clearing protein aggregates and attenuating downstream aggregation-dependent malfunction. In HD, autophagy has been shown to specifically mediate degradation of a polyQ-containing N-terminal fragments of huntingtin (Qin et al., 2003). Moreover, the induction of autophagy to ameliorate neurodegeneration in HD models has been extensively investigated (Sarkar et al., 2009). Our results extend these studies and suggest that caspase-generated cleavage products from polyQ disease proteins are cleared by autophagy and that posttranslational modifications may regulate this process. In accord with a significant role for autophagy in neurodegeneration, we found that SCA7 mice have increased autophagy in Purkinje cells as assessed by LC3-positive autophagosome number and LAMP-2 immunochemistry. The upregulation of autophagy is likely a compensatory response to the accumulating buildup of proteolytic ataxin-7-92Q cleavage products.
Like the “histone code” of histone modification, it is becoming increasingly clear that non-histone-associated proteins are subject to the same modifications, potentially with similar regulatory effects. For ataxin-7, we find that lysine modification has significant consequences regarding stability of the 1-266 fragment generated by caspase-7 cleavage. Since ataxin-7 is a member of the STAGA complex, which also contains the histone acetyltransferase GCN5, we hypothesized that ataxin-7 might be subject to GCN5-dependent acetylation. However, results from this study suggest that CBP, not GCN5, may be the relevant acetyltransferase targeting the proteolytic cleavage product of ataxin-7. CBP has long been implicated in polyglutamine-dependent neurodegeneration. Evidence that CBP is sequestered in polyglutamine aggregates, accompanied by histone hypoacetylation and transcriptional repression, has led to a model in which CBP depletion through sequestration in aggregates prevents its normal function in transcriptional activation of CREB-dependent gene expression (Rouaux et al., 2004). This model has been debated, although it was recently shown that CBP sequestration was not always accompanied by nuclear depletion, and that both were required for toxic effects (Jiang et al., 2006), perhaps explaining some of the previous conflicting results. In addition, overexpression of CBP has been shown to mitigate polyglutamine-dependent toxicity in neuronal models, supporting an important role for CBP function in neuronal stability. In this study, overexpression of CBP with ataxin-7 1-266 leads to a significant increase in the accumulation of this fragment, suggesting that it directly acetylates ataxin-7. This is consistent with Western blot analysis with acetylation antibodies in which the ataxin-7 1-266 is acetylated (data not shown). Of note, we found that GCN5 promoted the degradation of full-length ataxin-7, suggesting that acetylation regulates normal turnover by a distinct mechanism (data not shown).
Additionally, we find that the modification-dependent regulation of these fragment levels is decreased in the presence of an expanded polyglutamine tract; in this case, 92Q versus 10Q. This dampening effect may be one mechanism by which expanded polyQ fragments accumulate to aggregation-prone or toxic levels. That cleavage products of polyQ-containing proteins may be specifically regulated by posttranslational modification is a previously unreported mechanism. Moreover, the idea that polyQ expansion disrupts this regulation, contributing to fragment accumulation and toxicity, is also a novel explanation for the deleterious effects of polyQ expression on the cell.
Ataxin-7 belongs to the STAGA multiprotein acetylation complex; however, it remains unclear what the direct interactions are between this protein and the components of the acetylation/deacetylation machinery, which ultimately controls gene expression patterns. The critical role of deacetylation in ameliorating symptoms of SCA7 phenotype has been demonstrated by a number of research groups. Latouche et al. (2007) in an SCA7 Drosophila model demonstrated that administration of the HDAC inhibitor sodium butyrate was neuroprotective. Interestingly, a different group suggested that treatment with the same HDAC inhibitor relieved the expression of CRE/CBP transcription coactivators in a normal and expanded ataxin-7 cellular model (Ström et al., 2005). This observation further strengthened the critical role of ataxin-7 as a modulator of the acetylation/deacetylation balance, which is altered by the presence of CAG repeats in the SCA7 phenotype. In the present study, we examined whether there is a functional interaction of ataxin-7 and a specific member of the HDAC family. Screening of seven HDACs (2–8) suggested that HDAC7 predominantly regulates ataxin-7 fragment turnover and stability. The regulatory role of HDAC7 on ataxin-7 levels was further confirmed by disruption of HDAC7 expression using siRNA knockdown technology, which resulted in a buildup of ataxin-7 levels. Those findings may be particularly useful for future studies designing therapeutic agents associated with a specific class of HDACs to promote ataxin-7 clearance, and most importantly, restoring aberrant gene expression patterns caused by the expanded CAG repeats.
In summary, we demonstrate that N-terminal ataxin-7 fragments resulting from caspase cleavage are subject to regulated clearance. The regulatory mechanism appears to be posttranslational modification, as disruption of modification sites through mutation alters the fragment stability. Importantly, polyQ expansion partially inhibits the regulatory effects of posttranslational modification. We propose that the deregulation of proteolytic product turnover is an important mediator of polyQ-induced toxicity and cell death. This work may suggest that deacetylation of the ataxin-7 fragment, for example, by promoting HDAC activity, may ameliorate SCA7 by promoting fragment clearance. Our results also emphasize the importance of protein context in understanding the same posttranslational modification in polyglutamine expansion diseases. For ataxin-7 fragments, acetylation is found to stabilize the protein, while a recent study for huntingtin demonstrates that acetylation of mutant huntingtin increases the turnover of the protein (Jeong et al., 2009). Future work will evaluate the role of the proteases in the lysosome that are involved in the clearance of the ataxin-7 fragment.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS40251 (L.M.E.), NS40251 (L.M.E.), and EY14061 (A.R.L.) and the Nathan Shock Grant P30AG025708. S.M. was supported by NIH Training Grant T32 AG000266. We thank Caitlin Rugani for genotyping of SCA7 mice, Junli Zhang for carrying out injection of the SCA7-10Q transgene, Dr. Aleksey Kazantsev for the kind gift of CBP-MT, and Yingming Zhao for HA-SUMO1. We thank Juliette Gafni for suggestions on the work and manuscript.
References
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- Bett JS, Cook C, Petrucelli L, Bates GP. The ubiquitin-proteasome reporter GFPu does not accumulate in neurons of the R6/2 transgenic mouse model of Huntington's disease. PLoS ONE. 2009;4:e5128. doi: 10.1371/journal.pone.0005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:1950–1964. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 2005;14:679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- Chan HY, Warrick JM, Andriola I, Merry D, Bonini NM. Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum Mol Genet. 2002;11:2895–2904. doi: 10.1093/hmg/11.23.2895. [DOI] [PubMed] [Google Scholar]
- David G, Abbas N, Stevanin G, Dürr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- Del-Favero J, Krols L, Michalik A, Theuns J, Löfgren A, Goossens D, Wehnert A, Van den Bossche D, Van Zand K, Backhovens H, van Regenmorter N, Martin JJ, Van Broeckhoven C. Molecular genetic analysis of autosomal dominant cerebellar ataxia with retinal degeneration (ADCA type II) caused by CAG triplet repeat expansion. Hum Mol Genet. 1998;7:177–186. doi: 10.1093/hmg/7.2.177. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Ellerby LM, Andrusiak RL, Wellington CL, Hackam AS, Propp SS, Wood JD, Sharp AH, Margolis RL, Ross CA, Salvesen GS, Hayden MR, Bredesen DE. Cleavage of atrophin-1 at caspase site aspartic acid 109 modulates cytotoxicity. J Biol Chem. 1999a;274:8730–8736. doi: 10.1074/jbc.274.13.8730. [DOI] [PubMed] [Google Scholar]
- Ellerby LM, Hackam AS, Propp SS, Ellerby HM, Rabizadeh S, Cashman NR, Trifiro MA, Pinsky L, Wellington CL, Salvesen GS, Hayden MR, Bredesen DE. Kennedy's disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J Neurochem. 1999b;72:185–195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- Escaffit F, Vaute O, Chevillard-Briet M, Segui B, Takami Y, Nakayama T, Trouche D. Cleavage and cytoplasmic relocalization of histone deacetylase 3 are important for apoptosis progression. Mol Cell Biol. 2007;27:554–567. doi: 10.1128/MCB.00869-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, Smith AC, Martinez RA, Fine GC, Grote SK, Ware CB, Einum DD, Morrison RS, Ptacek LJ, Sopher BL, La Spada AR. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous Purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–4905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmüller A, Picaud S, Zoghbi HY, Trottier Y, Tora L, Devys D. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg M, Duyckaerts C, Dürr A, Cancel G, Gourfinkel-An I, Damier P, Faucheux B, Trottier Y, Hirsch EC, Agid Y, Brice A. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet. 1998;7:913–918. doi: 10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Poirier MA, Liang Y, Pei Z, Weiskittel CE, Smith WW, DeFranco DB, Ross CA. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis. 2006;23:543–551. doi: 10.1016/j.nbd.2006.04.011. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Fu YH, Sopher BL, Libby RT, Wang X, Li LY, Einum DD, Huang J, Possin DE, Smith AC, Martinez RA, Koszdin KL, Treuting PM, Ware CB, Hurley JB, Ptácek LJ, Chen S. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. 2001;31:913–927. doi: 10.1016/s0896-6273(01)00422-6. [DOI] [PubMed] [Google Scholar]
- Latouche M, Lasbleiz C, Martin E, Monnier V, Debeir T, Mouatt-Prigent A, Muriel MP, Morel L, Ruberg M, Brice A, Stevanin G, Tricoire H. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J Neurosci. 2007;27:2483–2492. doi: 10.1523/JNEUROSCI.5453-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yokota T, Gama V, Yoshida T, Gomez JA, Ishikawa K, Sasaguri H, Cohen HY, Sinclair DA, Mizusawa H, Matsuyama S. Bax-inhibiting peptide protects cells from polyglutamine toxicity caused by Ku70 acetylation. Cell death and differentiation. 2007;14:2058–2067. doi: 10.1038/sj.cdd.4402219. [DOI] [PubMed] [Google Scholar]
- Lindblad K, Savontaus ML, Stevanin G, Holmberg M, Digre K, Zander C, Ehrsson H, David G, Benomar A, Nikoskelainen E, Trottier Y, Holmgren G, Ptacek LJ, Anttinen A, Brice A, Schalling M. An expanded CAG repeat sequence in spinocerebellar ataxia type 7. Genome Res. 1996;6:965–971. doi: 10.1101/gr.6.10.965. [DOI] [PubMed] [Google Scholar]
- Matilla A, Gorbea C, Einum DD, Townsend J, Michalik A, van Broeckhoven C, Jensen CC, Murphy KJ, Ptácek LJ, Fu YH. Association of ataxin-7 with the proteasome subunit S4 of the 19S regulatory complex. Hum Mol Genet. 2001;10:2821–2831. doi: 10.1093/hmg/10.24.2821. [DOI] [PubMed] [Google Scholar]
- McMahon SJ, Pray-Grant MG, Schieltz D, Yates JR, 3rd, Grant PA. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci U S A. 2005;102:8478–8482. doi: 10.1073/pnas.0503493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Li L, Chin LS. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem. 2008;15:47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, Chait BT, La Spada AR, Roeder RG. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A. 2005;102:8472–8477. doi: 10.1073/pnas.0503505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12:3231–3244. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- Riley BE, Zoghbi HY, Orr HT. SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal. J Biol Chem. 2005;280:21942–21948. doi: 10.1074/jbc.M501677200. [DOI] [PubMed] [Google Scholar]
- Ross CA. Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron. 1997;19:1147–1150. doi: 10.1016/s0896-6273(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Loeffler JP, Boutillier AL. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol. 2004;68:1157–1164. doi: 10.1016/j.bcp.2004.05.035. [DOI] [PubMed] [Google Scholar]
- Sanders SL, Jennings J, Canutescu A, Link AJ, Weil PA. Proteomics of the eukaryotic transcription machinery: identification of proteins associated with components of yeast TFIID by multidimensional mass spectrometry. Mol Cell Biol. 2002;22:4723–4738. doi: 10.1128/MCB.22.13.4723-4738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- Stankovic-Valentin N, Deltour S, Seeler J, Pinte S, Vergoten G, Guérardel C, Dejean A, Leprince D. An acetylation/deacetylation-SUMOylation switch through a phylogenetically conserved psiKXEP motif in the tumor suppressor HIC1 regulates transcriptional repression activity. Mol Cell Biol. 2007;27:2661–2675. doi: 10.1128/MCB.01098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Ström AL, Forsgren L, Holmberg M. A role for both wild-type and expanded ataxin-7 in transcriptional regulation. Neurobiol Dis. 2005;20:646–655. doi: 10.1016/j.nbd.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Terashima T, Kawai H, Fujitani M, Maeda K, Yasuda H. SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. Neuroreport. 2002;13:2359–2364. doi: 10.1097/00001756-200212030-00038. [DOI] [PubMed] [Google Scholar]
- Thomas M, Dadgar N, Aphale A, Harrell JM, Kunkel R, Pratt WB, Lieberman AP. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem. 2004;279:8389–8395. doi: 10.1074/jbc.M311761200. [DOI] [PubMed] [Google Scholar]
- Tydlacka S, Wang CE, Wang X, Li S, Li XJ. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008;28:13285–13295. doi: 10.1523/JNEUROSCI.4393-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, He CY, Chou AH, Yeh TH, Chen YL, Li AH. Polyglutamine-expanded ataxin-7 decreases nuclear translocation of NF-kappaB p65 and impairs NF-kappaB activity by inhibiting proteasome activity of cerebellar neurons. Cell Signal. 2007;19:573–581. doi: 10.1016/j.cellsig.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, Loubser O, van Raamsdonk J, Singaraja R, Yang YZ, Gafni J, Bredesen D, Hersch SM, Leavitt BR, Roy S, Nicholson DW, Hayden MR. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J Neurosci. 2002;22:7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Gouw L, Propp S, Sopher BL, Taylor J, Lin A, Hermel E, Logvinova A, Chen SF, Chen S, Bredesen DE, Truant R, Ptacek LJ, La Spada AR, Ellerby LM. Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity and transcriptional dysregulation. J Biol Chem. 2007;282:30150–30160. doi: 10.1074/jbc.M705265200. [DOI] [PubMed] [Google Scholar]
- Young JE, Martinez RA, La Spada AR. Nutrient deprivation induces neuronal autophagy, and implicates reduced insulin signaling in neuroprotective autophagy activation. J Biol Chem. 2009;284:2363–2373. doi: 10.1074/jbc.M806088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander C, Takahashi J, El Hachimi KH, Fujigasaki H, Albanese V, Lebre AS, Stevanin G, Duyckaerts C, Brice A. Similarities between spinocerebellar ataxia type 7 (SCA7) cell models and human brain: proteins recruited in inclusions and activation of caspase-3. Hum Mol Genet. 2001;10:2569–2579. doi: 10.1093/hmg/10.22.2569. [DOI] [PubMed] [Google Scholar]
- Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, Li SH, Li XJ. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003;163:109–118. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]